
Receptor for Advanced Glycation End Product, Organ Crosstalk, and Pathomechanism Targets for Comprehensive Molecular Therapeutics in Diabetic Ischemic Stroke

 ,
,  ,
,
Abstract
:1. Introduction
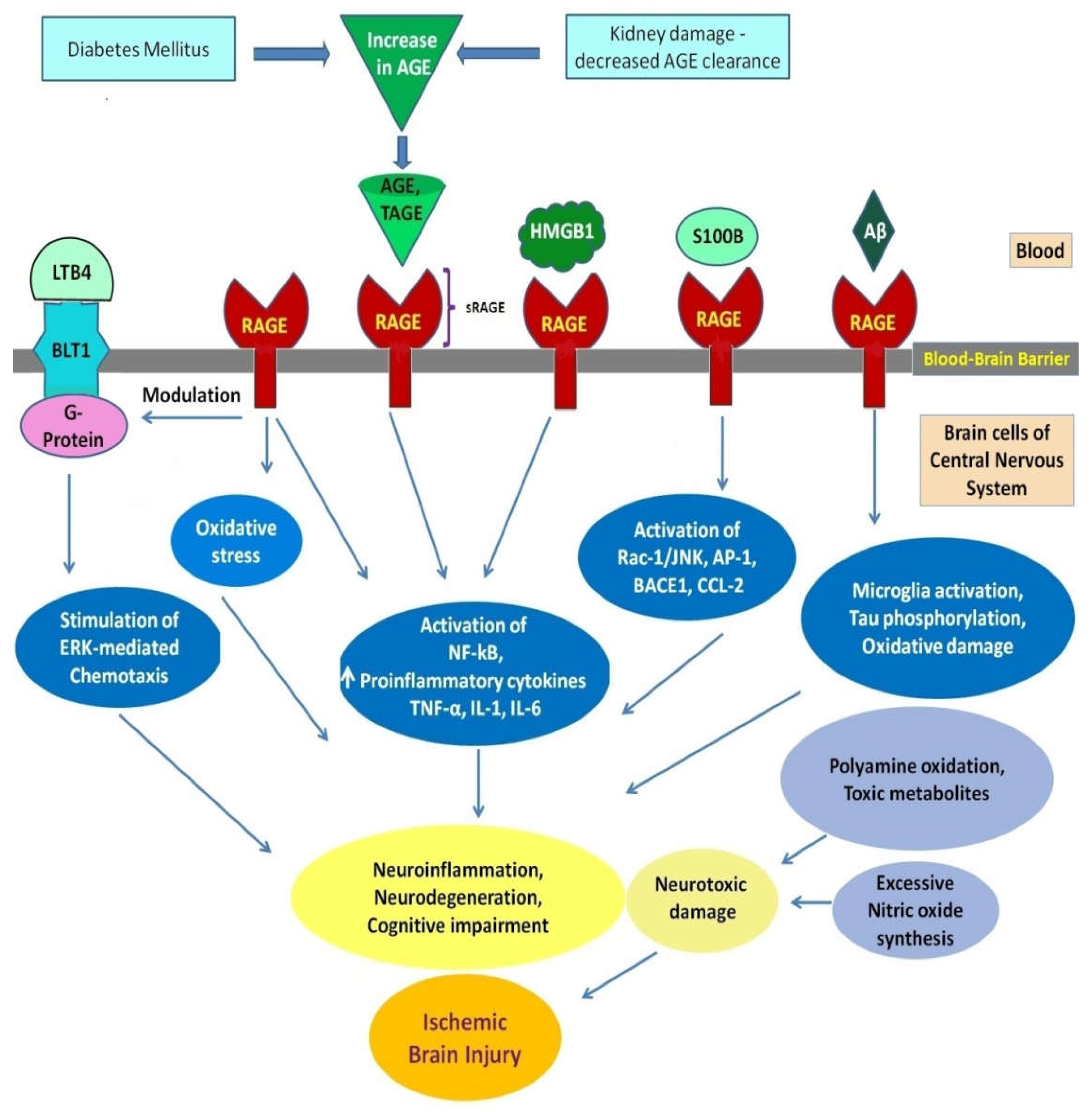
2. AGEs, Toxic AGEs, and RAGEs
3. Diabetic Ischemic Stroke and RAGE-Mediated Ischemic Brain Damage
- HMGB1 interacts with the receptors TLR-2 and TLR-4, which are expressed on monocytes through the adaptor protein myeloid differentiation factor 88 (MyD88) and elevates serum levels of TNF-α, IL-6, and IL-1β l, which leads to cerebral vessel occlusion.
- TNF-α and IL-1β, which are HMGB1-induced cytokines, can indirectly promote the upregulation of matrix metalloproteinase MMP-9. MMP upregulation causes a rise in infarct size, brain edema, and recombinant tissue plasminogen activator-induced bleeding, which hastens damage to the tight junction protein Occludin and other neurovascular substrates.
- HMGB1 stimulation of TNF-α, IL-1, IL-6, and IL-8 production induces the expression of inducible NOS (iNOS) during ischemic brain damage. The induction of iNOS and TNF-α occurs mainly in microglia. This produces an inflammatory response and BBB disruption, leading to brain infarction aggravation.
- RAGE expression, which is low in cells under physiological conditions, rises in response to an increase in HMGB1 ligand molecules, for which RAGE has a strong affinity. HMGB1 binding to upregulated RAGE leads to the activation of several signal-transduction pathways including Mitogen-activated protein kinases (MAPKs), phosphatidylinositol 3 kinase/protein kinase B (PI3K/Akt)-p38 kinase, SAPK/JNK, extracellular regulated protein kinases1/2 (ERK 1/2), Akt, Ras-related C3 botulinum toxin substrate (Rac), Cell division cycle 42 (Cdc42), and Just another kinase/signal transducer and activator of transcription 1 (JAK/STAT1)-mediated signal transduction pathways. Finally, these processes result in the translocation of nuclear factor kappa-light-chain-enhancer (NF-κB), which triggers the expression of inflammatory cytokines and chemokines that help immune cells mature, migrate, and express surface receptors, and cause neuritis [50].
4. AGE-RAGE System, Blood–Brain Barrier Dysfunction, Neuroinflammation and Neurodegeneration in Ischemic Stroke
- During thrombosis, amyloid beta (Aβ) peptides produced in blood vessels are discharged into the brain and momentarily accumulate there. During an ischemic stroke, they act as additional damaging factors by forming ion channels. RAGE is implicated in the neurotoxic immunoinflammatory cascades and in the amyloidogenic pathway. RAGE is the primary influx transporter for Aβ across the BBB. RAGE-soluble Aβ binding mediates the pathophysiologic cellular responses. The RAGE–Aβ interaction causes oxidative damage to RAGE-expressing neurons and activates microglia, which both directly and indirectly contributes to neuron death. RAGE inhibitors can prevent the production of cytokines and chemokines, oxidative stress, and Aβ BBB transport by blocking the pathophysiological effects of the RAGE–Aβ interaction in the afflicted vasculature [36,53].
- RAGE is connected to both independent neurotoxic immunoinflammatory cascades and the amyloidogenic pathway in neurodegenerative diseases [48]. RAGE is overexpressed in neurons, microglia, astrocytes, and the BBB vasculature when endogenous ligands such as AGE, S100, or Aβ bind to physiologically expressed RAGE [36,48]. RAGE is more prevalent at the BBB, which causes an influx of monocytes and Aβ into the brain. However, RAGE is more active in neurons, where it enhances Aβ-producing β-secretase enzyme (BACE1) activity, tau hyperphosphorylation, and neuroinflammation and impairs neuronal function. Ischemia-induced Aβ/tau pathology, similar to Alzheimer’s disease, is reported to be involved in post-stroke cognitive impairment [36].
