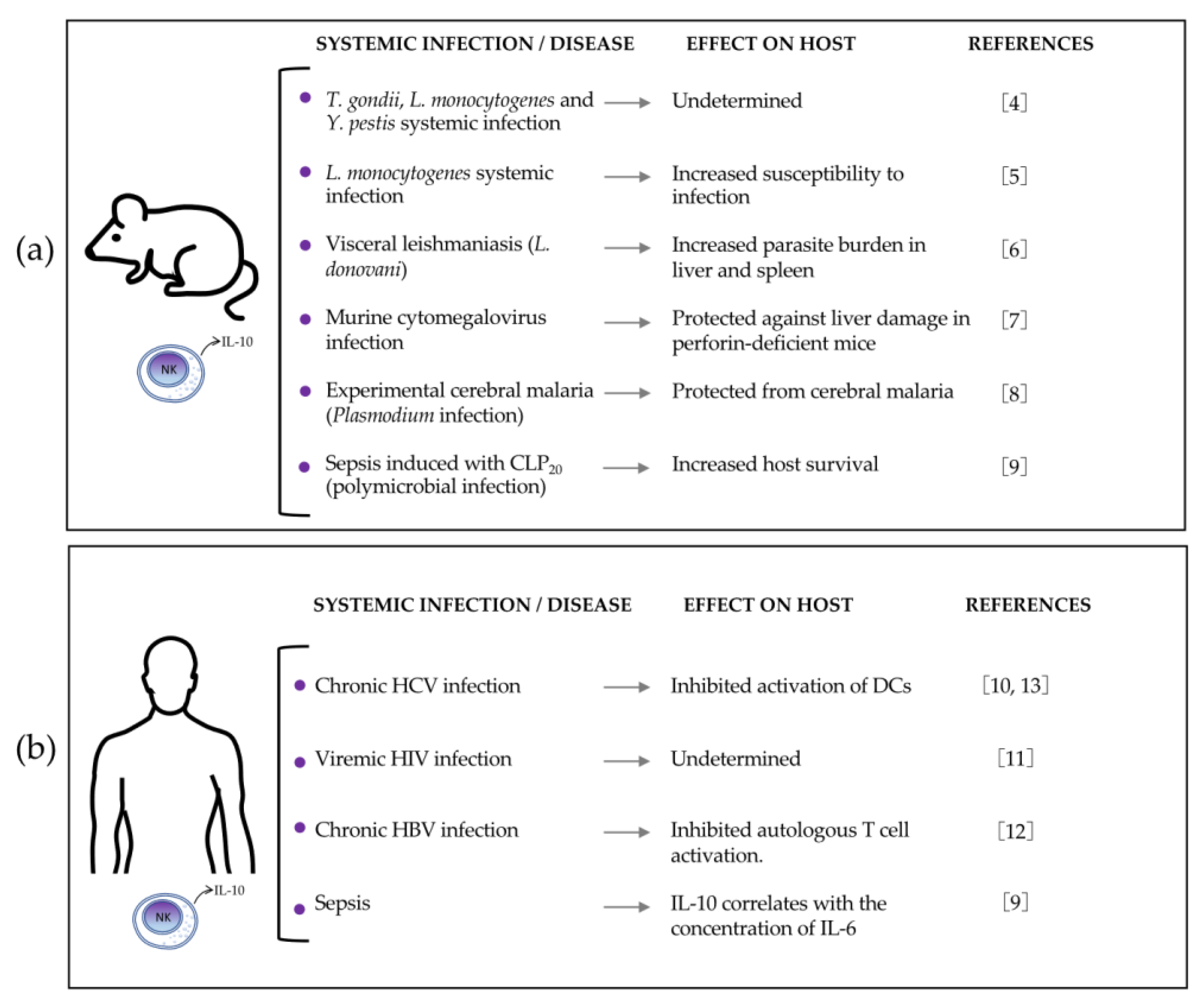
2.1. IL-10-Producing NK Cells during Listeria Monocytogenes (Lm) Systemic Infection
Lm is a Gram-positive intracellular bacterial pathogen that often contaminates food and causes infection, with different levels of severity. Most people who ingest the contaminated food develop mild gastrointestinal symptoms; however, in some individuals, Lm causes severe systemic infection and is propagated to peripheral organs (listeriosis), which often proves fatal. Although the incidence of severe listeriosis is low, Lm infections are a public health issue, owing to the frequency of outbreaks associated with contaminated food. The factors involved in the increased susceptibility of some individuals to severe infections are poorly understood, but systemic infections are mostly reported in the elderly, pregnant women, and immunocompromised individuals [
14,
15,
16]. Most of the knowledge regarding pathogenesis and immune response to Lm has been obtained from the murine model of listeriosis. In this animal model, systemic infection with Lm leads to an innate immune response characterized by a cytotoxic T lymphocyte response, the formation of Th1 effector cells, and the production of the pro-inflammatory cytokine IFNγ by activated NK cells and memory T cells [
17,
18,
19]. However, Perona-Wright et al. [
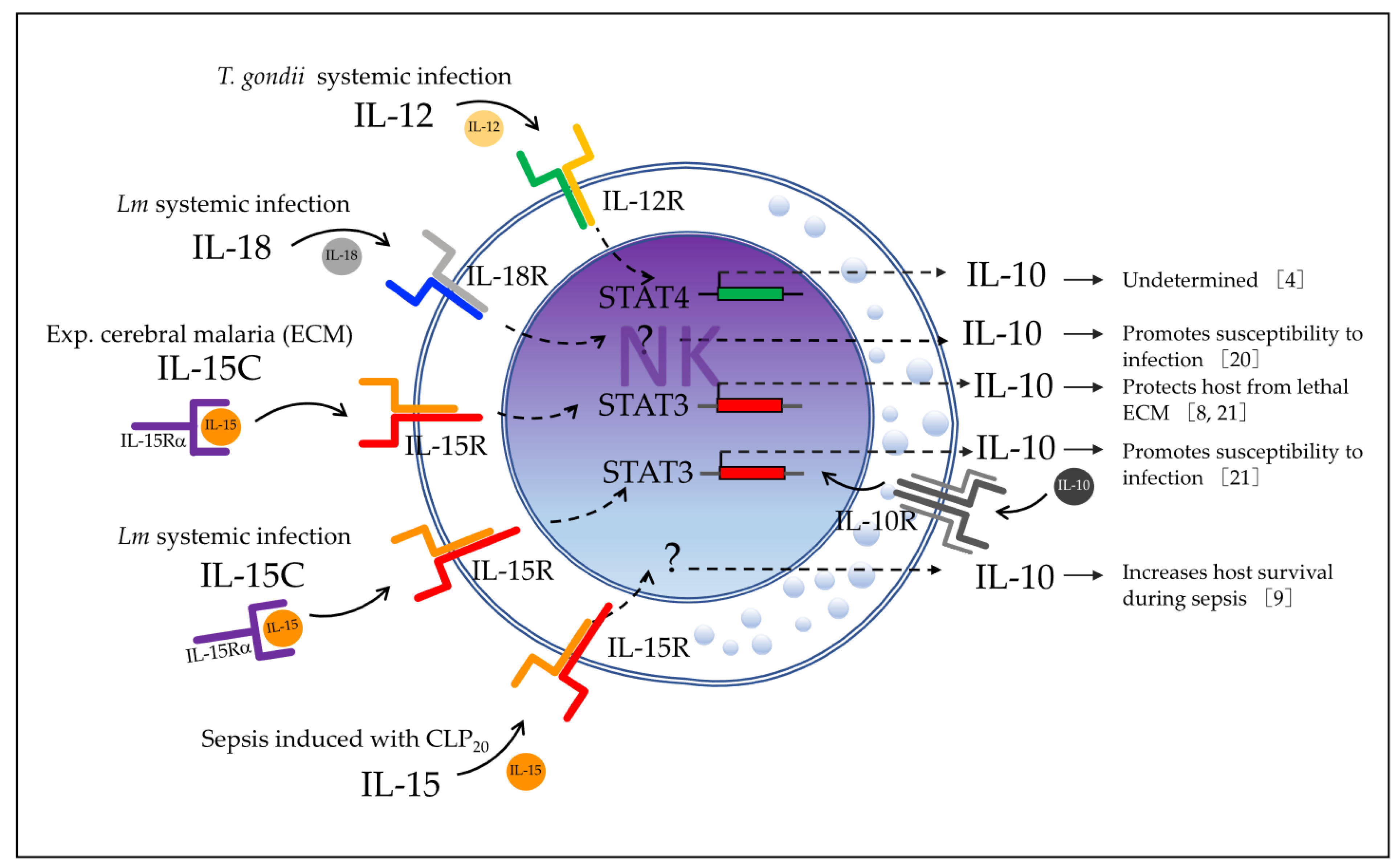
4] showed in 2009 that most circulating NK cells produce the anti-inflammatory cytokine IL-10 during systemic Lm infection. Using
Il10-eGFP-reporter mice (VertX, with genetic background C57BL/6J), the authors observed that 79% of blood NK cells became GFP-positive during acute infection with this bacterium.
