
New Insights into Microglial Mechanisms of Memory Impairment in Alzheimer’s Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
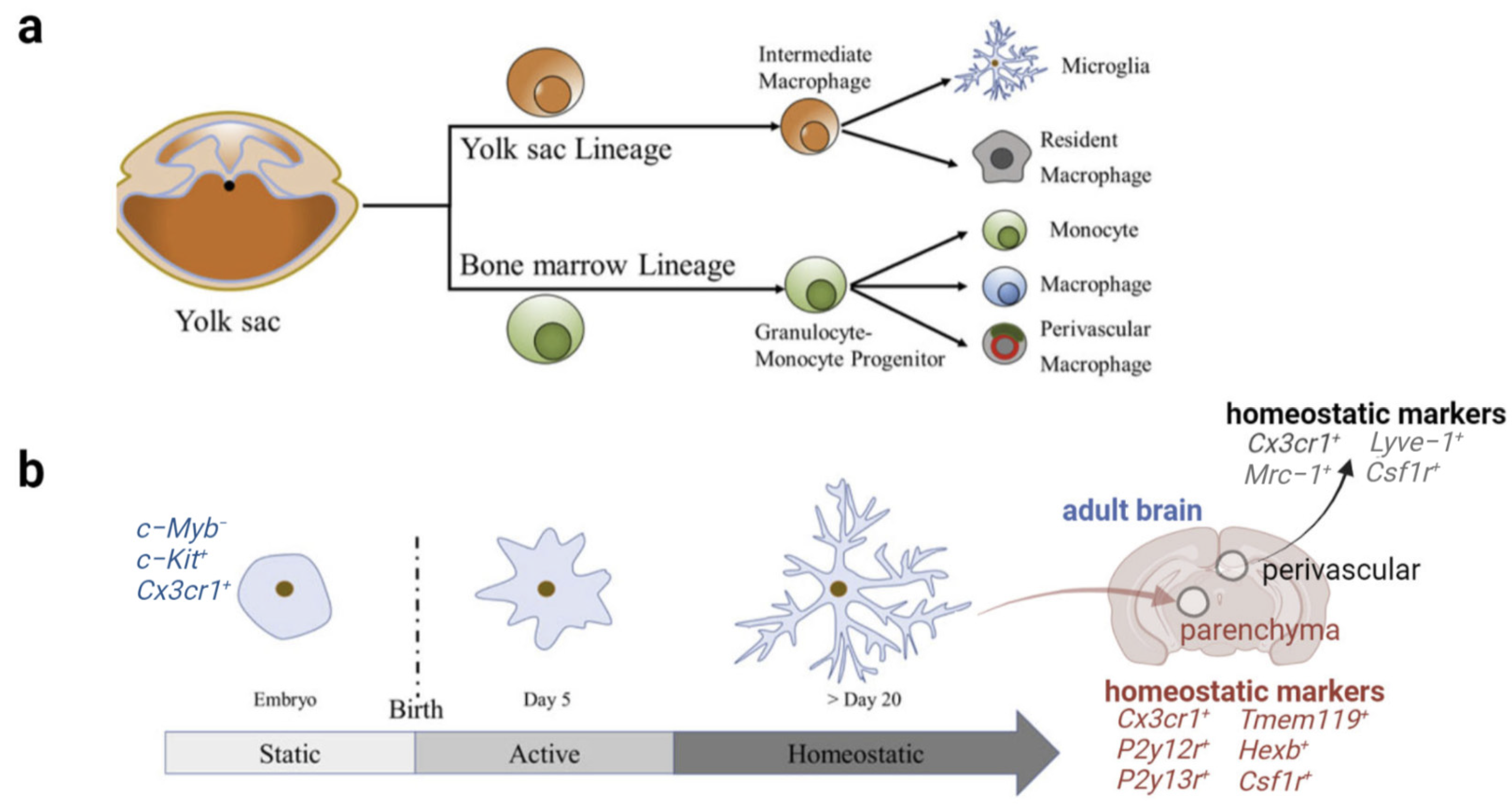
2. Origin and Physiological Function of Microglia
2.1. Origin and Morphology of Microglia
2.2. Multidimensional States and Phenotypes of Microglia
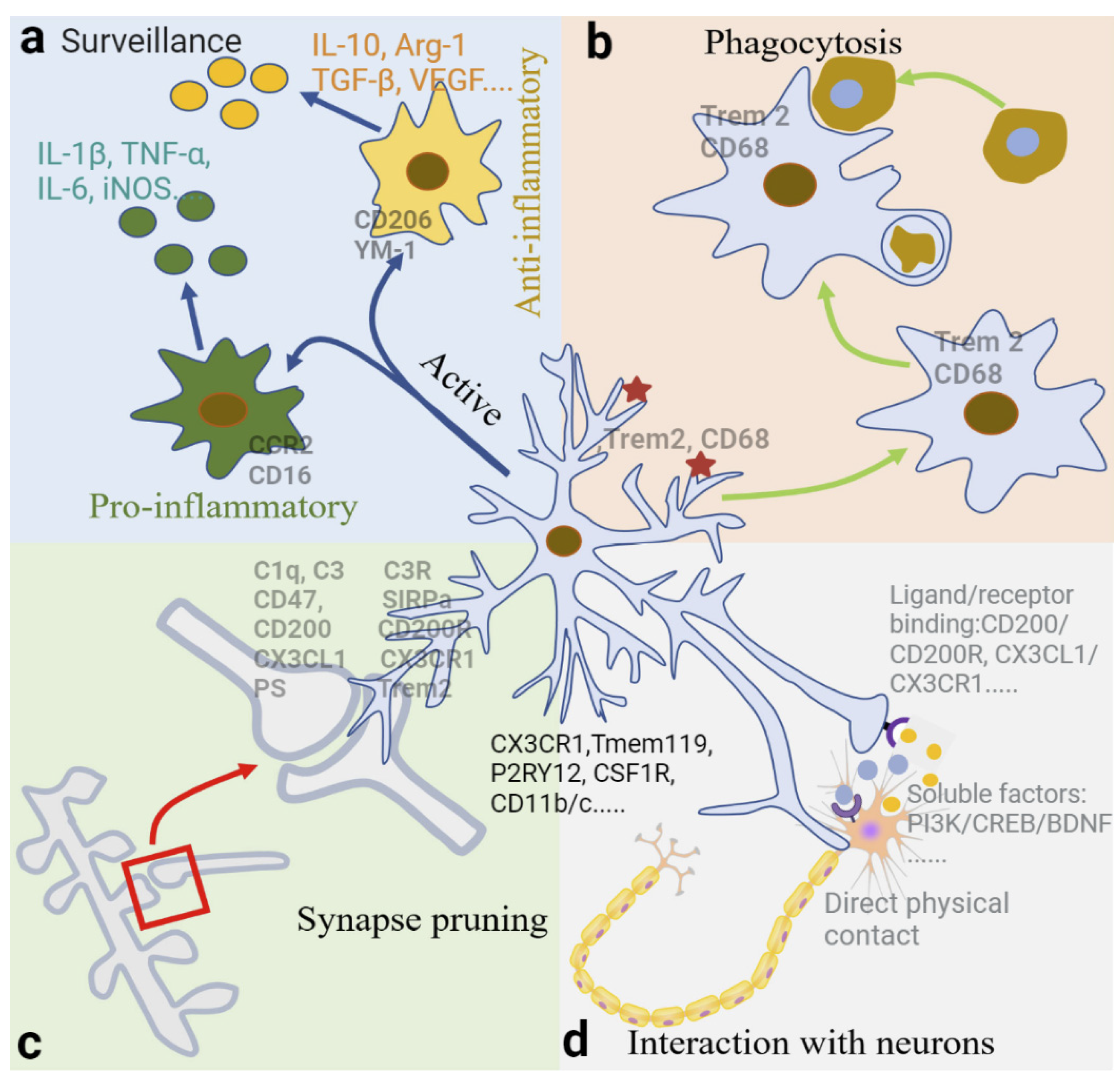
2.3. Physiological Functions of Microglia
2.3.1. Surveillance
2.3.2. Phagocytosis
2.3.3. Synapse Pruning and Elimination by Phagocytosis
2.3.4. Interaction between Neurons and Microglia
2.3.5. Maintenance of the NVU Integrity and the BBB Permeability
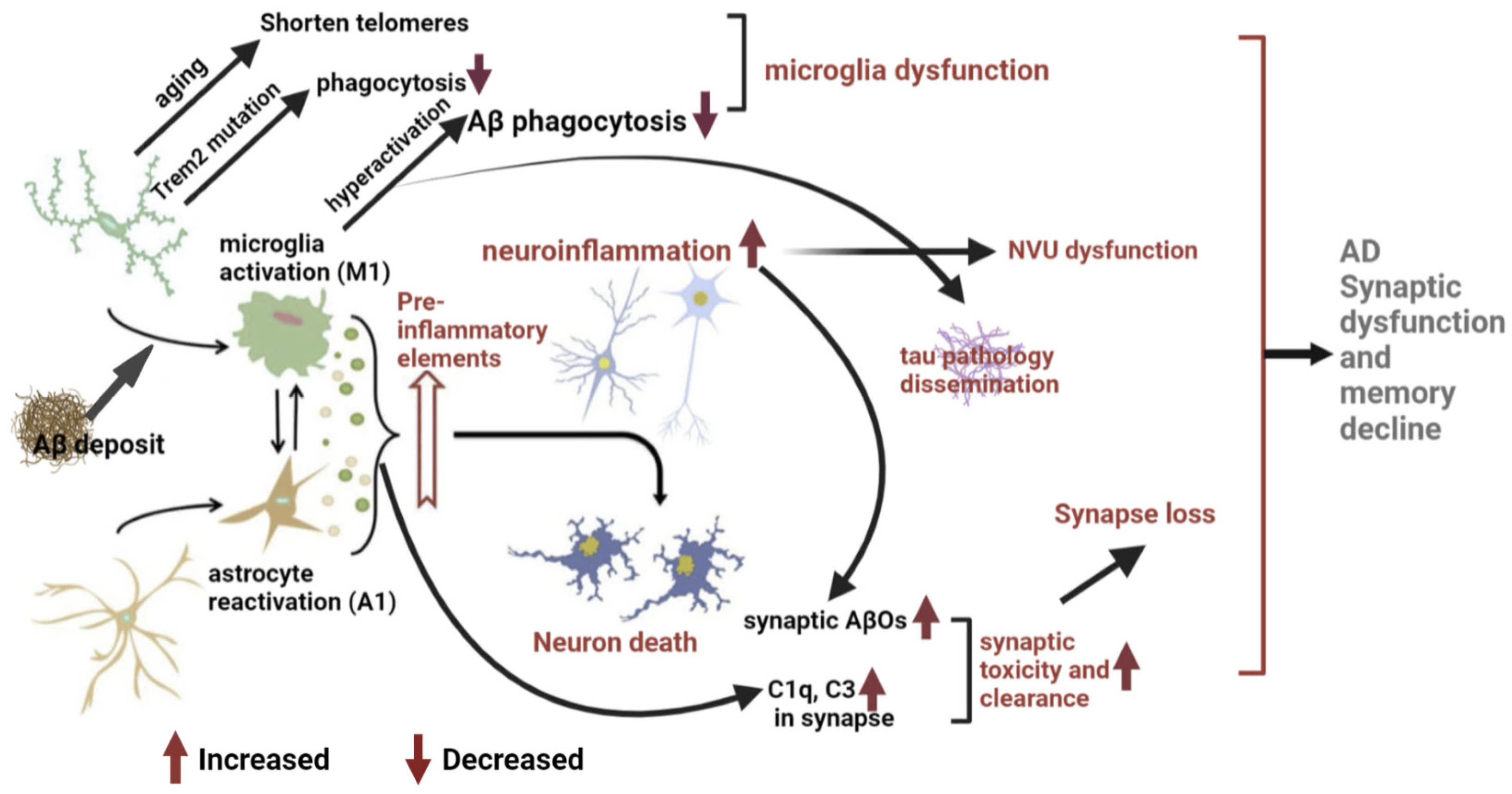
3. Microglial Mechanisms of AD Pathogenesis and Memory Impairment
3.1. Neuro-Inflammation Mechanism
3.2. Microglial Dysfunction Mechanism
3.3. Pathological Synaptic Loss and Dysfunction Mechanism
3.3.1. Synapse Loss Associated with Microglia
3.3.2. Aβ-Induced Synaptic Toxicity and Dysfunction
3.4. Microglia Drive Tau Pathology and Dissemination
3.5. Pathological NVU Dysfunction in AD
4. Clinical Implication
4.1. Inhibition of Neuro-Inflammation
4.2. Regulating Microglial Phenotype and Improving Microglial Dysfunction
4.3. Intervention of Microglia Priming
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hussain, R.; Zubair, H.; Pursell, S.; Shahab, M. Neurodegenerative Diseases: Regenerative Mechanisms and Novel Therapeutic Approaches. Brain Sci. 2018, 8, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.S.; Tan, Z.X.; Wu, L.Y.; Dong, F.; Zhang, F. The involvement of NLRP3 inflammasome in the treatment of Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101192. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Mobley, W.C. Exploring the Pathogenesis of Alzheimer Disease in Basal Forebrain Cholinergic Neurons: Converging Insights From Alternative Hypotheses. Front. Neurosci. 2019, 13, 446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowrishankar, S.; Yuan, P.; Wu, Y.; Schrag, M.; Paradise, S.; Grutzendler, J.; De Camilli, P.; Ferguson, S.M. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc. Natl. Acad. Sci. USA 2015, 112, E3699–E3708. [Google Scholar] [CrossRef] [Green Version]
- Gomes, B.A.Q.; Silva, J.P.B.; Romeiro, C.F.R.; Dos Santos, S.M.; Rodrigues, C.A.; Goncalves, P.R.; Sakai, J.T.; Mendes, P.F.S.; Varela, E.L.P.; Monteiro, M.C. Neuroprotective Mechanisms of Resveratrol in Alzheimer’s Disease: Role of SIRT1. Oxid. Med. Cell Longev. 2018, 2018, 8152373. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.L.; Gandhi, S. Crucial role of protein oligomerization in the pathogenesis of Alzheimer’s and Parkinson’s diseases. Febs. J. 2018, 285, 3631–3644. [Google Scholar] [CrossRef] [Green Version]
- Androuin, A.; Potier, B.; Nägerl, U.V.; Cattaert, D.; Danglot, L.; Thierry, M.; Youssef, I.; Triller, A.; Duyckaerts, C.; El Hachimi, K.H.; et al. Evidence for altered dendritic spine compartmentalization in Alzheimer’s disease and functional effects in a mouse model. Acta Neuropathol. 2018, 135, 839–854. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Spangenberg, E.E.; Green, K.N. Inflammation in Alzheimer’s disease: Lessons learned from microglia-depletion models. Brain Behav. Immun. 2017, 61, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wegiel, J.; Wang, K.C.; Imaki, H.; Rubenstein, R.; Wronska, A.; Osuchowski, M.; Lipinski, W.J.; Walker, L.C.; LeVine, H. The role of microglial cells and astrocytes in fibrillar plaque evolution in transgenic APP(SW) mice. Neurobiol. Aging 2001, 22, 49–61. [Google Scholar] [CrossRef]
