
Acetyltransferases GCN5 and PCAF Are Required for B Lymphocyte Maturation in Mice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Flow Cytometry
2.3. Class Switch Recombination
2.4. Western Blot
2.5. Statistics
3. Results
3.1. Generation of Mice Lacking GCN5 and PCAF in B Cells
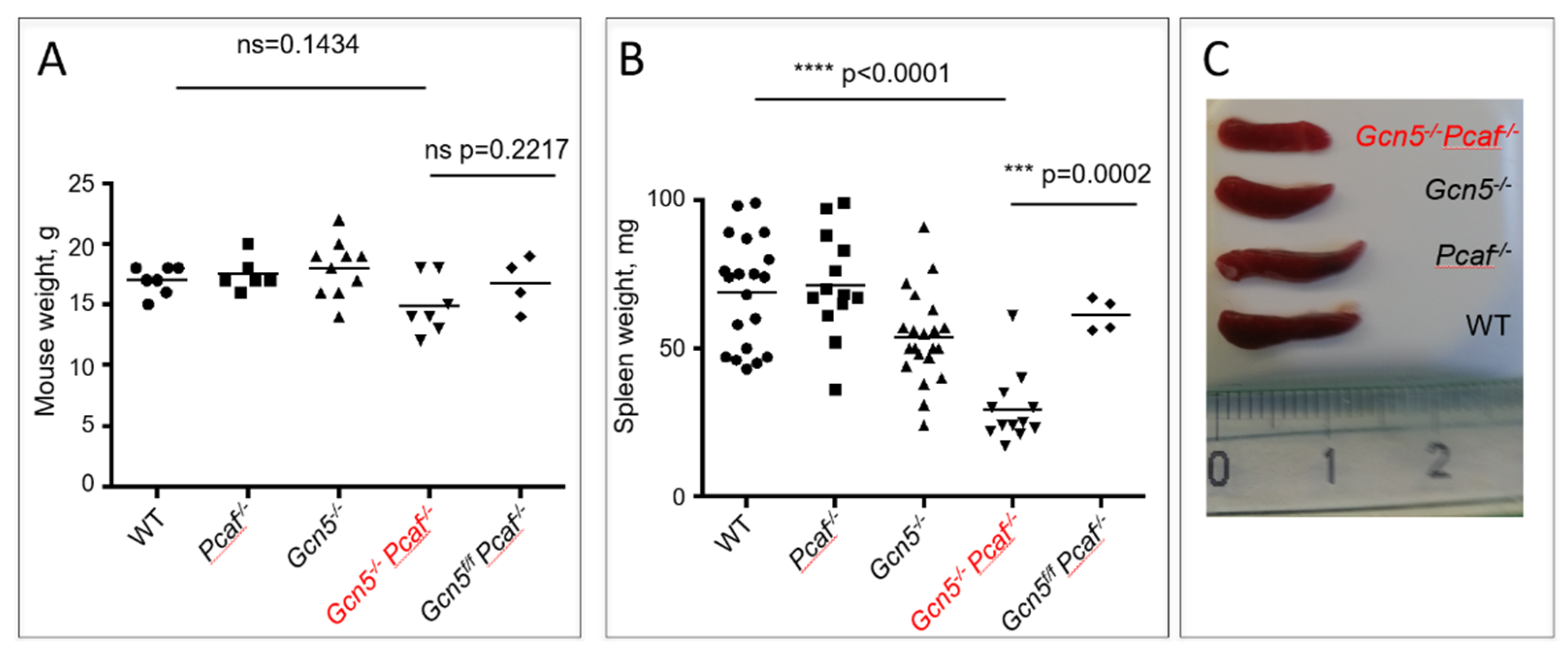
3.2. Mice Lacking GCN5 and PCAF in B Cells Possess Small Spleens
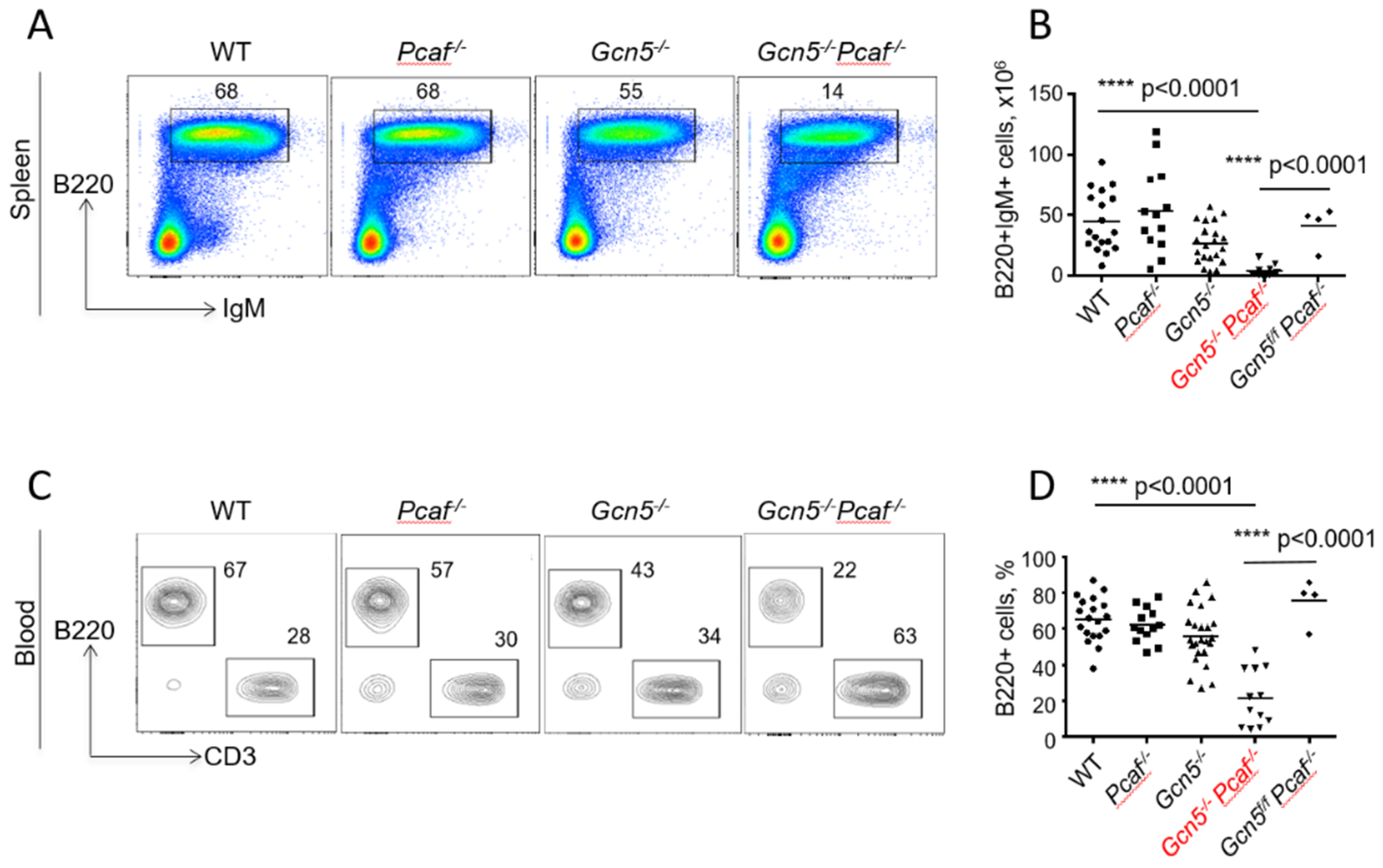
3.3. Mice Lacking GCN5 and PCAF in B Cells Possess Delayed B Lymphocyte Development
3.4. Inactivation of Gcn5 and Pcaf Results in a Reduced Proportion of B Cells in the Blood
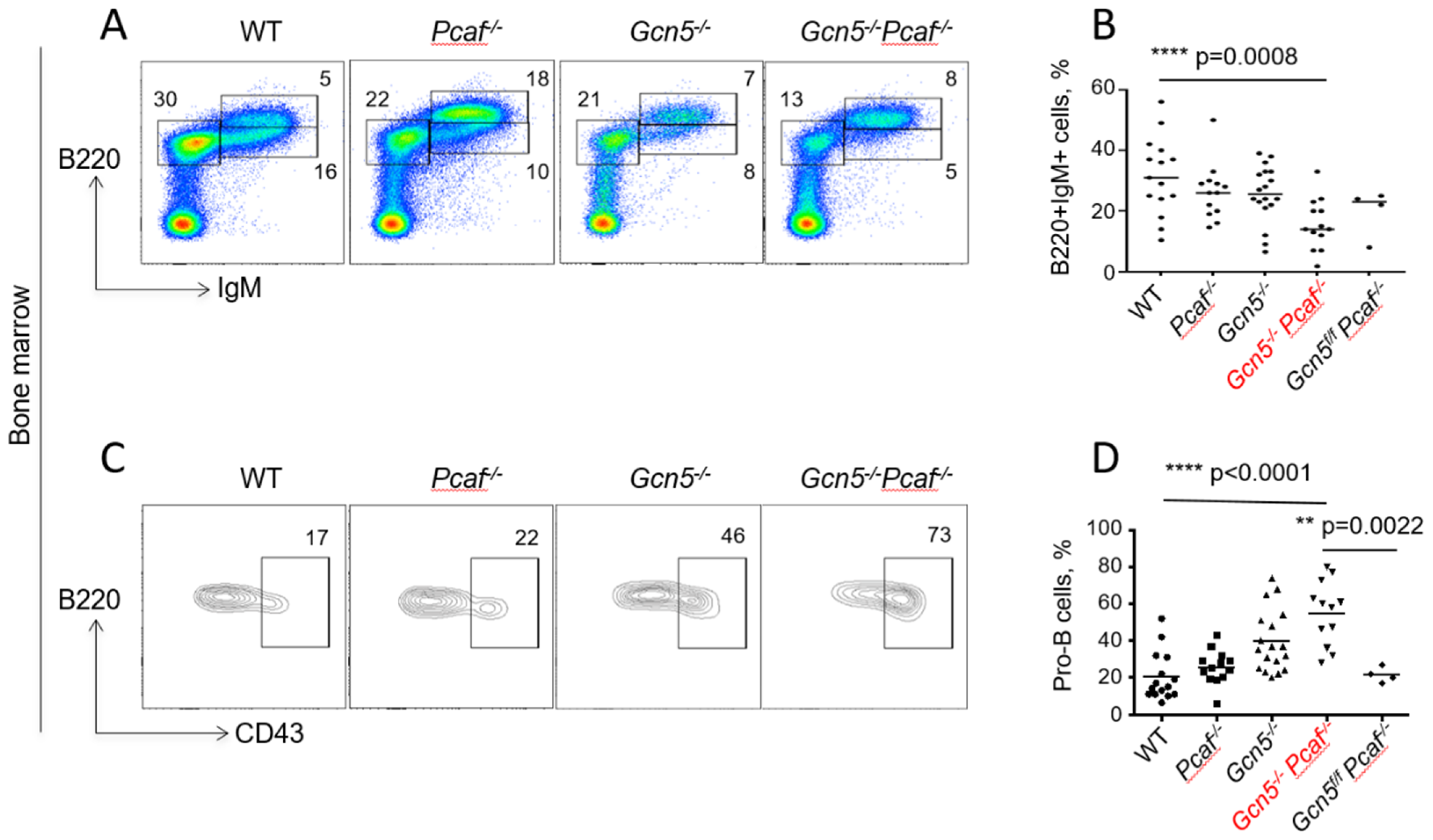
3.5. Inactivation of Gcn5 and Pcaf Results in Accumulation of Pro-B Cells in Bone Marrow
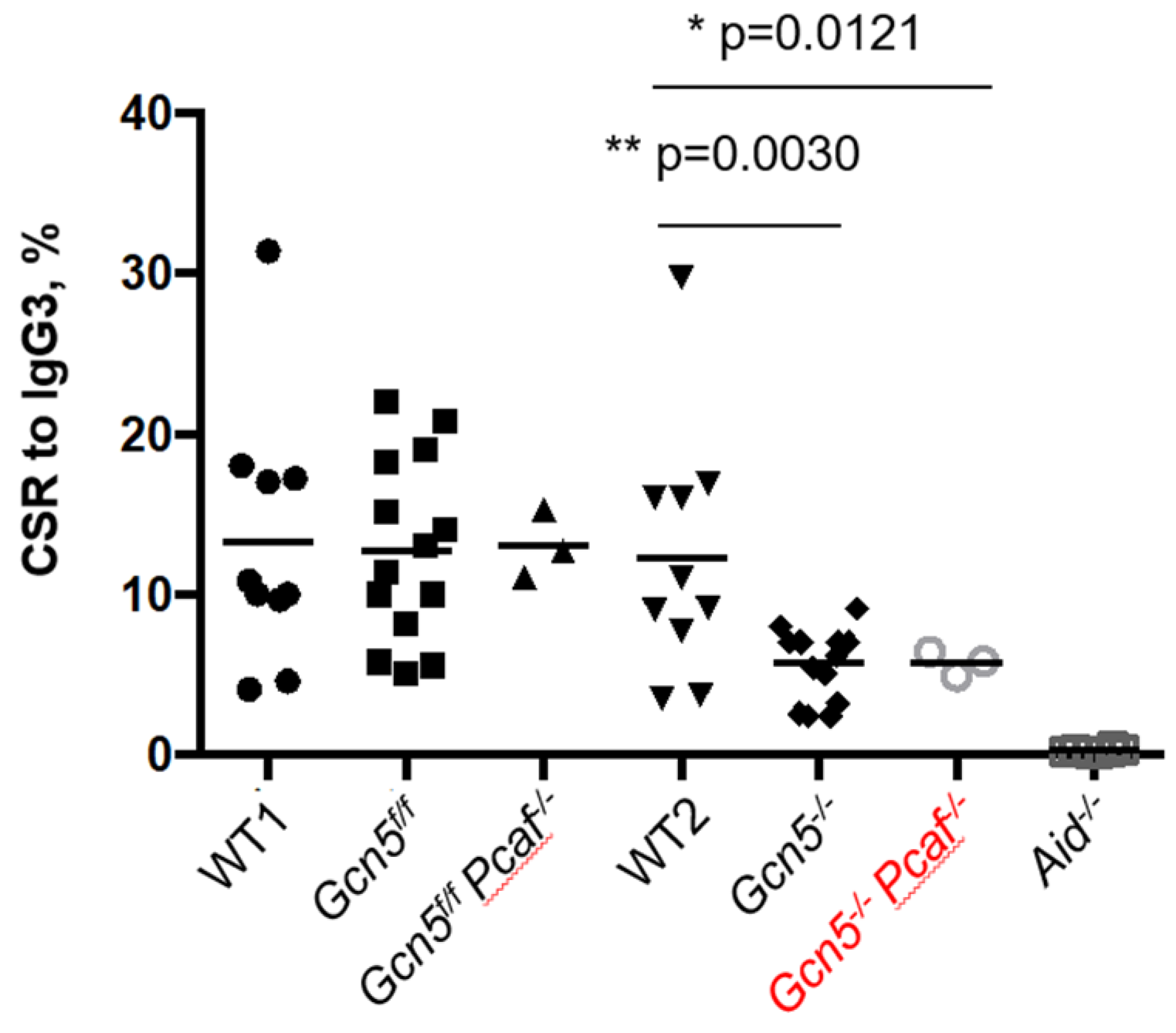
3.6. GCN5 Is Required for Robust Class Switch Recombination
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lescale, C.; Deriano, L. The RAG recombinase: Beyond breaking. Mech. Ageing Dev. 2017, 165, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Alt, F.W.; Zhang, Y.; Meng, F.L.; Guo, C.; Schwer, B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 2013, 152, 417–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.S.; Lee, B.J.; Zha, S. The recent advances in non-homologous end-joining through the lens of lymphocyte development. DNA Repair (Amst.) 2020, 94, 102874. [Google Scholar] [CrossRef]
- Castaneda-Zegarra, S.; Fernandez-Berrocal, M.; Tkachov, M.; Yao, R.; Upfold, N.L.E.; Oksenych, V. Genetic interaction between the non-homologous end-joining factors during B and T lymphocyte development: In vivo mouse models. Scand. J. Immunol. 2020, 92, e12936. [Google Scholar] [CrossRef]