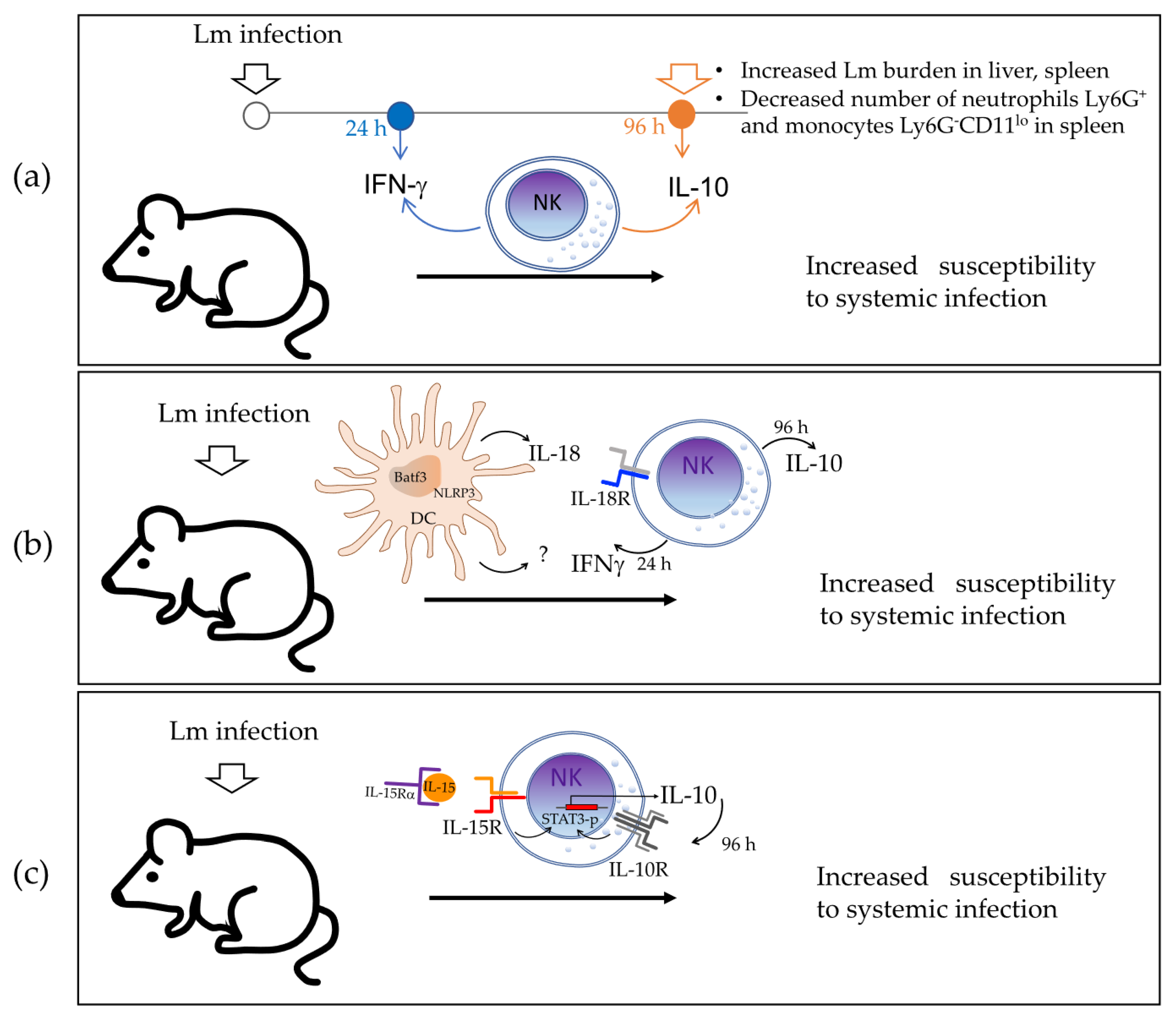
Later in 2016, Clark et al. [
5] found that NK cells produce IFNγ during the early phase of infection but rapidly switch to IL-10 production. Their results also suggested that IL-10 produced by NK cells increases susceptibility to systemic bacterium infection by reducing the number of Ly6G
+ neutrophils and Ly6G
−CD11
lo monocytes in the spleen of infected mice (
Figure 2a). They assessed IL-10 production using the tiger IL-10 GFP-reporter mouse model (where the 3′UTR of IL-10 remains unaltered) and found that GFP staining was selectively increased in NK cells from the spleen, liver, and blood of Lm-infected mice at 72 h post-infection, whereas the IL-10 GFP-reporter expression was not observed at 24 h post-infection. Intracellular staining for IFNγ showed that most NK cells indeed produce this pro-inflammatory cytokine at 24 h post-infection, but its levels declined by 96 h, which suggests the time-regulated dual function of NK cells during Lm systemic infection. The authors demonstrated that NK cells were the main source of IL-10 by performing adoptive transfer experiments in CD45.2
+ or CD45.1
+ B6.
il10−/− recipients that were infected with Lm and then received NK cells transferred from wild-type (wt) CD45.1 (B6) or from IL-10-deficient CD45.2 (B6.
il10−/−) mice. Results indicated that protein lysates of splenocytes from the B6.
il10−/− recipients of NK cells from wt but not from IL-10-deficient mice contained IL-10 protein at 96 h post-infection. In addition, at 96 h post-infection, the B6.
il10−/− mice that received wt NK cells showed an increased Lm burden in the liver and spleen (10- to 100-fold), compared with those that received IL-10-deficient NK cells, suggesting that IL-10 produced by NK cells increases susceptibility to systemic bacterium infection. The authors suggested that IL-10-producing NK cells may suppress the accumulation and activation of inflammatory myeloid cells based on their finding of an increased number of neutrophils Ly6G
+ and monocytes Ly6G
−CD11
lo in the spleen of infected B6.
il10−/− recipients, which was reduced when mice were transferred with wt NK cells capable of producing IL-10.