- d’Errico, P.; Ziegler-Waldkirch, S.; Aires, V.; Hoffmann, P.; Mezö, C.; Erny, D.; Monasor, L.S.; Liebscher, S.; Ravi, V.M.; Joseph, K.; et al. Microglia contribute to the propagation of Aβ into unaffected brain tissue. Nat. Neurosci. 2022, 25, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Edison, P.; Donat, C.K.; Sastre, M. In vivo Imaging of Glial Activation in Alzheimer’s Disease. Front. Neurol. 2018, 9, 625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQuade, A.; Blurton-Jones, M. Microglia in Alzheimer’s Disease: Exploring How Genetics and Phenotype Influence Risk. J. Mol. Biol. 2019, 431, 1805–1817. [Google Scholar] [CrossRef]
- Aloni, E.; Oni-Biton, E.; Tsoory, M.; Moallem, D.H.; Segal, M. Synaptopodin Deficiency Ameliorates Symptoms in the 3 × Tg Mouse Model of Alzheimer’s Disease. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 3983–3992. [Google Scholar] [CrossRef] [Green Version]
- Ohm, D.T.; Fought, A.J.; Martersteck, A.; Coventry, C.; Sridhar, J.; Gefen, T.; Weintraub, S.; Bigio, E.; Mesulam, M.M.; Rogalski, E.; et al. Accumulation of neurofibrillary tangles and activated microglia is associated with lower neuron densities in the aphasic variant of Alzheimer’s disease. Brain Pathol. 2021, 31, 189–204. [Google Scholar] [CrossRef] [PubMed]
- Sebastian Monasor, L.; Müller, S.A.; Colombo, A.V.; Tanrioever, G.; König, J.; Roth, S.; Liesz, A.; Berghofer, A.; Piechotta, A.; Prestel, M.; et al. Fibrillar Aβ triggers microglial proteome alterations and dysfunction in Alzheimer mouse models. eLife 2020, 9, 54083. [Google Scholar] [CrossRef] [PubMed]
- Triviño, J.J.; von Bernhardi, R. The effect of aged microglia on synaptic impairment and its relevance in neurodegenerative diseases. Neurochem. Int. 2021, 144, 104982. [Google Scholar] [CrossRef]
- Grubman, A.; Choo, X.Y.; Chew, G.; Ouyang, J.F.; Sun, G.; Croft, N.P.; Rossello, F.J.; Simmons, R.; Buckberry, S.; Landin, D.V.; et al. Transcriptional signature in microglia associated with Aβ plaque phagocytosis. Nat. Commun. 2021, 12, 3015. [Google Scholar] [CrossRef]
- Marioni, R.E.; Harris, S.E.; Zhang, Q.; McRae, A.F.; Hagenaars, S.P.; Hill, W.D.; Davies, G.; Ritchie, C.W.; Gale, C.R.; Starr, J.M.; et al. GWAS on family history of Alzheimer’s disease. Transl. Psychiatry 2018, 8, 99. [Google Scholar] [CrossRef] [Green Version]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e613. [Google Scholar] [CrossRef] [PubMed]
- Tay, T.L.; Savage, J.C.; Hui, C.W.; Bisht, K.; Tremblay, M. Microglia across the lifespan: From origin to function in brain development, plasticity and cognition. J. Physiol. 2017, 595, 1929–1945. [Google Scholar] [CrossRef] [Green Version]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y. The microglia-blood vessel interactions in the developing brain. Neurosci. Res. 2022, 24, 00248–00249. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Su, P.P.; Leff, J.; Gao, X.; Chen, J.; Guan, A.K.; Kalyanasundaram, G.; Ma, A.; Guan, Z. Distinct phases of adult microglia proliferation: A Myc-mediated early phase and a Tnfaip3-mediated late phase. Cell Discov. 2022, 8, 34. [Google Scholar] [CrossRef]