- Kumar, V.; Alt, F.W.; Oksenych, V. Functional overlaps between XLF and the ATM-dependent DNA double strand break response. DNA Repair (Amst.) 2014, 16, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Frock, R.L.; Sadeghi, C.; Meng, J.; Wang, J.L. DNA End Joining: G0-ing to the Core. Biomolecules 2021, 11, 1487. [Google Scholar] [CrossRef]
- Bordin, D.L.; Lirussi, L.; Nilsen, H. Cellular response to endogenous DNA damage: DNA base modifications in gene expression regulation. DNA Repair (Amst.) 2021, 99, 103051. [Google Scholar] [CrossRef] [PubMed]
- Saha, T.; Sundaravinayagam, D.; Di Virgilio, M. Charting a DNA Repair Roadmap for Immunoglobulin Class Switch Recombination. Trends Biochem. Sci. 2021, 46, 184–199. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Ba, Z.; Kyritsis, N.; Casellas, R.; Alt, F.W. Fundamental roles of chromatin loop extrusion in antibody class switching. Nature 2019, 575, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.; Ba, Z.; Liang, Z.; Dring, E.W.; Hu, H.; Lou, J.; Kyritsis, N.; Zurita, J.; Shamim, M.S.; et al. The fundamental role of chromatin loop extrusion in physiological V(D)J recombination. Nature 2019, 573, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Menolfi, D.; Zha, S. DNA-PKcs kinase activity orchestrates both end-processing and end-ligation. Trends Cell Biol. 2021, 12, 1–3. [Google Scholar] [CrossRef]
- Ragunathan, K.; Upfold, N.L.E.; Oksenych, V. Interaction between Fibroblasts and Immune Cells Following DNA Damage Induced by Ionizing Radiation. Int. J. Mol. Sci. 2020, 21, 8635. [Google Scholar] [CrossRef]
- Lee, H.S.; Park, J.H.; Kim, S.J.; Kwon, S.J.; Kwon, J. A cooperative activation loop among SWI/SNF, gamma-H2AX and H3 acetylation for DNA double-strand break repair. EMBO J. 2010, 29, 1434–1445. [Google Scholar] [CrossRef]