- Hyperglycemia-induced overexpression of mitochondrial superoxide in endothelial cells causes microvascular injury in diabetes mellitus. Superoxide overproduction can activate AGEs formation and protein kinase C (PKC) signaling. PKC induces BBB damage through the disruption of tight junction (TJ) proteins, phosphorylation of cytoplasmic adaptor zona occuldens-1 (ZO1), and enhanced expression of vascular endothelial growth factor. Upregulated and activated RAGEs induce oxidative stress and activate the NF-κB pathway. TNF-α, IL-6, and IL-1 transcription are increased when the NF-κB pathway is activated in vascular cells [18,36].
- The AGE–RAGE system may also participate in Apolipoprotein E-ε4 (APOE-ε4 allele)-associated pathological processes of dementia. Associations between higher skin autofluorescence due to AGEs, lower cognitive function, and APOE-ε4 status have been reported [57]. APOE-ε4 impacts the risk of dementia associated with stroke in patients, and both pre- and post-event dementia were found to be linked to APOE-ε4 homozygosity. The relationships were not related to the burden of the cerebrovascular system and may be explained by increased neurodegenerative disease or damage susceptibility [58].
5. Interplay of Leukotriene B4 Receptor 1 (BLT1) and RAGE in Ischemic Stroke
6. Neurotoxicity of AGEs Demonstrated by Animal and Human Models
7. Brain–Kidney Organ Crosstalk, Renal Dysfunction, and Plasma AGEs
8. Therapeutic Agents and Their Effects against the AGE–RAGE Axis and Other Key Targets in Diabetic Cardio-Cerebrovascular Complications
9. Myokines, Muscle-Organ Crosstalk, Neuromuscular Electrical Stimulation, and Improved Motor Recovery in Stroke Patients
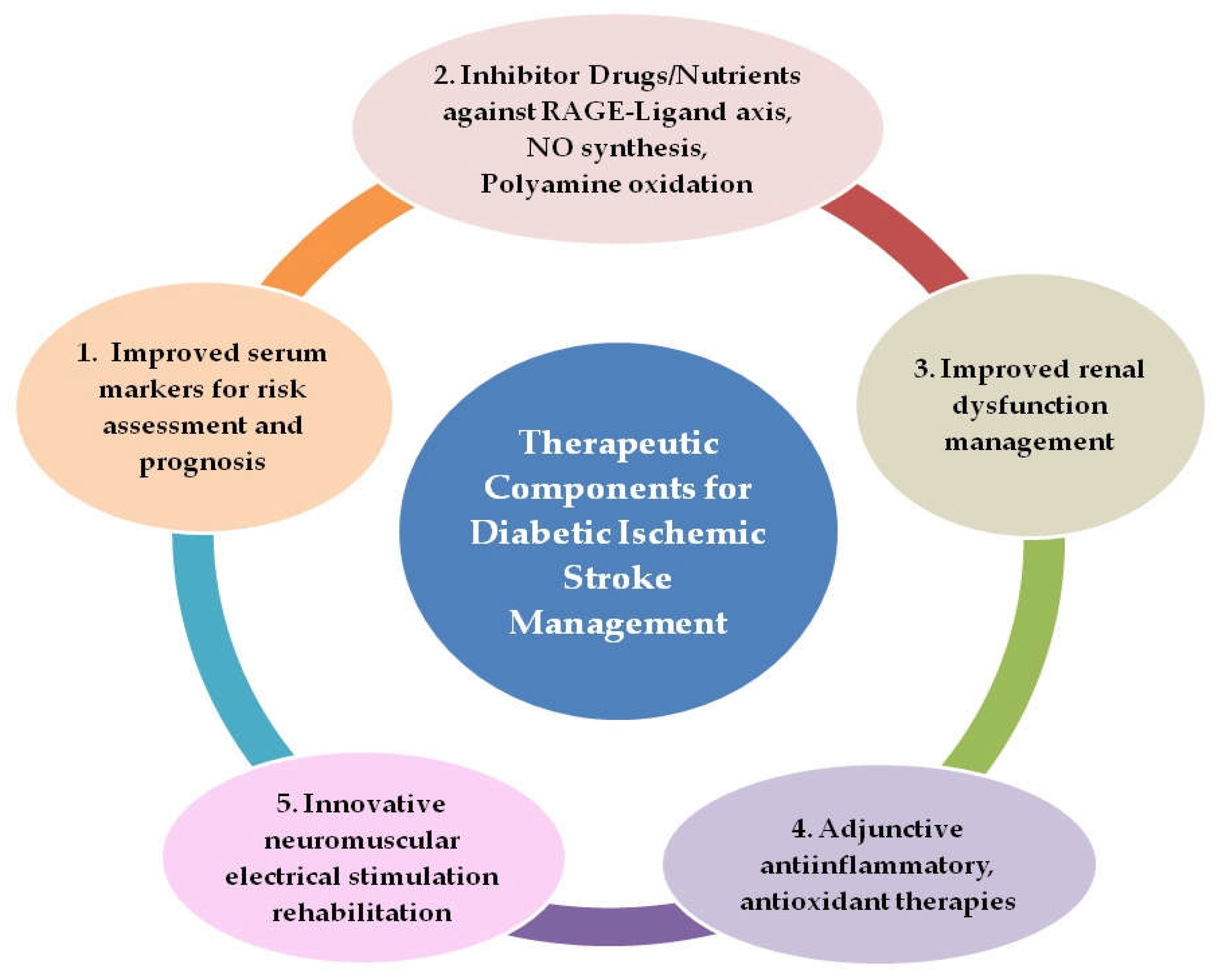
10. Plausible Comprehensive Therapeutic Strategies for Improved Management of Diabetic Ischemic Stroke
- Rapid screening of patients for serum AGEs, TAGEs, and risk stratification with point-of-care testing (POCT) devices validated for routine clinical use for monitoring disease progression and treatment effectiveness, as well as the employment of HMGB1 and CCL2 blood levels as prognostic markers for stroke patients.
- Administration of inhibitor drugs and nutrients against RAGE–ligand axes, NO synthesis, and polyamine oxidation as adjunctive therapy along with primary therapy.
- Improved management of renal dysfunction, the use of appropriate dialysate fluid to clear plasma AGEs more efficiently, and the formulation of renal dialysis modalities to improve clearance of low-molecular-weight fluorescent AGEs
- Anti-inflammatory therapy with agents from natural sources such as the plant flavanoid Quercetin or its analogues, which can target HMGB1–RAGE, LTB4–BLT1, and NO synthesis for selective neutralization of pathogenic immune signaling, tissue preservation, and neurological recovery. In addition, antioxidant therapy, which can combat not only the oxidative stress associated with hyperglycemia but also stress due to polyamine oxidation and NO synthesis, offering a key node in the preventive and therapeutic strategies for diabetes-related fatal cardio-cerebrovascular events.
- Neuromuscular stimulation interventions in diabetic ischemic stroke patients with possible employment of myokine Irisin as a biomarker for monitoring the impact of exercise type and amount.