- De Haas, A.H.; Boddeke, H.W.; Biber, K. Region-specific expression of immunoregulatory proteins on microglia in the healthy CNS. Glia 2008, 56, 888–894. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Smolders, S.M.; Kessels, S.; Vangansewinkel, T.; Rigo, J.M.; Legendre, P.; Brône, B. Microglia: Brain cells on the move. Prog. Neurobiol. 2019, 178, 101612. [Google Scholar] [CrossRef]
- Streit, W.J.; Graeber, M.B.; Kreutzberg, G.W. Functional plasticity of microglia: A review. Glia 1988, 1, 301–307. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Thion, M.S.; Ginhoux, F.; Garel, S. Microglia and early brain development: An intimate journey. Science 2018, 362, 185–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenz, K.M.; Nelson, L.H. Microglia and Beyond: Innate Immune Cells As Regulators of Brain Development and Behavioral Function. Front. Immunol. 2018, 9, 698. [Google Scholar] [CrossRef] [Green Version]
- Sierra, A.; Paolicelli, R.C.; Kettenmann, H. Cien Años de Microglía: Milestones in a Century of Microglial Research. Trends Neurosci. 2019, 42, 778–792. [Google Scholar] [CrossRef] [Green Version]
- Cornell, J.; Salinas, S.; Huang, H.Y.; Zhou, M. Microglia regulation of synaptic plasticity and learning and memory. Neural. Regen. Res. 2022, 17, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Kyrargyri, V.; Attwell, D.; Jolivet, R.B.; Madry, C. Analysis of Signaling Mechanisms Regulating Microglial Process Movement. Methods Mol. Biol. (Clifton N.J.) 2019, 2034, 191–205. [Google Scholar]
- Okajima, T.; Tsuruta, F. Microglial dynamics during brain development. Neural. Regen. Res. 2018, 13, 222–223. [Google Scholar] [PubMed]
- Uddin, M.S.; Lim, L.W. Glial cells in Alzheimer’s disease: From neuropathological changes to therapeutic implications. Ageing Res. Rev. 2022, 78, 101622. [Google Scholar] [CrossRef]
- Erny, D.; Prinz, M. How microbiota shape microglial phenotypes and epigenetics. Glia 2020, 68, 1655–1672. [Google Scholar] [CrossRef]
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2020, 98, 28–41. [Google Scholar] [CrossRef]
- Malik, B.R.; Maddison, D.C.; Smith, G.A.; Peters, O.M. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol. Brain 2019, 12, 100. [Google Scholar] [CrossRef] [PubMed]
- Van Weering, J.R.T.; Scheper, W. Endolysosome and Autolysosome Dysfunction in Alzheimer’s Disease: Where Intracellular and Extracellular Meet. CNS Drugs 2019, 33, 639–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, C.A.; Popescu, A.S.; Kitchener, E.J.A.; Allendorf, D.H.; Puigdellívol, M.; Brown, G.C. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J. Neurochem. 2021, 158, 621–639. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Aparicio, I.; Paris, I.; Sierra-Torre, V.; Plaza-Zabala, A.; Rodríguez-Iglesias, N.; Márquez-Ropero, M.; Beccari, S.; Huguet, P.; Abiega, O.; Alberdi, E.; et al. Microglia Actively Remodel Adult Hippocampal Neurogenesis through the Phagocytosis Secretome. J. Neurosci. Off. J. Soc. Neurosci. 2020, 40, 1453–1482. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.N.; Appel, B. Microglia phagocytose myelin sheaths to modify developmental myelination. Nat. Neurosci. 2020, 23, 1055–1066. [Google Scholar] [CrossRef]