Clark et al. [
20] reported in 2018 that the cytokine IL-18 promotes IL-10 production by NK cells in the murine model of systemic Lm infection, which is dependent on the expression of NOD-, LRR-, and pyrin domain-containing protein 3 (
Nlrp)3 and the basic leucine zipper transcriptional factor ATF-like 3 (
Batf3) by dendritic cells (DCs). The authors used in vivo models of Lm-infected B6.
Il18−/−, B6.
Il18r−/−, B6.
Nlrp3−/−, and B6.
Batf3−/− mice in which they found almost undetectable serum IL-10 and reduced Lm burdens in the spleen and liver compared to B6 mice at 72 h post-infection, showing the relevance of IL-18, IL-18R1, NLRP3, and Batf3 for IL-10 production and resistance to Lm systemic infection. In addition, compared with B6 mice, Lm-infected B6.
Il18−/−, B6.
Nlrp3−/−, and B6.
Batf3−/− mice showed reduced levels of serum IFNγ without any effect on the bacterial burden at 24 h post-infection, demonstrating that these factors also regulate IFNγ production during Lm infection. In their study, the authors assumed that NK cells were the main source of IL-10, based on their previous published work [
5].
In that study, the levels of serum IL-18 were also found to be increased in parallel with those of serum IL-10 in Lm-infected B6 mice at 72 h post-infection, whereas B6.Nlrp3−/− mice had significantly reduced serum IL-18, which indicated that NLRP3 expression is relevant for IL-18 production. The source of IL-18 was then determined to be DCs through in vitro experiments, where Lm-infected bone-marrow-derived DCs (BMDCs) produced IL-18, whereas B6.Nlrp3−/− BMDCs failed to release IL-18. The authors acknowledged that there are diverse cell types that may produce IL-18 that were not additionally tested. Evidence showed that supernatants from Lm-infected B6 BMDCs promoted NK cell IL-10 production, whereas supernatants from B6.Il18−/−, B6.Nlrp3−/−, and B6.Batf3−/− BMDCs did not. Recombinant IL-18 restored the ability of supernatants from B6.Il18−/− BMDCs to promote NK cell IL-10 production. However, the addition of recombinant IL-18 alone did not induce NK cell IL-10 production in the absence of conditioned BMDC supernatants. These data demonstrate that BMDC release of IL-18 together with an additional factor (or factors) elicit IL-10 production by NK cells. Any additional factors are not dependent on NLRP3 because the B6.Nlrp3−/− BMDC supernatants were able to promote IL-10 production by NK cells when recombinant IL-18 was added to the culture.
These data also verify that IL-18R is required by NK cells to produce IL-10 through in vitro experiments with B6.
Il10−/− BMDCs infected with Lm and cultured with purified splenic B6 or B6.
Il18r1−/− NK cells. In contrast to co-cultures with B6 NK cells, no IL-10 was produced in cultures containing only B6.
Il18r1−/− NK cells. In summary, IL-18 produced by
Nlrp3+Batf3
+ DCs, together with another DC factor(s), promotes IL-10 production by NK cells in Lm systemic infection (
Figure 2b).
In 2019, Clark et al. [
21] investigated which other factor(s) may be required for IL-10 production by NK cells during Lm systemic infection. Their new approach consisted of a model of conditionally mutant mice lacking Stat3, IL-15Rα, or IL-10Rα selectively in NK cells (NKSTAT3
−, NKIl10R
−, and NK15Rα
− mice, respectively) to investigate the requirements for NK cell IL-10 production during both Lm infection and IL-15 and IL-15Rα complex (IL-15C) treatment.
First, they showed that STAT3 activation was associated with IL-10 production by NK cells during Lm infection by detecting phosphorylated STAT3 (p-STAT3) in the lysates of NK cells purified from Lm-infected B6 mice at both 24 h and 72 h post-infection. However, the amount of p-STAT3 at 24 h was similar in NK cells from B6 and B6.Il10−/− mice, indicating that STAT3 activation occurs in NK cells at 24 h and 72 h post-infection, but that early activation is independent of IL-10 production. Using Lm-infected B6, NKSTAT3−, and B6.Il10−/− mice, they then showed that intrinsic STAT3 is required for the IL-10 NK cell response during systemic Lm infection. They found that serum IL-10 levels were increased at 72 h post-infection in infected B6 mice but not in infected NKSTAT3− and B6.Il10−/− mice, whereas the Lm burdens in the liver and spleen of NKSTAT3− and B6.Il10−/− mice were significantly reduced. The recruitment of inflammatory myeloid cells to the spleens of Lm-infected mice was increased in infected NKSTAT3− and B6.Il10−/− mice at 72 h post-infection, which correlates with improved resistance to Lm systemic infection.
Because IL-15C treatment induces IL-10 production by NK cells with a similar kinetics to the one observed during Lm infection, the authors used this model for tests. They found that the expression of IL-10 transcripts was significantly reduced in NK cells from NKSTAT3− compared with B6 mice treated with IL-15C, suggesting that intrinsic STAT3 is also required for the NK cell IL-10 response induced by IL-15C treatment. The authors then tested whether IL-15 directly affects the IL-10 production by NK cells using NK15Rα− mice. They found that levels of serum IL-10 and Lm burdens in the liver and spleen were reduced in Lm-infected NK15Rα− compared with B6 mice, indicating the relevance of the IL-15 receptor to IL-10 production by NK cells. Moreover, NK cells from NK15Rα− and B6 mice had similar amounts of total STAT3, but the amount of p-STAT3 was significantly reduced in the IL-15Rα-deficient NK cells at 72 h post-infection, which indicated that this receptor contributes to the STAT3 activation. Together, the data indicate that IL-15Rα contributes to STAT3 activation and IL-10 production in NK cells during Lm infection.
Finally, the authors examined STAT3 activation, IL-10 production, Lm burden, and inflammatory myeloid cell accumulation in the organs of IL-10Rα deficient-NK cells (NKIl10R
−) mice at 72 h post-infection with Lm. They found an expected high increase in p-STAT3 in the infected B6 mice, but the p-STAT3 response was weak in NK cells from NKIl10R
− mice at 72 h post-infection. Serum IL-10 and Lm burdens in the spleen and liver were significantly reduced, whereas more inflammatory myeloid cells accumulated in the spleens of NKIl10R
− mice compared with B6 mice at 72 h post-infection. These data indicated that expression of IL-10Rα by NK cells is critical for boost STAT3 activation up to 72 h post-infection and to stimulate IL-10 production in NK cells during Lm infection. In summary, evidence indicates that IL-15 signaling induces early STAT3 activation in NK cells, which is required for the IL-10 production response to Lm infection, which in turn feeds back through IL-10R on NK cells to promote sustained STAT3 activation and IL-10 production by NK cells observed at 72 h post-infection (
Figure 2c). This sustained IL-10 production by NK cells is associated with increased susceptibility to Lm systemic infection.