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Whiting, D.R.; Guariguata, L.; Weil, C.; Shaw, J. IDF Diabetes Atlas: Global Estimates of the Prevalence of Diabetes for 2011 and 2030. Diabetes Res. Clin. Pract. 2011, 94, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Vos, T.; Abajobir, A.A.; Abbafati, C.; Abbas, K.M.; Abate, K.H.; Abd-Allah, F.; Abdulle, A.M.; Abebo, T.A.; Abera, S.F.; Aboyans, V.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef] [Green Version]
- Kamalakannan, S.; Gudlavalleti, A.S.V.; Gudlavalleti, V.S.M.; Goenka, S.; Kuper, H. Incidence & Prevalence of Stroke in India: A Systematic Review. Indian J. Med. Res. 2017, 146, 175. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.; Mirzaei, M.; Anderson, L.; Leeder, S.R. The Epidemiology of Stroke in the Middle East and North Africa. J. Neurol. Sci. 2010, 295, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Sacco, R.L.; Kasner, S.E.; Broderick, J.P.; Caplan, L.R.; Connors, J.J.B.; Culebras, A.; Elkind, M.S.V.; George, M.G.; Hamdan, A.D.; Higashida, R.T.; et al. An Updated Definition of Stroke for the 21st Century: A Statement for Healthcare Professionals from the American Heart Association/American Stroke Association. Stroke 2013, 44, 2064–2089. [Google Scholar] [CrossRef] [Green Version]
- Boehme, A.K.; Esenwa, C.; Elkind, M.S.V. Stroke Risk Factors, Genetics, and Prevention. Circ. Res. 2017, 120, 472–495. [Google Scholar] [CrossRef]
- Donnan, G.A.; Fisher, M.; Macleod, M.; Davis, S.M. Stroke. Lancet 2008, 371, 1612–1623. [Google Scholar] [CrossRef]
- Adams, H.P.; Bendixen, B.H.; Kappelle, L.J.; Biller, J.; Love, B.B.; Gordon, D.L.; Marsh, E.E. Classification of Subtype of Acute Ischemic Stroke. Definitions for Use in a Multicenter Clinical Trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993, 24, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Tuttolomondo, A. Relationship between Diabetes and Ischemic Stroke: Analysis of Diabetes- Related Risk Factors for Stroke and of Specific Patterns of Stroke Associated with Diabetes Mellitus. J. Diabetes Metab. 2015, 6. [Google Scholar] [CrossRef]
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of Cardiovascular Disease in Type 2 Diabetes: A Systematic Literature Review of Scientific Evidence from across the World in 2007–2017. Cardiovasc. Diabetol. 2018, 17. [Google Scholar] [CrossRef]
- Rangel, É.B.; Rodrigues, C.O.; de Sá, J.R. Micro- and Macrovascular Complications in Diabetes Mellitus: Preclinical and Clinical Studies. J. Diabetes Res. 2019, 2019, 2161085. [Google Scholar] [CrossRef] [Green Version]
- Khan, R.; Yee Ooi, X.; Parvus, M.; Valdez, L.; Tsin, A. Advanced Glycation End Products: Formation, Role in Diabetic Complications, and Potential in Clinical Applications. Eye Foot Diabetes 2020. [Google Scholar] [CrossRef] [Green Version]
- Guerin-Dubourg, A.; Cournot, M.; Planesse, C.; Debussche, X.; Meilhac, O.; Rondeau, P.; Bourdon, E. Association between Fluorescent Advanced Glycation End-Products and Vascular Complications in Type 2 Diabetic Patients. BioMed Res. Int. 2017, 2017, 7989180. [Google Scholar] [CrossRef]
- Yamagishi, S.; Matsui, T. Role of Hyperglycemia-Induced Advanced Glycation End Product (AGE) Accumulation in Atherosclerosis. Ann. Vasc. Dis. 2018, 11, 253–258. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, M.N.; Farah, A.I.; Al-Qirim, T.M. The Cardiovascular Complications of Diabetes: A Striking Link through Protein Glycation. Rom. J. Intern. Med. 2020, 58, 188–198. [Google Scholar] [CrossRef]
- Perrone, A.; Giovino, A.; Benny, J.; Martinelli, F. Advanced Glycation End Products (AGEs): Biochemistry, Signaling, Analytical Methods, and Epigenetic Effects. Oxidative Med. Cell. Longev. 2020, 2020, 3818196. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M. Serum Levels of Toxic AGEs (TAGE) May Be a Promising Novel Biomarker for the Onset/Progression of Lifestyle-Related Diseases. Diagnostics 2016, 6, 23. [Google Scholar] [CrossRef]
- Takeuchi, M.; Takino, J.; Yamagishi, S. Involvement of the Toxic AGEs (TAGE)-RAGE System in the Pathogenesis of Diabetic Vascular Complications: A Novel Therapeutic Strategy. Curr. Drug Targets 2010, 11, 1468–1482. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Shimogaito, N.; Wu, X.; Kikuchi, S.; Yamagishi, S.; Takeuchi, M. Toxic Advanced Glycation End Products (TAGE) Theory in Alzheimer’s Disease. Am. J. Alzheimer’s Dis. Other Dement. 2006, 21, 197–208. [Google Scholar] [CrossRef]
- Kuzan, A. Toxicity of Advanced Glycation End Products (Review). Biomed. Rep. 2021, 14. [Google Scholar] [CrossRef]
- Takeuchi, M.; Sakasai-Sakai, A.; Takata, T.; Takino, J.; Koriyama, Y.; Kikuchi, C.; Furukawa, A.; Nagamine, K.; Hori, T.; Matsunaga, T. Intracellular Toxic AGEs (TAGE) Triggers Numerous Types of Cell Damage. Biomolecules 2021, 11, 387. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, G.A.; Meistrell, M.; Bloom, O.; Cockroft, K.M.; Bianchi, M.; Risucci, D.; Broome, J.; Farmer, P.; Cerami, A.; Vlassara, H. Neurotoxicity of Advanced Glycation End products during Focal Stroke and Neuroprotective Effects of Aminoguanidine. Proc. Natl. Acad. Sci. USA 1995, 92, 3744–3748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichiki, T.; Koga, T.; Okuno, T.; Saeki, K.; Yamamoto, Y.; Yamamoto, H.; Sakaguchi, M.; Yokomizo, T. Modulation of Leukotriene B4Receptor 1 Signaling by Receptor for Advanced Glycation End Products (RAGE). FASEB J. 2016, 30, 1811–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichiki, T.; Koga, T.; Yokomizo, T. Receptor for Advanced Glycation End Products Regulates Leukotriene B4 Receptor 1 Signaling. DNA Cell Biol. 2016, 35, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.J.; Ng, M.P.E.; Zhao, H.; Ng, G.J.L.; De Foo, C.; Wong, P.T.-H.; Seet, R.C.S. Early and Sustained Increases in Leukotriene B4 Levels Are Associated with Poor Clinical Outcome in Ischemic Stroke Patients. Neurotherapeutics 2019, 17, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Yan, T.; Chopp, M.; Venkat, P.; Chen, J. Brain-Kidney Interaction: Renal Dysfunction Following Ischemic Stroke. J. Cereb. Blood Flow Metab. 2020, 40, 246–262. [Google Scholar] [CrossRef]
- Shah, B.; Jagtap, P.; Sarmah, D.; Datta, A.; Raut, S.; Sarkar, A.; Bohra, M.; Singh, U.; Baidya, F.; Kalia, K.; et al. Cerebro-Renal Interaction and Stroke. Eur. J. Neurosci. 2020, 53, 1279–1299. [Google Scholar] [CrossRef]