- Gupta, N.; Shyamasundar, S.; Patnala, R.; Karthikeyan, A.; Arumugam, T.V.; Ling, E.A.; Dheen, S.T. Recent progress in therapeutic strategies for microglia-mediated neuroinflammation in neuropathologies. Expert Opin. Ther. Targets 2018, 22, 765–781. [Google Scholar] [CrossRef]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- Badimon, A.; Strasburger, H.J.; Ayata, P.; Chen, X.; Nair, A.; Ikegami, A.; Hwang, P.; Chan, A.T.; Graves, S.M.; Uweru, J.O.; et al. Negative feedback control of neuronal activity by microglia. Nature 2020, 586, 417–423. [Google Scholar] [CrossRef]
- Streit, W.J.; Xue, Q.S.; Tischer, J.; Bechmann, I. Microglial pathology. Acta Neuropathol. Commun. 2014, 2, 142. [Google Scholar] [CrossRef]
- Xie, L.; Zhang, N.; Zhang, Q.; Li, C.; Sandhu, A.F.; Iii, G.W.; Lin, S.; Lv, P.; Liu, Y.; Wu, Q.; et al. Inflammatory factors and amyloid β-induced microglial polarization promote inflammatory crosstalk with astrocytes. Aging 2020, 12, 22538–22549. [Google Scholar] [CrossRef]
- Polazzi, E.; Monti, B. Microglia and neuroprotection: From in vitro studies to therapeutic applications. Prog. Neurobiol. 2010, 92, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Bohlen, C.J.; Bennett, F.C.; Bennett, M.L. Isolation and Culture of Microglia. Curr. Protoc. Immunol. 2019, 125, e70. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [Green Version]
- Schwabenland, M.; Brück, W.; Priller, J.; Stadelmann, C.; Lassmann, H.; Prinz, M. Analyzing microglial phenotypes across neuropathologies: A practical guide. Acta Neuropathol. 2021, 142, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Favuzzi, E.; Huang, S.; Saldi, G.A.; Binan, L.; Ibrahim, L.A.; Fernández-Otero, M.; Cao, Y.; Zeine, A.; Sefah, A.; Zheng, K.; et al. GABA-receptive microglia selectively sculpt developing inhibitory circuits. Cell 2021, 184, 4048–4063.e4032. [Google Scholar] [CrossRef]
- Soteros, B.M.; Sia, G.M. Complement and microglia dependent synapse elimination in brain development. WIREs Mech. Dis. 2022, 14, e1545. [Google Scholar] [CrossRef]
- Liu, Y.J.; Spangenberg, E.E.; Tang, B.; Holmes, T.C.; Green, K.N.; Xu, X. Microglia Elimination Increases Neural Circuit Connectivity and Activity in Adult Mouse Cortex. J. Neurosci. Off. J. Soc. Neurosci. 2021, 41, 1274–1287. [Google Scholar] [CrossRef]
- Henry, R.J.; Ritzel, R.M.; Barrett, J.P.; Doran, S.J.; Jiao, Y.; Leach, J.B.; Szeto, G.L.; Wu, J.; Stoica, B.A.; Faden, A.I.; et al. Microglial Depletion with CSF1R Inhibitor During Chronic Phase of Experimental Traumatic Brain Injury Reduces Neurodegeneration and Neurological Deficits. J. Neurosci. Off. J. Soc. Neurosci. 2020, 40, 2960–2974. [Google Scholar] [CrossRef]
- Wu, T.; Dejanovic, B.; Gandham, V.D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Srinivasan, K.; Huntley, M.A.; Wang, Y.; Wang, T.M.; et al. Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Rep. 2019, 28, 2111–2123.e2116. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.R.; Zhang, J.; Steele, M.R.; Romero, C.O.; Kautzman, A.G.; Schafer, D.P.; Vetter, M.L. Complement Targets Newborn Retinal Ganglion Cells for Phagocytic Elimination by Microglia. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 2025–2040. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yue, H.; Hu, Z.; Shen, Y.; Ma, J.; Li, J.; Wang, X.D.; Wang, L.; Sun, B.; Shi, P.; et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science 2020, 367, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 2018, 48, 979–991.e978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott-Hewitt, N.; Perrucci, F.; Morini, R.; Erreni, M.; Mahoney, M.; Witkowska, A.; Carey, A.; Faggiani, E.; Schuetz, L.T.; Mason, S.; et al. Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. EMBO J. 2020, 39, e105380. [Google Scholar] [CrossRef]