2.2. IL-10-Producing NK Cells during Experimental Visceral Leishmaniasis
Leishmaniasis comprises a group of diseases caused by an intracellular protozoan of the genus
Leishmania that are transmitted by the bite of phlebotomine sandflies. Leishmaniasis is a major public health issue, with 350 million people living at risk of developing the disease in 98 countries and with an estimated 20,000–50,000 deaths annually worldwide [
22,
23]. There are three major forms of the disease, namely, cutaneous, mucocutaneous, and visceral, all of which depend on the infective
Leishmania species and the host immune response. Visceral leishmaniasis is the more severe form of the disease. This is characterized by parasite dissemination to internal organs, such as the liver, spleen, and bone marrow, with a frequently fatal prognosis without adequate treatment. The causal species for visceral leishmaniasis are
L. donovani and
L. infantum/chagasi [
22,
23]. It is known that protective immune response against
L. donovani infection consists of a granulomatous inflammatory response that restrains the parasite and is positively regulated by cytokines produced by both Th1 and Th2 and negatively regulated by IL-10 produced by Treg cells [
22,
23,
24,
25,
26].
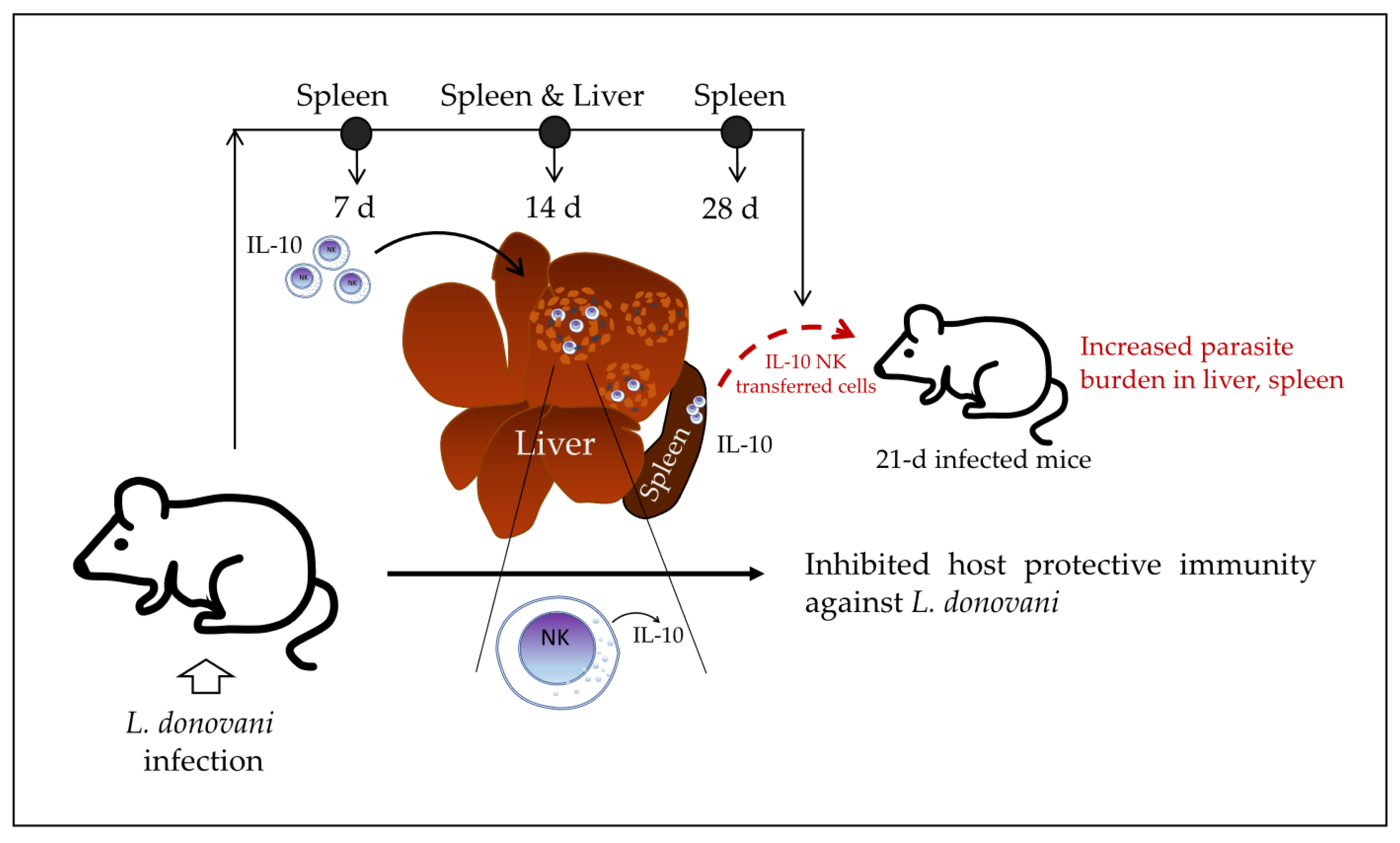
In 2008, Maroof et al. [
6] investigated the immunoregulatory role of NK cells in the murine model of experimental visceral leishmaniasis. They reported that IL-10-producing NK cells migrate into the spleen and hepatic granulomas of
L. donovani-infected mice and suppress host resistance to disseminated parasitosis, which is dependent on IL-10 production. Similar to what was found in the Lm systemic infection model, NK cells are able to produce IL-10 in the early infection phase, but sustained IL-10 production at the late phase of the infection is associated with the increased susceptibility of the host to the systemic infection. In the BALB/c mouse model of visceral leishmaniasis, the presence of liver granulomas is observed at 14–28 days post-infection. At 28 days post-infection, hepatic resistance becomes detectable (reduction of parasite burden in liver), whereas parasite numbers in the spleen are increasing. The authors first found that NK cells were the main source of IL-10 (mRNA) at 14 days post-infection with
L. donovani by analyzing the purified leucocyte population from the spleen of infected mice, whereas T CD4
+ and NK cells were the main sources of IL-10 production at 28 days post-infection. Infected mice were also found to have an accumulation of NK cells expressing IL-10 mRNA within immature and mature granulomas and within liver parenchyma. NK cells with high levels of IL-10 mRNA were also detected in the spleen and liver of infected mice by day 7 post-infection, indicating an early IL-10 production during
L. donovani infection.
To elucidate whether IL-10 produced by NK cells may affect the outcome of the infection, the authors generated CD45.1 B6.IL-10
+/+ and CD45.2 B6.IL-10
−/− mice, infected them with
L. donovani, and then purified splenic NK cells by sorting at 28 days post-infection. The purified NK cells were then transferred into 21-day-infected CD45.1 B6.IL-10
+/+ recipient mice. They found that the transferred IL-10
+/+ NK cells increased the parasite burden in the spleen and liver of recipient mice, whereas the transferred IL-10
−/− NK cells did not have an effect on the parasite burden of recipient mice compared with control mice without transferred cells. The authors suggested that transferred NK cells with the ability to produce IL-10 may suppress hepatic resistance to
L. donovani in the infected recipient mice; however, the impact of the IL-10-producing NK cells from the recipient mice themselves cannot be tested in this model. Perhaps a model of IL-10
−/− or NK-depleted NK cell recipient mice should be used to test the specific impact of the transferred IL-10
+/+ NK cells on parasite burden. Together, the data indicate that NK cells produce IL-10 at early stages of visceral leishmaniasis in mice and continue producing IL-10 at a higher concentration over the course of infection while migrating into granulomas in the liver of infected mice. At late stages of infection, these IL-10-producing NK cells may induce an increased parasite burden in 21-day-infected recipient mice, which suggests that they inhibit host protective immunity against
L. donovani (
Figure 3).