- Liu, H.; Zheng, H.; Wu, P.; Liu, C.-F.; Wang, D.; Li, H.; Meng, X.; Wang, Y.; Cao, Y.; Wang, Y.; et al. Estimated Glomerular Filtration Rate, Anemia and Outcomes in Patients with Ischemic Stroke. Ann. Transl. Med. 2020, 8, 2. [Google Scholar] [CrossRef]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced Glycoxidation and Lipoxidation End Products (AGEs and ALEs): An Overview of Their Mechanisms of Formation. Free. Radic. Res. 2013, 47, 3–27. [Google Scholar] [CrossRef] [Green Version]
- Sajithlal, G.B.; Chithra, P.; Chandrakasan, G. Advanced Glycation End Products Induce Crosslinking of Collagen in Vitro. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 1998, 1407, 215–224. [Google Scholar] [CrossRef]
- Münch, G.; Keis, R.; Weßels, A.; Riederer, P.; Bahner, U.; Heidland, A.; Niwa, T.; Lemke, H.-D.; Schinzel, R. Determination of Advanced Glycation End Products in Serum by Fluorescence Spectroscopy and Competitive ELISA. Clin. Chem. Lab. Med. 1997, 35. [Google Scholar] [CrossRef]
- MohdAshraf, J.; Ahmad, S.; Choi, I.; Ahmad, N.; Farhan, M.; Tatyana, G.; Shahab, U. Recent Advances in Detection of AGEs: Immunochemical, Bioanalytical and Biochemical Approaches. IUBMB Life 2015, 67, 897–913. [Google Scholar] [CrossRef]
- Leńska-Mieciek, M.; Korczak-Kowalska, G.; Bocian, K.; Fiszer, U. Pentosidine, Advanced Glycation End Product, in Acute Ischaemic Stroke Patients with and without Atrial Rhythm Disturbances. Neurol. Neurochir. Pol. 2020, 54, 323–328. [Google Scholar] [CrossRef]
- Filipov, A.; Fuchshuber, H.; Kraus, J.; Ebert, A.D.; Sandikci, V.; Alonso, A. Measuring of Advanced Glycation End Products in Acute Stroke Care: Skin Autofluorescence as a Predictor of Ischemic Stroke Outcome in Patients with Diabetes Mellitus. J. Clin. Med. 2022, 11, 1625. [Google Scholar] [CrossRef]
- Cheng, C.; Tsuneyama, K.; Kominami, R.; Shinohara, H.; Sakurai, S.; Yonekura, H.; Watanabe, T.; Takano, Y.; Yamamoto, H.; Yamamoto, Y. Expression Profiling of Endogenous Secretory Receptor for Advanced Glycation End Products in Human Organs. Mod. Pathol. 2005, 18, 1385–1396. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Pan, B.-S.; Sun, G.-C.; Sun, X.; Sun, F.-Y. Diabetes Synergistically Exacerbates Poststroke Dementia and Tau Abnormality in Brain. Neurochem. Int. 2010, 56, 955–961. [Google Scholar] [CrossRef]
- Tang, S.C.; Wang, Y.C.; Li, Y.I.; Li, H.C.; Manzanero, S.; Hsieh, Y.H.; Phipps, S.; Hu, C.J.; Chiou, H.Y.; Huang, Y.S.; et al. Functional role of soluble receptor for advanced glycation end products in stroke. Arter. Thromb. Vasc. Biol. 2013, 33, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Arancio, O.; Zhang, H.P.; Chen, X.; Lin, C.; Trinchese, F.; Puzzo, D.; Liu, S.; Hegde, A.; Yan, S.F.; Stern, A.; et al. Rage Potentiates Aβ-Induced Perturbation of Neuronal Function in Transgenic Mice. EMBO J. 2004, 23, 4096–4105. [Google Scholar] [CrossRef] [Green Version]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the Receptor for Advanced Glycation End Products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Zhai, D.-X.; Kong, Q.-F.; Xu, W.-S.; Bai, S.-S.; Peng, H.-S.; Zhao, K.; Li, G.-Z.; Wang, D.-D.; Sun, B.; Wang, J.-H.; et al. Rage Expression Is up-Regulated in Human Cerebral Ischemia and pMCAO Rats. Neurosci. Lett. 2008, 445, 117–121. [Google Scholar] [CrossRef]
- Willeit, P.; Tschiderer, L.; Allara, E.; Reuber, K.; Seekircher, L.; Gao, L.; Liao, X.; Lonn, E.; Gerstein, H.C.; Yusuf, S.; et al. Carotid intima-media thickness progression as surrogate marker for cardiovascular risk: Meta-analysis of 119 clinical trials involving 100 667 patients. Circulation 2020, 142, 621–642. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, Y.; Ohkubo, T. Hypertension with Diabetes Mellitus: Significance from an Epidemiological Perspective for Japanese. Hypertens. Res. 2017, 40, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, M. Hypertension with Diabetes Mellitus: Physiology and Pathology. Hypertens. Res. 2018, 41, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.; Mishra, M.; Tiwari, A.; Bisen, P.S.; Goswamy, H.M.; Prasad, G.B.K.S. Prevalence of Dyslipidemia and Hypertension in Indian Type 2 Diabetic Patients with Metabolic Syndrome and Its Clinical Significance. Osong Public Health Res. Perspect. 2014, 5, 169–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Liu, B.; Zhao, Q.; Jin, P.; Hua, F.; Zhang, Z.; Liu, Y.; Zan, K.; Cui, G.; Ye, X. Bone Marrow Stromal Cells Inhibits HMGB1-Mediated Inflammation after Stroke in Type 2 Diabetic Rats. Neuroscience 2016, 324, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Richard Seidu, A.; Sackey, M.; Su, Z.; Xu, H. Pivotal Neuroinflammatory and Therapeutic Role of High Mobility Group Box 1 in Ischemic Stroke. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Liu, B.; Yang, C.; Chen, H.; Eunice, D.; Yuan, Z. Acute Hyperglycemia Worsens Ischemic Stroke-Induced Brain Damage via High Mobility Group Box-1 in Rats. Brain Res. 2013, 1535, 148–155. [Google Scholar] [CrossRef]
- Carty, M.; Bowie, A.G. Evaluating the Role of Toll-like Receptors in Diseases of the Central Nervous System. Biochem. Pharmacol. 2011, 81, 825–837. [Google Scholar] [CrossRef]
- Ding, Q.; Keller, J.N. Evaluation of Rage Isoforms, Ligands, and Signaling in the Brain. Biochim. Et Biophys. Acta (BBA) Mol. Cell Res. 2005, 1746, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Zeng, Z.; Jin, T.; Zhang, H.; Xiong, X.; Gu, L. The Role of High Mobility Group Box 1 in Ischemic Stroke. Front. Cell. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Shichita, T.; Sugiyama, Y.; Ooboshi, H.; Sugimori, H.; Nakagawa, R.; Takada, I.; Iwaki, T.; Okada, Y.; Iida, M.; Cua, D.J.; et al. Pivotal Role of Cerebral Interleukin-17–Producing ΓδT Cells in the Delayed Phase of Ischemic Brain Injury. Nat. Med. 2009, 15, 946–950. [Google Scholar] [CrossRef]
- Muhammad, S.; Barakat, W.; Stoyanov, S.; Murikinati, S.; Yang, H.; Tracey, K.J.; Bendszus, M.; Rossetti, G.; Nawroth, P.P.; Bierhaus, A.; et al. The HMGB1 Receptor RAGE Mediates Ischemic Brain Damage. J. Neurosci. 2008, 28, 12023–12031. [Google Scholar] [CrossRef] [Green Version]
- Zlokovic, B.V. Blood–Brain Barrier and Neurovascular Mechanisms of Neurodegeneration and Injury. Encycl. Neurosci. 2009, 265–271. [Google Scholar] [CrossRef]
- Horie, K.; Miyata, T.; Yasuda, T.; Takeda, A.; Yasuda, Y.; Maeda, K.; Sobue, G.; Kurokawa, K. Immunohistochemical Localization of Advanced Glycation End Products, Pentosidine, and Carboxymethyllysine in Lipofuscin Pigments of Alzheimer’s Disease and Aged Neurons. Biochem. Biophys. Res. Commun. 1997, 236, 327–332. [Google Scholar] [CrossRef]