- Gunner, G.; Cheadle, L.; Johnson, K.M.; Ayata, P.; Badimon, A.; Mondo, E.; Nagy, M.A.; Liu, L.; Bemiller, S.M.; Kim, K.W.; et al. Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat. Neurosci. 2019, 22, 1075–1088. [Google Scholar] [CrossRef]
- Lehrman, E.K.; Wilton, D.K.; Litvina, E.Y.; Welsh, C.A.; Chang, S.T.; Frouin, A.; Walker, A.J.; Heller, M.D.; Umemori, H.; Chen, C.; et al. CD47 Protects Synapses from Excess Microglia-Mediated Pruning during Development. Neuron 2018, 100, 120–134.e126. [Google Scholar] [CrossRef] [Green Version]
- Sato-Hashimoto, M.; Nozu, T.; Toriba, R.; Horikoshi, A.; Akaike, M.; Kawamoto, K.; Hirose, A.; Hayashi, Y.; Nagai, H.; Shimizu, W.; et al. Microglial SIRPα regulates the emergence of CD11c(+) microglia and demyelination damage in white matter. eLife 2019, 8, e42025. [Google Scholar] [CrossRef]
- Sun, H.; He, X.; Tao, X.; Hou, T.; Chen, M.; He, M.; Liao, H. The CD200/CD200R signaling pathway contributes to spontaneous functional recovery by enhancing synaptic plasticity after stroke. J. Neuroinflamm. 2020, 17, 171. [Google Scholar] [CrossRef]
- Raghuraman, R.; Karthikeyan, A.; Wei, W.L.; Dheen, S.T.; Sajikumar, S. Activation of microglia in acute hippocampal slices affects activity-dependent long-term potentiation and synaptic tagging and capture in area CA1. Neurobiol. Learn. Mem. 2019, 163, 107039. [Google Scholar] [CrossRef] [PubMed]
- Brunialti, E.; Villa, A.; Mekhaeil, M.; Mornata, F.; Vegeto, E.; Maggi, A.; Di Monte, D.A.; Ciana, P. Inhibition of microglial β-glucocerebrosidase hampers the microglia-mediated antioxidant and protective response in neurons. J. Neuroinflamm. 2021, 18, 220. [Google Scholar] [CrossRef]
- Akiyoshi, R.; Wake, H.; Kato, D.; Horiuchi, H.; Ono, R.; Ikegami, A.; Haruwaka, K.; Omori, T.; Tachibana, Y.; Moorhouse, A.J.; et al. Microglia Enhance Synapse Activity to Promote Local Network Synchronization. eNeuro 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Baalman, K.; Marin, M.A.; Ho, T.S.; Godoy, M.; Cherian, L.; Robertson, C.; Rasband, M.N. Axon initial segment-associated microglia. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 2283–2292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cserép, C.; Pósfai, B.; Lénárt, N.; Fekete, R.; László, Z.I.; Lele, Z.; Orsolits, B.; Molnár, G.; Heindl, S.; Schwarcz, A.D.; et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 2020, 367, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Ronzano, R.; Roux, T.; Thetiot, M.; Aigrot, M.S.; Richard, L.; Lejeune, F.X.; Mazuir, E.; Vallat, J.M.; Lubetzki, C.; Desmazières, A. Microglia-neuron interaction at nodes of Ranvier depends on neuronal activity through potassium release and contributes to remyelination. Nat. Commun. 2021, 12, 5219. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Feng, D.; Huang, A.; Yan, W.; Chen, D. CD200 dysfunction in neuron contributes to synaptic deficits and cognitive impairment. Biochem. Biophys. Res. Commun. 2019, 516, 1053–1059. [Google Scholar] [CrossRef]