- Jin, Q.; Yu, L.R.; Wang, L.; Zhang, Z.; Kasper, L.H.; Lee, J.E.; Wang, C.; Brindle, P.K.; Dent, S.Y.; Ge, K. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011, 30, 249–262. [Google Scholar] [CrossRef]
- Oksenych, V.; Kumar, V.; Liu, X.; Guo, C.; Schwer, B.; Zha, S.; Alt, F.W. Functional redundancy between the XLF and DNA-PKcs DNA repair factors in V(D)J recombination and nonhomologous DNA end joining. Proc. Natl. Acad. Sci. USA 2013, 110, 2234–2239. [Google Scholar] [CrossRef] [Green Version]
- Xing, M.; Bjoras, M.; Daniel, J.A.; Alt, F.W.; Oksenych, V. Synthetic lethality between murine DNA repair factors XLF and DNA-PKcs is rescued by inactivation of Ku70. DNA Repair (Amst.) 2017, 57, 133–138. [Google Scholar] [CrossRef]
- Xing, M.; Oksenych, V. Genetic interaction between DNA repair factors PAXX, XLF, XRCC4 and DNA-PKcs in human cells. FEBS Open Bio 2019, 9, 1315–1326. [Google Scholar] [CrossRef] [Green Version]
- Abramowski, V.; Etienne, O.; Elsaid, R.; Yang, J.; Berland, A.; Kermasson, L.; Roch, B.; Musilli, S.; Moussu, J.P.; Lipson-Ruffert, K.; et al. PAXX and Xlf interplay revealed by impaired CNS development and immunodeficiency of double KO mice. Cell Death Differ. 2018, 25, 444–452. [Google Scholar] [CrossRef] [Green Version]
- Balmus, G.; Barros, A.C.; Wijnhoven, P.W.; Lescale, C.; Hasse, H.L.; Boroviak, K.; le Sage, C.; Doe, B.; Speak, A.O.; Galli, A.; et al. Synthetic lethality between PAXX and XLF in mammalian development. Genes Dev. 2016, 30, 2152–2157. [Google Scholar] [CrossRef] [Green Version]
- Castaneda-Zegarra, S.; Xing, M.; Gago-Fuentes, R.; Saeterstad, S.; Oksenych, V. Synthetic lethality between DNA repair factors Xlf and Paxx is rescued by inactivation of Trp53. DNA Repair (Amst.) 2019, 73, 164–169. [Google Scholar] [CrossRef]