- Sasaki, N.; Fukatsu, R.; Tsuzuki, K.; Hayashi, Y.; Yoshida, T.; Fujii, N.; Koike, T.; Wakayama, I.; Yanagihara, R.; Garruto, R.; et al. Advanced Glycation End Products in Alzheimer’s Disease and Other Neurodegenerative Diseases. Am. J. Pathol. 1998, 153, 1149–1155. [Google Scholar] [CrossRef]
- Li, X.-H.; Du, L.-L.; Cheng, X.-S.; Jiang, X.; Zhang, Y.; Lv, B.-L.; Liu, R.; Wang, J.-Z.; Zhou, X.-W. Glycation Exacerbates the Neuronal Toxicity of β-Amyloid. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Mooldijk, S.S.; Licher, S.; Waqas, K.; Ikram, M.K.; Uitterlinden, A.G.; Zillikens, M.C.; Ikram, M.A. Assessment of Advanced Glycation End Products and Receptors and the Risk of Dementia. JAMA Netw. Open 2021, 4, e2033012. [Google Scholar] [CrossRef]
- Pendlebury, S.T.; Poole, D.; Burgess, A.; Duerden, J.; Rothwell, P.M. APOE-ε4 Genotype and Dementia before and after Transient Ischemic Attack and Stroke. Stroke 2020, 51, 751–758. [Google Scholar] [CrossRef]
- Hao, J.; Feng, Y.; Xu, X.; Li, L.; Yang, K.; Dai, G.; Gao, W.; Zhang, M.; Fan, Y.; Yin, T.; et al. Plasma Lipid Mediators Associate with Clinical Outcome after Successful Endovascular Thrombectomy in Patients with Acute Ischemic Stroke. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef]
- Yokomizo, T.; Nakamura, M.; Shimizu, T. Leukotriene Receptors as Potential Therapeutic Targets. J. Clin. Investig. 2018, 128, 2691–2701. [Google Scholar] [CrossRef]
- Takeuchi, M.; Yamagishi, S. TAGE (Toxic AGEs) Hypothesis in Various Chronic Diseases. Med. Hypotheses 2004, 63, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced Glycation End Products. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.Q.; Mou, R.T.; Feng, D.X.; Wang, Z.; Chen, G. The role of nitric oxide in stroke. Med. Gas Res. 2017, 7, 194–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wierońska, J.M.; Cieślik, P.; Kalinowski, L. Nitric Oxide-dependent pathways as critical factors in the consequences and recovery after brain ischemic hypoxia. Biomolecules 2021, 11, 1097. [Google Scholar] [CrossRef]
- Bucala, R.; Tracey, K.J.; Cerami, A. Advanced Glycosylation Products Quench Nitric Oxide and Mediate Defective Endothelium-Dependent Vasodilatation in Experimental Diabetes. J. Clin. Investig. 1991, 87, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Grzebyk, E.; Piwowar, A. Inhibitory Actions of Selected Natural Substances on Formation of Advanced Glycation Endproducts and Advanced Oxidation Protein Products. BMC Complement. Altern. Med. 2016, 16. [Google Scholar] [CrossRef] [Green Version]
- Alshehri, B.; Vijayakumar, R.; Senthilkumar, S.; Ismail, A.; Abdel-Hadi, A.; Choudhary, R.K.; Albenasy, K.S.; Banawas, S.; Alaidarous, M.A.; Manikandan, P. Therapeutic Potential of Nitric Oxide Synthase Inhibitor from Natural Sources for the Treatment of Ischemic Stroke. Saudi J. Biol. Sci. 2022, 29, 984–991. [Google Scholar] [CrossRef]
- Murray, S.T.; Dunston, T.T.; Woster, P.M.; Casero, R.A. Polyamine Catabolism and Oxidative Damage. J. Biol. Chem. 2018, 293, 18736–18745. [Google Scholar] [CrossRef] [Green Version]
- Bourourou, M.; Gouix, E.; Melis, N.; Friard, J.; Heurteaux, C.; Tauc, M.; Blondeau, N. Inhibition of EIF5A Hypusination Pathway as a New Pharmacological Target for Stroke Therapy. J. Cereb. Blood Flow Metab. 2020, 41, 1080–1090. [Google Scholar] [CrossRef]
- Masuko, T.; Takao, K.; Samejima, K.; Shirahata, A.; Igarashi, K.; Casero, R.A.; Kizawa, Y.; Sugita, Y. N1-Nonyl-1,4-Diaminobutane Ameliorates Brain Infarction Size in Photochemically Induced Thrombosis Model Mice. Neurosci. Lett. 2018, 672, 118–122. [Google Scholar] [CrossRef]
- Kaze, A.D.; Jaar, B.G.; Fonarow, G.C.; Echouffo-Tcheugui, J.B. Diabetic Kidney Disease and Risk of Incident Stroke among Adults with Type 2 Diabetes. BMC Med. 2022, 20. [Google Scholar] [CrossRef] [PubMed]
- Kratochvilová, M.; Zakiyanov, O.; Kalousová, M.; Kříha, V.; Zima, T.; Tesař, V. Associations of Serum Levels of Advanced. Glycation End Products with Nutrition Markers and Anemia in Patients with Chronic Kidney Disease. Ren. Fail. 2011, 33, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Dozio, E.; Vettoretti, S.; Caldiroli, L.; Nerini-Molteni, S.; Tacchini, L.; Ambrogi, F.; Messa, P.; Corsi Romanelli, M.M. Advanced Glycation End Products (AGE) and Soluble Forms of AGE Receptor: Emerging Role as Mortality Risk Factors in CKD. Biomedicines 2020, 8, 638. [Google Scholar] [CrossRef] [PubMed]
- Indyk, D.; Bronowicka-Szydełko, A.; Gamian, A.; Kuzan, A. Advanced Glycation End Products and Their Receptors in Serum of Patients with Type 2 Diabetes. Sci. Rep. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Gerdemann, A.; Wagner, Z.; Solf, A.; Bahner, U.; Heidland, A.; Vienken, J.; Schinzel, R. Plasma levels of advanced glycation end products during haemodialysis, haemodiafiltration and haemofiltration: Potential importance of dialysate quality. Nephrol. Dial. Transplant. 2002, 17, 1045–1049. [Google Scholar] [CrossRef] [Green Version]
- Hong Sheng, C.; Ha, T.S.; Khalid, A.K. Therapeutic Agents Targeting at AGE-RAGE Axis for the Treatment of Diabetes and Cardiovascular Disease: A Review of Clinical Evidence. Clin. Diabetes Res. 2017, 1. [Google Scholar] [CrossRef]
- Yang, P.; Feng, J.; Peng, Q.; Liu, X.; Fan, Z. Advanced Glycation End Products: Potential Mechanism and Therapeutic Target in Cardiovascular Complications under Diabetes. Oxid. Med. Cell. Longev. 2019, 2019, 9570616. [Google Scholar] [CrossRef]
- Bongarzone, S.; Savickas, V.; Luzi, F.; Gee, A.D. Targeting the Receptor for Advanced Glycation Endproducts (RAGE): A Medicinal Chemistry Perspective. J. Med. Chem. 2017, 60, 7213–7232. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.M.; Yee, L.T.L.; Thallas, V.; Lassila, M.; Candido, R.; Jandeleit-Dahm, K.A.; Thomas, M.C.; Burns, W.C.; Deemer, E.K.; Thorpe, S.R.; et al. Advanced Glycation End Product Interventions Reduce Diabetes-Accelerated Atherosclerosis. Diabetes 2004, 53, 1813–1823. [Google Scholar] [CrossRef] [Green Version]
- Ha, C.H.; Kim, S.; Chung, J.; An, S.H.; Park, S.; Choi, D.; Kwon, K. Inhibitory effect of soluble RAGE in disturbed flow-induced atherogenesis. Int. J. Mol. Med. 2013, 32, 373–380. [Google Scholar] [CrossRef]
- Shimizu, Y.; Harashima, A.; Munesue, S.; Oishi, M.; Hattori, T.; Hori, O.; Kitao, Y.; Yamamoto, H.; Leerach, N.; Nakada, M.; et al. Neuroprotective Effects of Endogenous Secretory Receptor for Advanced Glycation End-Products in Brain Ischemia. Aging Dis. 2020, 11, 547. [Google Scholar] [CrossRef]