2.3. IL-10-Producing NK Cells in Experimental Cerebral Malaria (ECM)
In humans, cerebral malaria is a severe complication of
Plasmodium falciparum infection, leading to a high mortality rate in patients with malaria, mainly young children [
27]. The pathogenesis of cerebral malaria is not completely understood, but a consistent characteristic is the sequestration of infected erythrocytes within cerebral blood vessels, which may lead to endothelial damage, disruption of the vessel wall, myelin and axonal damage, and breakdown of the blood–brain barrier [
28]. There have been several strategies to elucidate the mechanisms underlying the pathogenesis of cerebral malaria, such as the analysis of clinical case series and case–control studies, post-mortem analysis, and experimental murine models [
29]. ECM is an accepted murine model, where several events similar to those occurring in human cerebral malaria are replicated when susceptible mouse strains, such as C57BL/6, are infected with
Plasmodium berghei ANKA (PbA) [
30].
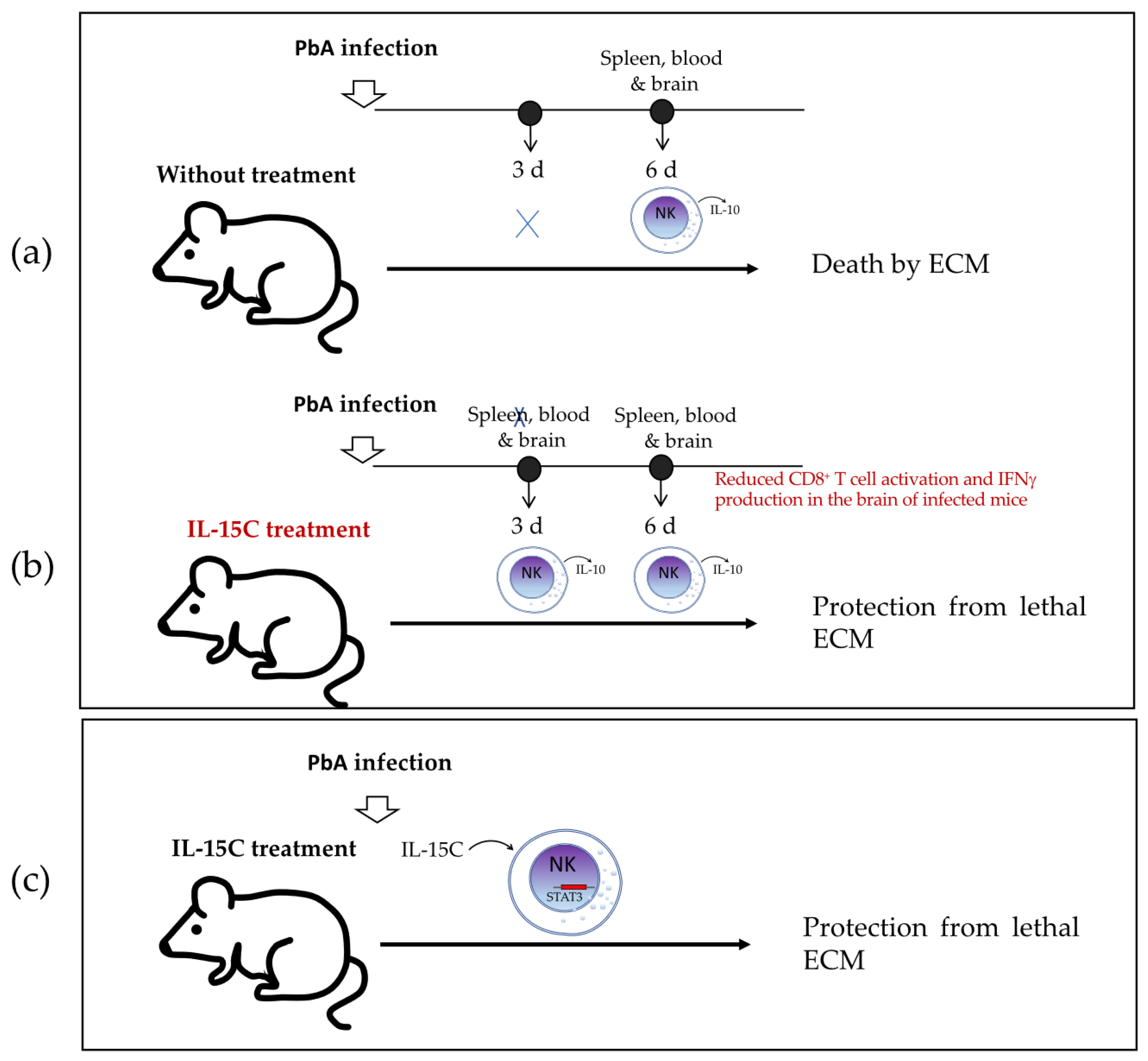
Burrack et al. [
8] investigated in 2018 whether cytokine/cytokine receptor complex therapy (with IL-15/IL-15R or IL-2/IL-2R) modulates the immune cell activation in ECM. They reported that only IL-15 complex therapy (IL-15C) prevents mice from death by ECM in PbA-infected C57BL/6 mice, which occurs through stimulation of NK cells to produce IL-10. They first treated C57BL/6 mice with IL-15C or IL-2C prior to PbA infection and found that prophylactic treatment with IL-15C, but not IL-2C, prevented lethal ECM in most of the infected mice. They then used
Il10-eGFP-reporter mice (VertX, with genetic background C57BL/6J) to determine that on day 3 in the spleen, blood, and brain of infected mice following IL-15C treatment, the cell population within IL-10
GFP+ cells consisted of a majority of NK cells. IL-10
GFP+ NK cells were also the main population in the spleen, blood, and brain of untreated mice but only until day 6 post-infection, when there was death from ECM, suggesting that IL-15C treatment induces early IL-10 production that may be related to the protective effect. Furthermore, they demonstrated that IL-10 was necessary for IL-15C-mediated protection from ECM by comparing
Il10−/− mice with wild-type (wt) mice, the latter of which were protected from PbA infection-induced ECM after IL-15C treatment, whereas
Il10−/− mice were not. Finally, the authors investigated whether IL-10 production by NK cells is essential for the IL-15C-mediated protection by transferring NK cells from IL-15C-treated wt or
Il10−/− mice into wt recipients on day 2 post-PbA infection; they found that the transfer of wt NK cells protected against ECM, while most PbA-infected mice receiving
Il10−/− NK cells died, similar to untreated controls. Because NK cell adoptive transfer had no effect on parasitemia levels, they suggested that the protective role of IL-10 produced by NK cells was related to the development of cerebral malaria but not to the control of the infection itself. In summary, the findings of the study indicated that NK cells produce IL-10 during ECM in mice infected with PbA, but that this induction occurs too late to prevent the fatal outcome (
Figure 4a), whereas the treatment with IL-15C promotes early IL-10 production by NK cells that is sufficient to protect mice from the fatal outcome (
Figure 4b).