- Lescale, C.; Lenden Hasse, H.; Blackford, A.N.; Balmus, G.; Bianchi, J.J.; Yu, W.; Bacoccina, L.; Jarade, A.; Clouin, C.; Sivapalan, R.; et al. Specific Roles of XRCC4 Paralogs PAXX and XLF during V(D)J Recombination. Cell Rep. 2016, 16, 2967–2979. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Shao, Z.; Jiang, W.; Lee, B.J.; Zha, S. PAXX promotes KU accumulation at DNA breaks and is essential for end-joining in XLF-deficient mice. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Hung, P.J.; Chen, B.R.; George, R.; Liberman, C.; Morales, A.J.; Colon-Ortiz, P.; Tyler, J.K.; Sleckman, B.P.; Bredemeyer, A.L. Deficiency of XLF and PAXX prevents DNA double-strand break repair by non-homologous end joining in lymphocytes. Cell Cycle 2017, 16, 286–295. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Alt, F.W.; Frock, R.L. PAXX and XLF DNA repair factors are functionally redundant in joining DNA breaks in a G1-arrested progenitor B-cell line. Proc. Natl. Acad. Sci. USA 2016, 113, 10619–10624. [Google Scholar] [CrossRef] [Green Version]
- Castaneda-Zegarra, S.; Zhang, Q.; Alirezaylavasani, A.; Fernandez-Berrocal, M.; Yao, R.; Oksenych, V. Leaky severe combined immunodeficiency in mice lacking non-homologous end joining factors XLF and MRI. Aging (Albany NY) 2020, 12, 23578–23597. [Google Scholar] [CrossRef]
- Hung, P.J.; Johnson, B.; Chen, B.R.; Byrum, A.K.; Bredemeyer, A.L.; Yewdell, W.T.; Johnson, T.E.; Lee, B.J.; Deivasigamani, S.; Hindi, I.; et al. MRI Is a DNA Damage Response Adaptor during Classical Non-homologous End Joining. Mol. Cell 2018, 71, 332–342. [Google Scholar] [CrossRef]
- Zha, S.; Guo, C.; Boboila, C.; Oksenych, V.; Cheng, H.L.; Zhang, Y.; Wesemann, D.R.; Yuen, G.; Patel, H.; Goff, P.H.; et al. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature 2011, 469, 250–254. [Google Scholar] [CrossRef] [Green Version]