- Mirmiranpour, H.; Mousavizadeh, M.; Noshad, S.; Ghavami, M.; Ebadi, M.; Ghasemiesfe, M.; Nakhjavani, M.; Esteghamati, A. Comparative Effects of Pioglitazone and Metformin on Oxidative Stress Markers in Newly Diagnosed Type 2 Diabetes Patients: A Randomized Clinical Trial. J. Diabetes Complicat. 2013, 27, 501–507. [Google Scholar] [CrossRef]
- Sakata, K.; Hayakawa, M.; Yano, Y.; Tamaki, N.; Yokota, N.; Eto, T.; Watanabe, R.; Hirayama, N.; Matsuo, T.; Kuroki, K.; et al. Efficacy of alogliptin, a dipeptidyl peptidase-4 inhibitor, on glucose parameters, the activity of the advanced glycation end product (AGE)—Receptor for AGE (RAGE) axis and albuminuria in Japanese type 2 diabetes. Diabetes Metab. Res. Rev. 2013, 29, 624–630. [Google Scholar] [CrossRef]
- Jinnouchi, Y.; Yamagishi, S.; Takeuchi, M.; Ishida, S.; Jinnouchi, Y.; Jinnouchi, J.; Imaizumi, T. Atorvastatin Decreases Serum Levels of Advanced Glycation End Products (AGEs) in Patients with Type 2 Diabetes. Clin. Exp. Med. 2006, 6, 191–193. [Google Scholar] [CrossRef]
- Kawai, T.; Takei, I.; Tokui, M.; Funae, O.; Miyamoto, K.; Tabata, M.; Hirata, T.; Saruta, T.; Shimada, A.; Itoh, H. Effects of Epalrestat, an Aldose Reductase Inhibitor, on Diabetic Peripheral Neuropathy in Patients with Type 2 Diabetes, in Relation to Suppression of Nɛ-Carboxymethyl Lysine. J. Diabetes Its Complicat. 2010, 24, 424–432. [Google Scholar] [CrossRef]
- Contreras, C.L.; Guzman-Rosiles, I.; Del Castillo, D.; Gomez-Ojeda, A.; Garay-Sevilla, M.E. Advanced glycation end products (AGEs) and sRAGE levels after benfotiamine treatment in diabetes mellitus type 2. FASEB J. 2017, 31, 646.32. [Google Scholar] [CrossRef]
- Williams, M.E.; Bolton, W.K.; Khalifah, R.G.; Degenhardt, T.P.; Schotzinger, R.J.; McGill, J.B. Effects of Pyridoxamine in Combined Phase 2 Studies of Patients with Type 1 and Type 2 Diabetes and Overt Nephropathy. Am. J. Nephrol. 2007, 27, 605–614. [Google Scholar] [CrossRef]
- Noori, N.; Tabibi, H.; Hosseinpanah, F.; Hedayati, M.; Nafar, M. Effects of Combined Lipoic Acid and Pyridoxine on Albuminuria, Advanced Glycation End-Products, and Blood Pressure in Diabetic Nephropathy. Int. J. Vitam. Nutr. Res. 2013, 83, 77–85. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, W.; Jia, Q.; Feng, Z.; Guo, J.; Han, X.; Liu, Y.; Shang, H.; Wang, Y.; Liu, W.J. High Dose Vitamin E Attenuates Diabetic Nephropathy via Alleviation of Autophagic Stress. Front. Physiol. 2019, 9, 1939. [Google Scholar] [CrossRef] [Green Version]
- Zafar, H.; Sheikh, M.A.; Hussain, F.; Maan, M.A. Inhibition of protein glycation and advanced glycation end products by ascorbic acid. Afr. J. Biotechnol. 2012, 11, 11309–11314. [Google Scholar] [CrossRef]
- Umadevi, S.; Gopi, V.; Vellaichamy, E. Inhibitory Effect of Gallic Acid on Advanced Glycation End Products Induced Up-Regulation of Inflammatory Cytokines and Matrix Proteins in H9C2 (2-1) Cells. Cardiovasc. Toxicol. 2013, 13, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, L.-S.; Dong, L.-L. Inhibitory Effect of Polysaccharides from Pumpkin on Advanced Glycation End-Products Formation and Aldose Reductase Activity. Food Chem. 2012, 130, 821–825. [Google Scholar] [CrossRef]
- Rao, A.R.; Veeresham, C.; Asres, K. In vitro and in vivo inhibitory activities of four indian medicinal plant extracts and their major components on rat aldose reductase and generation of advanced glycation endproducts. Phytother. Res. 2013, 27, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Ni, M.; Zhang, G.; Liao, Y.; Hu, X.; Zhang, Y.; Gong, D. The Inhibition of Oleanolic Acid on Protein Non-Enzymatic Glycation. LWT 2020, 125, 109253. [Google Scholar] [CrossRef]
- Wu, D.; Wen, W.; Qi, C.-L.; Zhao, R.-X.; Lü, J.-H.; Zhong, C.-Y.; Chen, Y.-Y. Ameliorative Effect of Berberine on Renal Damage in Rats with Diabetes Induced by High-Fat Diet and Streptozotocin. Phytomedicine 2012, 19, 712–718. [Google Scholar] [CrossRef]
- Bae, O.-N.; Serfozo, K.; Baek, S.-H.; Lee, K.Y.; Dorrance, A.; Rumbeiha, W.; Fitzgerald, S.D.; Farooq, M.U.; Naravelta, B.; Bhatt, A.; et al. Safety and Efficacy Evaluation of Carnosine, an Endogenous Neuroprotective Agent for Ischemic Stroke. Stroke 2013, 44, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Agustin, A.; Safitri, A.; Fatchiyah, F. An in Silico Approach Reveals the Potential Function of Cyanidin-3-o-Glucoside of Red Rice in Inhibiting the Advanced Glycation End Products (AGES)-Receptor (RAGE) Signaling Pathway. Acta Inform. Med. 2020, 28, 170. [Google Scholar] [CrossRef]
- Ivanova, S.; Batliwalla, F.; Mocco, J.; Kiss, S.; Huang, J.; Mack, W.; Coon, A.; Eaton, J.W.; Al-Abed, Y.; Gregersen, P.K.; et al. Neuroprotection in cerebral ischemia by neutralization of 3-aminopropanal. PNAS 2002, 99, 5579–5584. [Google Scholar] [CrossRef] [Green Version]
- Danielisova, V.; Nemethova, M.; Burda, J. The Protective Effect of Aminoguanidine on Cerebral Ischemic Damage in the Rat Brain. Physiol. Res. 2004, 53, 533–540. [Google Scholar]
- Al-kuraishy, H.M.; Al-Gareeb, A.I.; Alblihed, M.; Cruz-Martins, N.; Batiha, G.E.-S. COVID-19 and Risk of Acute Ischemic Stroke and Acute Lung Injury in Patients with Type II Diabetes Mellitus: The Anti-Inflammatory Role of Metformin. Front. Med. 2021, 8, 644295. [Google Scholar] [CrossRef]
- Nash, R.J.; Kato, A.; Yu, C.-Y.; Fleet, G.W. Iminosugars as Therapeutic Agents: Recent Advances and Promising Trends. Future Med. Chem. 2011, 3, 1513–1521. [Google Scholar] [CrossRef]
- Yang, L.-F.; Shimadate, Y.; Kato, A.; Li, Y.-X.; Jia, Y.-M.; Fleet, G.W.J.; Yu, C.-Y. Synthesis and Glycosidase Inhibition of N-Substituted Derivatives of 1,4-Dideoxy-1,4-Imino-D-Mannitol (DIM). Org. Biomol. Chem. 2020, 18, 999–1011. [Google Scholar] [CrossRef]
- Chennaiah, A.; Dahiya, A.; Dubbu, S.; Vankar, Y.D. A Stereoselective Synthesis of an IminoGlycal: Application in the Synthesis of (-)-1-Epi -Adenophorine and a Homoimindosugar. Eur. J. Org. Chem. 2019, 2019, 2089. [Google Scholar] [CrossRef] [Green Version]
- Chennaiah, A.; Bhowmick, S.; Vankar, Y.D. Conversion of Glycals into Vicinal-1,2-Diazides and 1,2-(or 2,1)-Azidoacetates Using Hypervalent Iodine Reagents and Me3SiN3. Application in the Synthesis of N-Glycopeptides, Pseudo-Trisaccharides and an Iminosugar. RSC Adv. 2017, 7, 41755–41762. [Google Scholar] [CrossRef] [Green Version]
- Rajasekaran, P.; Ande, C.; Vankar, Y.D. Synthesis of (5,6 & 6,6)-Oxa-Oxa Annulated Sugars as Glycosidase Inhibitors from 2-Formyl Galactal Using Iodocyclization as a Key Step. Arkivoc 2022, 2022, 5–23. [Google Scholar] [CrossRef]