In addition, the authors investigated the effect on CD8
+ T-cell activation of treatment with IL-15C, which migrates into the brain during the development of ECM. They used flow cytometry,
Nr4a1GFP reporter mice, GAP50
41-48/D
b tetramers, and
IfngYFP reporter mice to evaluate activation of antigen-specific CD8
+ T cells.
Nr4a1 encodes Nur77, which is expressed in direct proportion to the strength of TCR activation; GAP50 is the glideosome-associated protein 50 from
Plasmodium that functions as a highly immunogenic CD8
+ TCR epitope in C57BL/6J mice; and IFNγ is associated with ECM immunopathology. They found that the number of Nr4a1
GFP+ GAP50-specific CD8
+ T cells was significantly reduced in the brains of IL-15C-treated mice (6 days post-infection) compared with untreated mice. Using the
IfngYFP reporter mice, they found that IL-15C-treated mice had significantly reduced frequencies of
IfngYFP+ total and GAP50-specific CD8
+ T cells and reduced
IfngYFP mean fluorescence intensity (MFI) compared with untreated mice, although the total number of
IfngYFP+ cells was not different between the groups. These data indicate that IL-15C treatment reduces CD8
+ T-cell activation and IFNγ production in the brain of infected mice, although it is not known whether the NK cells that produce IL-10 have any direct role in this reduced T-cell activation (
Figure 4b).
Clark et al. [
21], in addition to investigating the factors required for NK cell IL-10 production in Lm infection as reviewed in
Section 2.1, also evaluated the impact of NK cell intrinsic STAT3 on the protective effect of IL-15C treatment in ECM. They confirmed that IL-15C treatment protects mice against lethal ECM and found that STAT3-deficient NK cells (NKSTAT3
−) mice succumbed to ECM regardless of receiving IL-15C treatment. These findings indicate that NK cell STAT3 is required for the survival-promoting effects of IL-15C treatment in ECM (
Figure 4c).
2.4. IL-10-Producing NK Cells during Murine Cytomegalovirus (MCMV) Infection
The human cytomegalovirus (HCMV) is a double-stranded DNA beta-herpesvirus with a high prevalence in the general population. After the establishment of infection, HCMV becomes latent and persists throughout the lifetimes of healthy asymptomatic persons. The reactivation of latent HCMV is usually asymptomatic but results in the production of infectious virions that are transmitted via the saliva, urine, and other body fluids. During active infection of immunocompetent individuals, HCMV is found in the salivary glands, blood, kidneys, and liver. In normal individuals, subclinical hepatitis is frequently associated with HCMV infection, which may be caused by the host inflammatory response or by direct virus cytopathogenicity [
31]. Although HCMV infection is usually controlled by the immune system in healthy individuals, it can be life-threatening for immunocompromised patients, such as transplant recipients, AIDS individuals, and neonates [
31,
32,
33].
Mouse animal models of MCMV infection have been used to investigate the pathogenesis of HCMV and the role of the immune response components because they share several common features. During MCMV infection, the virus replicates in organs such as the spleen and liver, whereas NK cells and macrophages are recruited in the early phases of infection, and pro-inflammatory INFγ production by NK cells is known to function in containing MCMV infection spread [
34,
35,
36]. CD4
+ T cells and CD8
+ T cells are subsequently recruited into the infected organs to participate in the pathogen clearance [
37,
38]. This protective immune response must be regulated to avoid damage to organs, such as the liver [
36,
39]. Ali et al. [
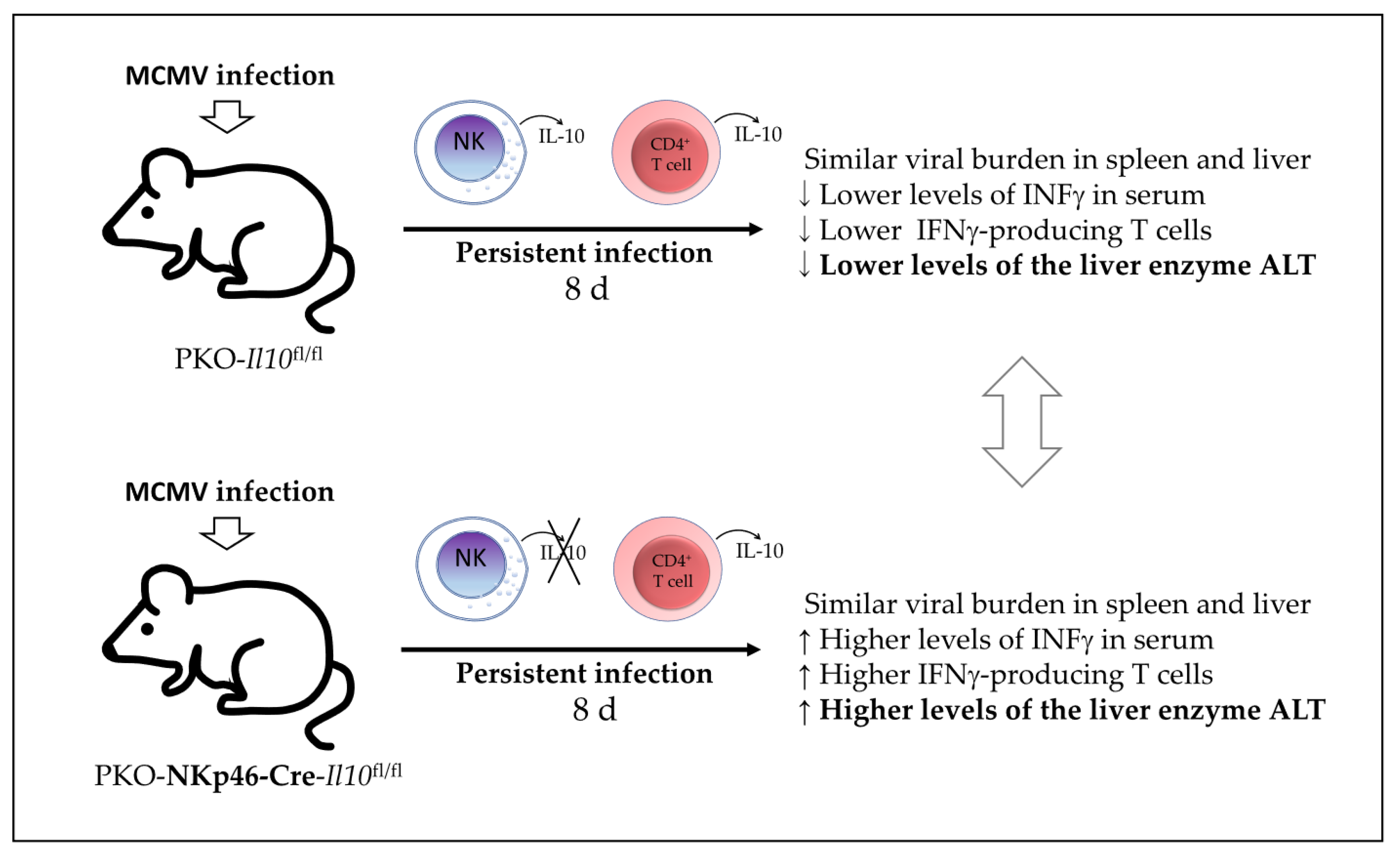
7] investigated in 2019 whether IL-10-producing NK cell have a dual role in the regulation of inflammatory responses during MCMV infection. Using a model of IL-10 GFP-reporter mice, they first found that NK cells in the peripheral blood are the major producers of IL-10 GFP during the early stages of MCMV infection, with a peak observed on day 4 post-infection. Using an NK cell-specific IL-10-deficient mouse (NKp46-Cre-Il10
fl/fl) and their control Il10
fl/fl mice, they found that the presence of NK cell-derived IL-10 did not affect the viral clearance in the spleen, liver damage, and the production of IFNγ by T cells in the spleen of infected mice. Although the authors were able to eliminate IL-10-producing NK cells in the NKp46-Cre-Il10