- Beck, C.; Castaneda-Zegarra, S.; Huse, C.; Xing, M.; Oksenych, V. Mediator of DNA Damage Checkpoint Protein 1 Facilitates V(D)J Recombination in Cells Lacking DNA Repair Factor XLF. Biomolecules 2019, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.R.; Wang, Y.; Shen, Z.J.; Bennett, A.; Hindi, I.; Tyler, J.K.; Sleckman, B.P. The RNF8 and RNF168 Ubiquitin Ligases Regulate Pro- and Anti-Resection Activities at Broken DNA Ends During Non-Homologous End Joining. DNA Repair (Amst.) 2021, 108, 103217. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, W.; Dubois, R.L.; Yamamoto, K.; Wolner, Z.; Zha, S. Overlapping functions between XLF repair protein and 53BP1 DNA damage response factor in end joining and lymphocyte development. Proc. Natl. Acad. Sci. USA 2012, 109, 3903–3908. [Google Scholar] [CrossRef] [Green Version]
- Oksenych, V.; Alt, F.W.; Kumar, V.; Schwer, B.; Wesemann, D.R.; Hansen, E.; Patel, H.; Su, A.; Guo, C. Functional redundancy between repair factor XLF and damage response mediator 53BP1 in V(D)J recombination and DNA repair. Proc. Natl. Acad. Sci. USA 2012, 109, 2455–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Edmondson, D.G.; Evrard, Y.A.; Wakamiya, M.; Behringer, R.R.; Roth, S.Y. Loss of Gcn5l2 leads to increased apoptosis and mesodermal defects during mouse development. Nat. Genet. 2000, 26, 229–232. [Google Scholar] [CrossRef]
- Lin, W.; Srajer, G.; Evrard, Y.A.; Phan, H.M.; Furuta, Y.; Dent, S.Y. Developmental potential of Gcn5(-/-) embryonic stem cells in vivo and in vitro. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2007, 236, 1547–1557. [Google Scholar] [CrossRef]
- Rickert, R.C.; Roes, J.; Rajewsky, K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 1997, 25, 1317–1318. [Google Scholar] [CrossRef]