- Crema, A.; Bassolino, M.; Guanziroli, E.; Colombo, M.; Blanke, O.; Serino, A.; Micera, S.; Molteni, F. Neuromuscular Electrical Stimulation Restores Upper Limb Sensory-Motor Functions and Body Representations in Chronic Stroke Survivors. Med 2022, 3, 58–74.e10. [Google Scholar] [CrossRef]
- Sanchis-Gomar, F.; Lopez-Lopez, S.; Romero-Morales, C.; Maffulli, N.; Lippi, G.; Pareja-Galeano, H. Neuromuscular Electrical Stimulation: A New Therapeutic Option for Chronic Diseases Based on Contraction-Induced Myokine Secretion. Front. Physiol. 2019, 10, 1463. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, B.K.; Febbraio, M.A. Muscles, Exercise and Obesity: Skeletal Muscle as a Secretory Organ. Nat. Rev. Endocrinol. 2012, 8, 457–465. [Google Scholar] [CrossRef]
- Piccirillo, R. Exercise-Induced Myokines with Therapeutic Potential for Muscle Wasting. Front. Physiol. 2019, 10, 289. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Huang, W.; Peng, J.; Zhu, T.-T.; Sun, X.-L.; Zhou, X.-Y.; Yang, H.; Xiong, J.-F.; He, H.-Q.; Xu, Y.-H.; et al. Irisin Alleviates Advanced Glycation End Products-Induced Inflammation and Endothelial Dysfunction via Inhibiting ROS-NLRP3 Inflammasome Signaling. Inflammation 2017, 41, 260–275. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, C.; Guo, J.; Chen, Y.; Meng, C. The Neuroprotective Effect of Irisin in Ischemic Stroke. Front. Aging Neurosci. 2020, 12, 588958. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Guo, P.; Jin, Z.; Li, X.; Yang, X.; Tang, C.; Wang, Y.; Ke, J. Serum Levels of Irisin Predict Short-Term Outcomes in Ischemic Stroke. Cytokine 2019, 122, 154303. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Chen, L.; Tang, J.; Chen, Y.; Wang, L. The Role of CCL2/CCR2 Axis in Cerebral Ischemia-Reperfusion Injury and Treatment: From Animal Experiments to Clinical Trials. Int. J. Mol. Sci. 2022, 23, 3485. [Google Scholar] [CrossRef] [PubMed]
- Echouffo-Tcheugui, J.B.; Xu, H.; Matsouaka, R.A.; Xian, Y.; Schwamm, L.H.; Smith, E.E.; Bhatt, D.L.; Hernandez, A.F.; Heidenreich, P.A.; Fonarow, G.C. Diabetes and Long-Term Outcomes of Ischaemic Stroke: Findings from Get with the Guidelines-Stroke. Eur. Heart J. 2018, 39, 2376–2386. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| AGEs | |
|---|---|
| Cross-linking | Non-cross-linking |
| Fluorescent: Vesperlysine, Pentosidine, Crossline | Non-fluorescent: N-fructosyl-lysine (FL) N carboxyethyl-lysine (CEL) N-carboxymethyllysine (CML) PyrralineImidazolone |
| Non-fluorescent: Imadazolium dilysine crosslinks Alkyl formyl glycosyl pyrroles Arginine-lysineimidazole crosslinks | |
| Toxic AGEs Glyceraldehyde-derived AGE-2 Glycolaldehyde-derived AGE-3 Acetaldehyde-derived AA-AGE | |
| Investigational Therapeutic Agent, Category | Model | Result | References |
|---|---|---|---|
| Aminoguanidine, Dicarbonyl scavenger AGE cross link breaker | Rat model of focal cerebral ischemia | Aminoguanidine attenuated infarct volume in AGE-treated animals in dose- and time-related manner with cerebral blood flow. | [22] |
| ALT-711 (Alagebrium), AGE cross-link breaker and Aminoguanidine | Mouse, Streptozotocin- induced diabetic apolipoprotein E–deficient (apoE-/-) | ALT-711 and Aminoguanidine reduced vascular AGEs-CML, CEL accumulation, skin collagen solubility and attenuated atherosclerosis. | [79] |
| Soluble RAGE, Competitive inhibitor of RAGE | Cultured human umbilical vein endothelial cells, (HUVECs) and mouse model of partial carotid artery ligation | sRAGE significantly inhibited oscillatory shear stress (OSS)-induced expression of RAGE and HMGB1 in HUVECs. RAGE expression was markedly elevated in the vicinity of atherosclerotic plaque, and administration of sRAGE inhibited plaque development. | [80] |
| endogenous secretory RAGE (esRAGE), Decoy splicing variant RAGE | RAGE knockout, wild-type and human esRAGE overexpressing transgenic, mice | In the BBB system, esRAGE transfer from vascular to the brain side was shown to be RAGE-dependent. esRAGE served as a decoy to prevent neuronal cell death induced by ischemia. | [81] |
| Therapeutic Agent, Category | Model | Result | References |
|---|---|---|---|
| Metformin, Hypoglycemic drug Sulphonylurea, Hypoglycemic drug Acetylsalicylic acid/Aspirin, Anti-inflammatory drug Acarbose, AGE inhibitor with chelating properties, inhibitor ofα-glucosidase Clopidogrel, Glycation-preventing inhibitor of excessive platelet aggregation | in vitro, serum of DM patients with complications, ischemic stroke Single-arm T2DM, hypertension | SR-AI scavenging receptor concentration was significantly reduced in metformin-treated diabetic patients. Correlation was found between ischemic stroke and Melibiose-derived glycation product (MAGE) content. Significant association was found between sulphonylurea intake and a higher sRAGE concentration due to counteraction of effects of AGE formation by sulphonylurea. Aspirin use was significantly associated with decreased total AGE fluorescence which confirmed its effectiveness on glycation inhibition. Significant correlation was found between acarbose intake and fluorescence of total soluble AGEs or soluble pentosidine. Clopidogrel lowered protein-bound AGE and protein-bound Pentosidine fluorescence due to its action of inhibiting fluorescent AGE formation. | [74] |
| Hypoglycemic drugs: Pioglitazone/Thiazolidinediones (TZD) and Metformin Dipeptidyl peptidase 4 (DPP4) inhibitor/ Alogliptin | RCT, T2 DM Single-arm, T2DM | Pioglitazone proved to be superior in amelioration of oxidative stress in comparison with no medication. Pioglitazone and metformin were equally effective in advanced oxidation protein products (AOPP) and AGE decrements. sRAGE concentrations decreased with alogliptin treatment and was associated with HbAlc concentration changes. Albuminuria was reduced after the treatment. | [82,83] |
| Atorvastatin, Lipid-lowering drug | RCT, T2DM, hypercholesterolemia | Atorvastatin significantly reduced AGE, total cholesterol, LDL and triglycerides levels. | [84] |
| Epalrestat, Aldose reductase inhibitor | Observational, diabetic peripheral neuropathy (DPN) | Epalrestat decreased CML and slowed the progression of peripheral diabetic neuropathy. | [85] |
| Vitamin | Model | Result | References |
|---|---|---|---|
| Benfotiamine Lipid-soluble precursor of Thiamine | RCT T2DM | Benfotiamine group had significantly decreased CML-AGE levels and placebo group had significantly decreased sRAGE levels. | [86] |
| Pyridoxamine, a broad inhibitor of advanced glycation | RCT DM, overt nephropathy | Pyridoxamine reduced the serum AGEs-CML and CEL in addition to TGF-β1and urine creatinine levels. There were no differences in urinary albumin excretion. | [87] |