fl/fl mice, the levels of IL-10 were not altered in the serum at day 4 post-infection, indicating another source of IL-10 in this model during acute MCMV infection. Therefore, during acute MCMV infection in these mice, IL-10 production by NK cells is not required for pathogen clearance nor control of the IFNγ-mediated T-cell response. However, the authors tested another model by producing PKO-IL-10 GFP mice and PKO-NKp46-Cre-Il10
fl/fl mice and found that perforin-deficient (PKO) mice could not clear the MCMV-infected cells efficiently and subsequently suffered from persistent infection. In this PKO murine model of persistent infection, they found that NK cells and CD4
+ T cells produced high levels of IL-10 GFP for a longer time (until day 8 post-infection). Meanwhile, infected PKO-NKp46-Cre-Il10
fl/fl showed an overall reduction in the serum IL-10 levels and increased serum INFγ levels and liver damage (as determined by increased levels of the enzyme alanine aminotransferase or ALT) compared with their infected control littermates PKOIl10
fl/fl, although they had similar virus titers in their spleens and livers. Therefore, the authors reported that NK cell-derived IL-10 prevents liver damage and potentially regulates INFγ production in a model of perforin-deficient mice suffering from persistent MCMV infection (
Figure 5). However, the potential participation of CD4
+ T-cell-derived IL-10 in this MCMV infection model was not discussed in this study.
2.5. IL-10-Producing NK Cells during Sepsis
The Third International Consensus Definition for Sepsis defines sepsis as a life-threatening organ dysfunction caused by dysregulated host response to infection [
40]. Sepsis is a relevant public health problem with a high incidence and is one of the leading causes of death worldwide [
41,
42]. Sepsis comprises complex immune and physiological responses to systemic infection that are still poorly understood and can lead to organ dysfunction and a high risk of death [
43]. It is known that dysfunctional innate and adaptive immunity simultaneously elicit inflammatory and anti-inflammatory responses that can exist synchronously, and the sustained effect leads to organ injury [
44]. Over 100 clinical trials targeting the pro-inflammatory component of the cytokine storm in sepsis have failed, indicating that targeting only the pro-inflammatory components is not the right approach for addressing the dualistic/synchronous pro- and anti-inflammatory response in sepsis [
45]. NK cells are known for their classic pro-inflammatory function in sepsis, but their dual role as immunoregulatory factors in sepsis has been poorly explored.
Jensen et al. [
9] in 2021 investigated the role of IL-10-producing NK cells using a cecal ligation and puncture (CLP) model of sepsis induction. They hypothesized that NK cells may have a dual role as a classic pro-inflammatory and as a novel anti-inflammatory effector during the biphasic inflammatory states of sepsis. The authors used a CLP model where the severity of the septic events was modulated by varying the number of cecal punctures: CLP
50 (two punctures) and CLP
20 (one puncture) were performed to evaluate mortality and morbidity in high and low disease severity, respectively. They first depleted the total NK cells from mice using anti-NK1.1-depleting antibodies (depletion that lasted up to 10 days) and found that NK-depleted animals who underwent CLP
50 had increased mortality and an increased scope of 28 plasma cytokines, compared with NK-replete mice. In addition, the concentration of the three cytokines associated with sepsis severity (IL-6, IL-1β, and IFNγ) was maintained in NK-depleted mice over an extended duration without any increases in magnitude, suggesting a NK cell-limiting effect on the duration and scope of the cytokine storm. Because the authors found an elevated IL-15 concentration in the plasma of NK-depleted mice, they tested the effect of IL-15 on splenocyte cultures, which showed increased IFNγ production during the first 24 h and a subsequent increased IL-10 production by NK cells after 48 h of culture, indicating a time-regulated dual response to IL-15. They then showed that the NK cells of mice that underwent CL
50 and CLP
20 had expressed IL-10 mRNA.