- Srinivas, S.; Watanabe, T.; Lin, C.S.; William, C.M.; Tanabe, Y.; Jessell, T.M.; Costantini, F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 2001, 1, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Dewan, A.; Xing, M.; Lundbaek, M.B.; Gago-Fuentes, R.; Beck, C.; Aas, P.A.; Liabakk, N.B.; Saeterstad, S.; Chau, K.T.P.; Kavli, B.M.; et al. Robust DNA repair in PAXX-deficient mammalian cells. FEBS Open Bio 2018, 8, 442–448. [Google Scholar] [CrossRef]
- Gago-Fuentes, R.; Xing, M.; Saeterstad, S.; Sarno, A.; Dewan, A.; Beck, C.; Bradamante, S.; Bjoras, M.; Oksenych, V. Normal development of mice lacking PAXX, the paralogue of XRCC4 and XLF. FEBS Open Bio 2018, 8, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Castaneda-Zegarra, S.; Huse, C.; Rosand, O.; Sarno, A.; Xing, M.; Gago-Fuentes, R.; Zhang, Q.; Alirezaylavasani, A.; Werner, J.; Ji, P.; et al. Generation of a Mouse Model Lacking the Non-Homologous End-Joining Factor Mri/Cyren. Biomolecules 2019, 9, 798. [Google Scholar] [CrossRef] [Green Version]
- Starnes, L.M.; Su, D.; Pikkupeura, L.M.; Weinert, B.T.; Santos, M.A.; Mund, A.; Soria, R.; Cho, Y.W.; Pozdnyakova, I.; Kubec Hojfeldt, M.; et al. A PTIP-PA1 subcomplex promotes transcription for IgH class switching independently from the associated MLL3/MLL4 methyltransferase complex. Genes Dev. 2016, 30, 149–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, D.; Vanhee, S.; Soria, R.; Gyllenback, E.J.; Starnes, L.M.; Hojfeldt, M.K.; Pedersen, G.K.; Yuan, J.; Daniel, J.A. PTIP chromatin regulator controls development and activation of B cell subsets to license humoral immunity in mice. Proc. Natl. Acad. Sci. USA 2017, 114, E9328–E9337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boboila, C.; Oksenych, V.; Gostissa, M.; Wang, J.H.; Zha, S.; Zhang, Y.; Chai, H.; Lee, C.S.; Jankovic, M.; Saez, L.M.; et