| α-lipoic acid plus Pyridoxine | RCT Diabetic nephropathy | The AGEs-Pentosidine and CML were significantly decreased in the supplemented group.Urinary albumin, serum malondialdehyde (MDA), and systolic blood pressure significantly decreased in the supplemented group compared to the placebo group. Serum NO was increased in the supplemented group compared to the placebo group. | [88] |
| Vitamin E | Invitro, Diabetic nephropathy patients | AGE-Bovine serum albumin exposure enhanced cellular secretion of the renal tubular injury markers Hepatitis A virus cellular receptor 1 (HAVCR1) and Lipocalin 2(LCN2) which was significantly reduced by vitamin E treatment. | [89] |
| Ascorbic acid/ Vitamin C | Plasma levels, T2DM | Antiglycation role of ascorbic acid was evident as increasing the ascorbic acid concentrations greatly diminished protein glycations and inhibited AGE in dose-dependent manner. | [90] |
| Natural Compound | Model | Result | References |
|---|---|---|---|
| Gallic acid, Polyphenol | Cell culture H9C2 (2-1), heart | Significant attenuated expressions of RAGE and other cytokines were found in Gallic acid pre-treated cells. | [91] |
| Cucurbita, Pumpkin Polysaccharides (PPs) | in vitro | Inhibitory effects of PPs on AGEs formation were higher and stronger than the positive control, Aminoguanidine. | [92] |
| Terpenoids: Ursolic acid Oleanolic acid (OA) | Rat, in vitro and in vivo in vitro | Urosolic acid showed the most potent Aldose reductase inhibitory action and suppressed RAGE expression. OA almost completely inhibited AGE formation. | [93,94] |
| Berberine, Alkaloid | Rat model of diabetes | Berberine inhibited AGE accumulation, and improved antioxidant capacity with protective effects against diabeticrenal damage. | [95] |
| Carnosine, Pleiotropic dipeptide | Rat, ex vivobrain homogenates and primary neuronal/astrocytic cultures | Carnosine treatment exhibited significant cerebroprotection against histological and functional damage in both permanent and transient ischemic models. | [96] |
| Cyanidin-3-O-glucoside Anthocyanin of red rice | in silico analysis | Cyanidin-3-O-glucoside could bind to RAGE at the same residue as AGEs -Argypirimidine and Pyrralline, which indicated that it might have a biological function as a competitive inhibitor of AGEs-RAGE interactions through AGEs-cyanidin-3-O-glucoside-RAGE complex establishment. | [97] |
| Investigational Therapeutic Agents, Category | Model | Result | Ref. |
|---|---|---|---|
| HMGB1 axis antagonists: Soluble RAGE (sRAGE); recombinant A neutralising anti-HMGB1 antibody | Ischemic stroke (IS) patients and C57BL/6J mouse model of focal ischemic stroke Mouse model of cerebral ischemia | Within 48 h of IS, sRAGE and HMGB1 plasma levels considerably rose. Recombinant sRAGE significantly reduced immune cell infiltration, enhanced mouse injury outcome, and ameliorated the negative effects of recombinant HMGB1. Ischemic brain damage was ameliorated by a neutralising anti-HMGB1 antibody and HMGB1 box A, an antagonist of HMGB1 at RAGE. Infarct size was decreased by soluble RAGE and genetic RAGE deficiency. | [37,52] |
| BLT Receptor antagonist LY255283 | Ischemic stroke patients, Rat stroke model | Compared to early post-stroke levels, plasma LTB4 levels surged quickly, roughly doubling in just 24 h. LY255283 reduced the size of the infarct. | [59] |
| Polyamine-eIF5A-hypusine Axis inhibitors: N1-guanyl-1,7-diaminoheptane (GC7) N1-Nonyl-1,4-diaminobutane (C9-4) N-2-mercaptopropionyl glycine (N-2-MPG) | in vivo transient focal cerebral ischemia mouse model Photochemically induced thrombosis model mice in vitroscreening of clinically approved drugs in rat model of middle cerebral artery occlusion, neuronal cell line (HTB11), glial cell line (HTB14) | A single GC7 pre- or post-treatment significantly reduced infarct volume post-stroke. Post-treatment GC7 significantly improved motor and cognitive post-stroke deficits. C9-4 reduced the volume of brain infarction significantly, demonstrated to be potent inhibitor of polyamine-oxidizing enzymes. in vitro, N-2-MPG significantly inhibited cytotoxicity of 3-aminopropanal (reactive catabolite ofpolyamines) and in rats it reduced infarct volume, even when the agent was administered after ischemia onset. | [69,70,98] |
| Nitric oxide synthase antagonists: Aminoguanidine (AG) Quercetin and its analogues | Rat model cerebral ischemia Validated Homology modeling for human inducible NO synthase (NOS), Molecular docking studies | NO production via inducible NOS was suppressed by Aminoguanidine. NO contributed to delay in recovery from brain neuronal damage in hippocampus following ischemic brain injury. Highly favourable interactions of Quercetin and its derivatives occurred on NOS involving ligand–protein and docking scores. Quercetine and derivatives were found to be suitable molecules for testing as anti-stroke agents. | [67,99] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rao, N.L.; Kotian, G.B.; Shetty, J.K.; Shelley, B.P.; Dmello, M.K.; Lobo, E.C.; Shankar, S.P.; Almeida, S.D.; Shah, S.R. Receptor for Advanced Glycation End Product, Organ Crosstalk, and Pathomechanism Targets for Comprehensive Molecular Therapeutics in Diabetic Ischemic Stroke. Biomolecules 2022, 12, 1712. https://doi.org/10.3390/biom12111712
Rao NL, Kotian GB, Shetty JK, Shelley BP, Dmello MK, Lobo EC, Shankar SP, Almeida SD, Shah SR. Receptor for Advanced Glycation End Product, Organ Crosstalk, and Pathomechanism Targets for Comprehensive Molecular Therapeutics in Diabetic Ischemic Stroke. Biomolecules. 2022; 12(11):1712. https://doi.org/10.3390/biom12111712
Chicago/Turabian StyleRao, Nivedita L., Greeshma B. Kotian, Jeevan K. Shetty, Bhaskara P. Shelley, Mackwin Kenwood Dmello, Eric C. Lobo, Suchetha Padar Shankar, Shellette D. Almeida, and Saiqa R. Shah. 2022. "Receptor for Advanced Glycation End Product, Organ Crosstalk, and Pathomechanism Targets for Comprehensive Molecular Therapeutics in Diabetic Ischemic Stroke" Biomolecules 12, no. 11: 1712. https://doi.org/10.3390/biom12111712
APA StyleRao, N. L., Kotian, G. B., Shetty, J. K., Shelley, B. P., Dmello, M. K., Lobo, E. C., Shankar, S. P., Almeida, S. D., & Shah, S. R. (2022). Receptor for Advanced Glycation End Product, Organ Crosstalk, and Pathomechanism Targets for Comprehensive Molecular Therapeutics in Diabetic Ischemic Stroke. Biomolecules, 12(11), 1712. https://doi.org/10.3390/biom12111712