- Zlotnik, A.; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Choi, J.; Yoon, B.E. Neuron-Glia Interactions in Neurodevelopmental Disorders. Cells 2020, 9, 2176. [Google Scholar] [CrossRef]
- Watson, A.E.S.; Goodkey, K.; Footz, T.; Voronova, A. Regulation of CNS precursor function by neuronal chemokines. Neurosci. Lett. 2020, 715, 134533. [Google Scholar] [CrossRef]
- Saw, G.; Krishna, K.; Gupta, N.; Soong, T.W.; Mallilankaraman, K.; Sajikumar, S.; Dheen, S.T. Epigenetic regulation of microglial phosphatidylinositol 3-kinase pathway involved in long-term potentiation and synaptic plasticity in rats. Glia 2020, 68, 656–669. [Google Scholar] [CrossRef]
- Lin, L.; Chen, X.; Zhou, Q.; Huang, P.; Jiang, S.; Wang, H.; Deng, Y. Synaptic structure and alterations in the hippocampus in neonatal rats exposed to lipopolysaccharide. Neurosci. Lett. 2019, 709, 134364. [Google Scholar] [CrossRef] [PubMed]
- Vainchtein, I.D.; Chin, G.; Cho, F.S.; Kelley, K.W.; Miller, J.G.; Chien, E.C.; Liddelow, S.A.; Nguyen, P.T.; Nakao-Inoue, H.; Dorman, L.C.; et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 2018, 359, 1269–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitu, V.; Biundo, F.; Stanley, E.R. Colony stimulating factors in the nervous system. Semin. Immunol. 2021, 54, 101511. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.U.; Ying, Y.; Li, Y.; Eyo, U.B.; Chen, T.; Zheng, J.; Umpierre, A.D.; Zhu, J.; Bosco, D.B.; Dong, H.; et al. Neuronal network activity controls microglial process surveillance in awake mice via norepinephrine signaling. Nat. Neurosci. 2019, 22, 1771–1781. [Google Scholar] [CrossRef] [PubMed]
- Soto-Rojas, L.O.; Campa-Córdoba, B.B.; Harrington, C.R.; Salas-Casas, A.; Hernandes-Alejandro, M.; Villanueva-Fierro, I.; Bravo-Muñoz, M.; Garcés-Ramírez, L.; De La Cruz-López, F.; Ontiveros-Torres, M.; et al. Insoluble Vascular Amyloid Deposits Trigger Disruption of the Neurovascular Unit in Alzheimer’s Disease Brains. Int. J. Mol. Sci. 2021, 22, 3654. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Gamdzyk, M.; Lenahan, C.; Tang, J.; Tan, S.; Zhang, J.H. The Dual Role of Microglia in Blood-Brain Barrier Dysfunction after Stroke. Curr. Neuropharmacol. 2020, 18, 1237–1249. [Google Scholar] [CrossRef]
- Fu, A.K.; Hung, K.W.; Yuen, M.Y.; Zhou, X.; Mak, D.S.; Chan, I.C.; Cheung, T.H.; Zhang, B.; Fu, W.Y.; Liew, F.Y.; et al. IL-33 ameliorates Alzheimer’s disease-like pathology and cognitive decline. Proc. Natl. Acad. Sci. USA 2016, 113, E2705–E2713. [Google Scholar] [CrossRef] [Green Version]
- Morgan, A.R.; Touchard, S.; Leckey, C.; O’Hagan, C.; Nevado-Holgado, A.J.; Consortium, N.; Barkhof, F.; Bertram, L.; Blin, O.; Bos, I.; et al. Inflammatory biomarkers in Alzheimer’s disease plasma. Alzheimers Dement. 2019, 15, 776–787. [Google Scholar] [CrossRef]
- Nordengen, K.; Kirsebom, B.E.; Henjum, K.; Selnes, P.; Gisladottir, B.; Wettergreen, M.; Torsetnes, S.B.; Grontvedt, G.R.; Waterloo, K.K.; Aarsland, D.; et al. Glial activation and inflammation along the Alzheimer’s disease continuum. J. Neuroinflamm. 2019, 16, 46. [Google Scholar] [CrossRef] [Green Version]
- Ju Hwang, C.; Choi, D.Y.; Park, M.H.; Hong, J.T. NF-κB as a Key Mediator of Brain Inflammation in Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets 2019, 18, 3–10. [Google Scholar] [CrossRef]