al. Robust chromosomal DNA repair via alternative end-joining in the absence of X-ray repair cross-complementing protein 1 (XRCC1). Proc. Natl. Acad. Sci. USA 2012, 109, 2473–2478. [Google Scholar] [CrossRef] [Green Version]
- Gago-Fuentes, R.; Oksenych, V. Non-Homologous End Joining Factors XLF, PAXX and DNA-PKcs Maintain the Neural Stem and Progenitor Cell Population. Biomolecules 2020, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Farria, A.T.; Plummer, J.B.; Salinger, A.P.; Shen, J.; Lin, K.; Lu, Y.; McBride, K.M.; Koutelou, E.; Dent, S.Y.R. Transcriptional Activation of MYC-Induced Genes by GCN5 Promotes B-cell Lymphomagenesis. Cancer Res. 2020, 80, 5543–5553. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Kondo, S.; Sugai, M.; Nazarea, M.; Imamura, S.; Honjo, T. High frequency class switching of an IgM+ B lymphoma clone CH12F3 to IgA+ cells. Int. Immunol. 1996, 8, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oksenych, V.; Su, D.; Daniel, J.A. Acetyltransferases GCN5 and PCAF Are Required for B Lymphocyte Maturation in Mice. Biomolecules 2022, 12, 61. https://doi.org/10.3390/biom12010061
Oksenych V, Su D, Daniel JA. Acetyltransferases GCN5 and PCAF Are Required for B Lymphocyte Maturation in Mice. Biomolecules. 2022; 12(1):61. https://doi.org/10.3390/biom12010061
Chicago/Turabian StyleOksenych, Valentyn, Dan Su, and Jeremy A. Daniel. 2022. "Acetyltransferases GCN5 and PCAF Are Required for B Lymphocyte Maturation in Mice" Biomolecules 12, no. 1: 61. https://doi.org/10.3390/biom12010061
APA StyleOksenych, V., Su, D., & Daniel, J. A. (2022). Acetyltransferases GCN5 and PCAF Are Required for B Lymphocyte Maturation in Mice. Biomolecules, 12(1), 61. https://doi.org/10.3390/biom12010061