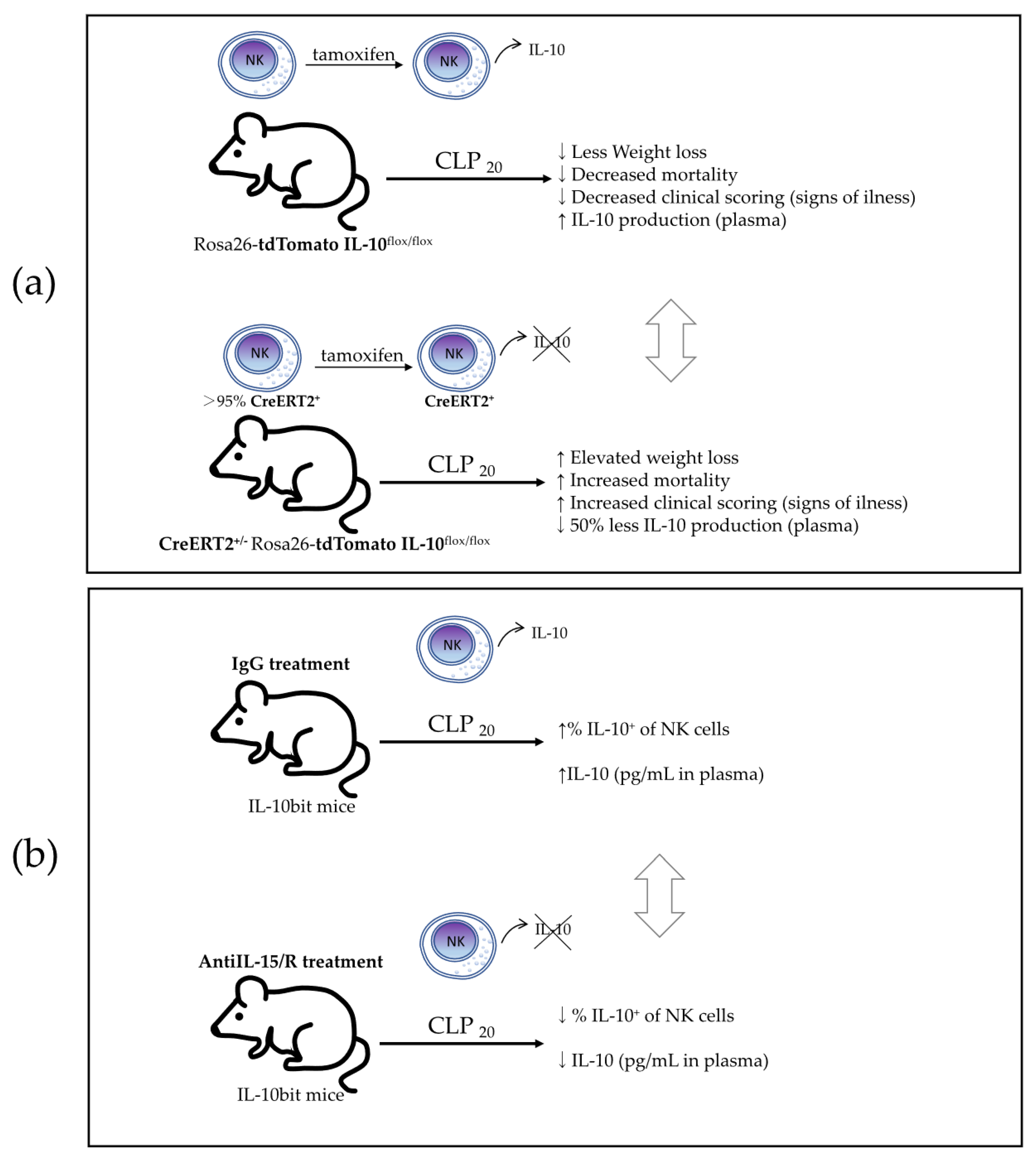
The authors finally proved that NK cell-derived IL-10 is critical for host survival during sepsis using a model of NCR1-CreERT2
+/2 Rosa26-tdTomato IL-10
flox/flox mice along with Rosa26-tdTomato IL-10
flox/flox littermates that were treated with tamoxifen to knock out IL-10 expression in NK cells expressing the CreERT2 (
Figure 6a). Mice whose NK cells lacked IL-10 (NCR1-CreERT2
+/2 Rosa26-tdTomato IL-10
flox/flox) had elevated weight loss, increased mortality, and prolonged signs of illness after undergoing CLP
20-induced sepsis. Surprisingly, they did not test any cytokines in this model to verify that NK cell-derived IL-10 limits the scope and duration of the cytokine storm, as suggested in their previous study using a model of total NK-depleted mice. Additionally, the authors showed that NK cells produce IL-10 in an IL-15-dependent manner by using IL-10bit mice (which express Thy1.1 during active IL-10 transcription) treated with either anti-IL-15/R-blocking antibodies or control IgG. In the mice treated with anti-IL-15/R-blocking antibodies after they underwent CLP
20, they found a reduced frequency of IL-10-producing NK cells, but not a reduced frequency or number of total NK cells, and a reduced concentration of plasma IL-10 (
Figure 6b), indicating the necessity of IL-15 for IL-10 production by NK cells. To translate these findings to human disease, the authors showed an increased production of IL-10 by NK cells in septic patients, together with an elevated concentration of IL-10, IFNγ, and IL-6 in plasma, compared to healthy controls. They also found that the IL-10 concentration was positively correlated with the IL-6 concentration and negatively correlated with the concentration of IFNγ, suggesting a potential regulation role of the pro-inflammatory signals. This work reported the production of the immunosuppressive cytokine IL-10 produced by NK cells in a murine model of sepsis and in human sepsis disease, revealing a potential immunoregulatory mechanism of NK cells during sepsis that may have a crucial role in the survival of the host.
2.6. IL-10-Producing NK Cells during Lung Infections
The airways are one of the primary routes for pathogens to enter the lungs and body and therefore require an effective immune response to prevent pathogens from spreading. However, since the lungs are sensitive organs in which the essential exchange of breathing gases takes place, any inflammatory response must be tightly regulated in order to eliminate pathogens but prevent immune diseases and chronic inflammation [
46]. The lungs contain the second highest percentage of NK cells in the body, which play an important role in the inflammatory response to lung infections. NK cells are known to migrate from the blood into the lungs within hours of inflammatory signals and secrete pro-inflammatory cytokines, such as IFNγ, but this inflammatory response is rapidly controlled by IL-10 and transforming growth factor (TGFβ) produced by alveolar macrophages [
47]. However, it was unclear whether the NK cells resident in the lungs could play an immunoregulatory role through the production of IL-10.
Clark et al. [
48] showed in 2020 that IL-10 produced by NK cells in the lung has a negative immunoregulatory role during sublethal
Streptococcus pneumoniae (
S. pneumoniae) infection, promoting bacterial burden in the lung. First, they used tiger IL-10 GFP-reporter mice to determine that NK cells from the blood and lung, but not other lung cells, are the main source of IL-10 after 72 h of intranasal infection with sublethal doses of
S. pneumoniae. They then performed adoptive transfer experiments with CD45.2
+ or CD45.1
+ B6.
il10−/− recipients that were infected with
S. pneumoniae and then received NK cells from wild-type (wt) CD45.1 (B6) or from IL-10-deficient CD45.2 (B6.
il10−/−) mice. Donor NK cells were injected directly into the lungs of IL-10-deficient recipient mice infected with
S. pneumoniae at 24 h post-infection, and bacterial burdens, IL-10, and lung myeloid cell populations were analyzed at 96 h post-infection. Results indicated that compared with recipients of IL-10-deficient NK cells, the mice that received wt NK cells had increased bacterial burdens in the lung but not other systemic tissues. In addition, the total number of neutrophils and alveolar macrophages in mice that received IL-10-deficient NK cells was higher compared with wt NK cells, suggesting that IL-10 produced by NK cells in the lung limits the recruitment of myeloid cells to the lung.
On the other hand, Bortell et al. [
49] discovered in 2021 that
S. pneumoniae infection in the lung together with oral coinfection with Lm increase susceptibility to systemic Lm infection in a manner dependent on IL-10 production by NK cells. The IL-10-deficient NK cell Ncr1-iCre-Il10
fl/fl mouse strain and wt B6 mice were infected with Lm peroral (PO) or coinfected with
S. pneumoniae intratracheal (IT) + Lm PO, and the spleens, livers, and lungs were harvested at 3 days post-infection. Lm burdens in the spleens, livers, and lungs of coinfected Ncr1-iCre-Il10
fl/fl mice were significantly lower than in coinfected B6 mice, whereas Ncr1-iCre-Il10
fl/fl mice infected with only Lm PO did not show any difference compared with Lm PO-infected wt B6, indicating the necessity of coinfections. The author also found that upon co-infection, the Ncr1-iCre-Il10
fl/fl mice significantly increased the number of neutrophils in both spleen and lungs when compared to B6-coinfected mice.
Taken together, data from both studies indicated that NK cells are the main source of IL-10 in the lung during S. pneumoniae infection, which has a negative immunoregulatory role, promoting susceptibility to S. pneumoniae dissemination in the lung and oral Lm coinfections.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