- Velásquez, E.; Szeitz, B.; Gil, J.; Rodriguez, J.; Palkovits, M.; Renner, É.; Hortobágyi, T.; Döme, P.; Nogueira, F.C.; Marko-Varga, G.; et al. Topological Dissection of Proteomic Changes Linked to the Limbic Stage of Alzheimer’s Disease. Front. Immunol. 2021, 12, 750665. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, H.; Liu, B.; Zhang, Y.; Pan, X.; Yu, X.Y.; Shen, Z.; Song, Y.H. Inflammasomes as therapeutic targets in human diseases. Signal Transduct. Target. 2021, 6, 247. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Fan, L.; Khawaja, R.R.; Liu, B.; Zhan, L.; Kodama, L.; Chin, M.; Li, Y.; Le, D.; Zhou, Y.; et al. Microglial NF-κB drives tau spreading and toxicity in a mouse model of tauopathy. Nat. Commun. 2022, 13, 1969. [Google Scholar] [CrossRef] [PubMed]
- Baj, T.; Seth, R. Role of Curcumin in Regulation of TNF-α Mediated Brain Inflammatory Responses. Recent Pat. Inflamm. Allergy Drug Discov. 2018, 12, 69–77. [Google Scholar] [CrossRef]
- Liu, X.G. Normalization of Neuroinflammation: A New Strategy for Treatment of Persistent Pain and Memory/Emotional Deficits in Chronic Pain. J. Inflamm. Res. 2022, 15, 5201–5233. [Google Scholar] [CrossRef]
- Schmid, A.W.; Lynch, M.A.; Herron, C.E. The effects of IL-1 receptor antagonist on beta amyloid mediated depression of LTP in the rat CA1 in vivo. Hippocampus 2009, 19, 670–676. [Google Scholar] [CrossRef]
- Rajesh, Y.; Kanneganti, T.D. Innate Immune Cell Death in Neuroinflammation and Alzheimer’s Disease. Cells 2022, 11, 3654. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Uddin, M.S.; Tewari, D.; Mamun, A.A.; Kabir, M.T.; Niaz, K.; Wahed, M.I.I.; Barreto, G.E.; Ashraf, G.M. Circadian and sleep dysfunction in Alzheimer’s disease. Ageing Res. Rev. 2020, 60, 101046. [Google Scholar] [CrossRef]
- Bisht, K.; Sharma, K.; Tremblay, M.E. Chronic stress as a risk factor for Alzheimer’s disease: Roles of microglia-mediated synaptic remodeling, inflammation, and oxidative stress. Neurobiol. Stress 2018, 9, 9–21. [Google Scholar] [CrossRef]
- Caruso, G.; Caraci, F.; Jolivet, R.B. Pivotal role of carnosine in the modulation of brain cells activity: Multimodal mechanism of action and therapeutic potential in neurodegenerative disorders. Prog. Neurobiol. 2019, 175, 35–53. [Google Scholar] [CrossRef]
- Iulita, M.F.; Bistue Millon, M.B.; Pentz, R.; Aguilar, L.F.; Do Carmo, S.; Allard, S.; Michalski, B.; Wilson, E.N.; Ducatenzeiler, A.; Bruno, M.A.; et al. Differential deregulation of NGF and BDNF neurotrophins in a transgenic rat model of Alzheimer’s disease. Neurobiol. Dis. 2017, 108, 307–323. [Google Scholar] [CrossRef]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nisticò, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef]
- Iulita, M.F.; Do Carmo, S.; Ower, A.K.; Fortress, A.M.; Flores Aguilar, L.; Hanna, M.; Wisniewski, T.; Granholm, A.C.; Buhusi, M.; Busciglio, J.; et al. Nerve growth factor metabolic dysfunction in Down’s syndrome brains. Brain 2014, 137, 860–872. [Google Scholar] [CrossRef] [Green Version]
- Cuello, A.C.; Pentz, R.; Hall, H. The Brain NGF Metabolic Pathway in Health and in Alzheimer’s Pathology. Front. Neurosci. 2019, 13, 62. [Google Scholar] [CrossRef] [Green Version]
- Bruno, M.A.; Mufson, E.J.; Wuu, J.; Cuello, A.C. Increased matrix metalloproteinase 9 activity in mild cognitive impairment. J. Neuropathol. Exp. Neurol. 2009, 68, 1309–1318. [Google Scholar] [CrossRef]
- Anderton, H.; Wicks, I.P.; Silke, J. Cell death in chronic inflammation: Breaking the cycle to treat rheumatic disease. Nat. Rev. Rheumatol. 2020, 16, 496–513. [Google Scholar] [CrossRef]
- Lee, S.H.; Meilandt, W.J.; Xie, L.; Gandham, V.D.; Ngu, H.; Barck, K.H.; Rezzonico, M.G.; Imperio, J.; Lalehzadeh, G.; Huntley, M.A.; et al. Trem2 restrains the enhancement of tau accumulation and neurodegeneration by β-amyloid pathology. Neuron 2021, 109, 1283–1301.e1286. [Google Scholar] [CrossRef]
- Lecca, D.; Jung, Y.J.; Scerba, M.T.; Hwang, I.; Kim, Y.K.; Kim, S.; Modrow, S.; Tweedie, D.; Hsueh, S.C.; Liu, D.; et al. Role of chronic neuroinflammation in neuroplasticity and cognitive function: A hypothesis. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2022, 2, 12610. [Google Scholar] [CrossRef]
- Harry, G.J.; Kraft, A.D. Neuroinflammation and microglia: Considerations and approaches for neurotoxicity assessment. Expert Opin Drug Metab. Toxicol. 2008, 4, 1265–1277. [Google Scholar] [CrossRef]
- Zhong, L.; Xu, Y.; Zhuo, R.; Wang, T.; Wang, K.; Huang, R.; Wang, D.; Gao, Y.; Zhu, Y.; Sheng, X.; et al. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 1365. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031.e1027. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Chen, Y.; Xu, C.; Zhang, H.; Lin, C. TLR4 Targeting as a Promising Therapeutic Strategy for Alzheimer Disease Treatment. Front. Neurosci. 2020, 14, 602508. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef] [Green Version]
- Ellwanger, D.C.; Wang, S.; Brioschi, S.; Shao, Z.; Green, L.; Case, R.; Yoo, D.; Weishuhn, D.; Rathanaswami, P.; Bradley, J.; et al. Prior activation state shapes the microglia response to antihuman TREM2 in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2017742118. [Google Scholar] [CrossRef]
- Flanary, B.E.; Sammons, N.W.; Nguyen, C.; Walker, D.; Streit, W.J. Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res. 2007, 10, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Desjardins, M.; Huber, L.A.; Parton, R.G.; Griffiths, G. Biogenesis of phagolysosomes proceeds through a sequential series of interactions with the endocytic apparatus. J. Cell Biol. 1994, 124, 677–688. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Walter, S.; Stagi, M.; Cherny, D.; Letiembre, M.; Schulz-Schaeffer, W.; Heine, H.; Penke, B.; Neumann, H.; Fassbender, K. LPS receptor (CD14): A receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain 2005, 128, 1778–1789. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Yang, D.S.; Goulbourne, C.N.; Im, E.; Stavrides, P.; Pensalfini, A.; Chan, H.; Bouchet-Marquis, C.; Bleiwas, C.; Berg, M.J.; et al. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat. Neurosci. 2022, 25, 688–701. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef] [Green Version]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- McFarland, K.N.; Chakrabarty, P. Microglia in Alzheimer’s Disease: A Key Player in the Transition Between Homeostasis and Pathogenesis. Neurother. J. Am. Soc. Exp. Neurother. 2022, 19, 186–208. [Google Scholar] [CrossRef]
- Rubio-Araiz, A.; Finucane, O.M.; Keogh, S.; Lynch, M.A. Anti-TLR2 antibody triggers oxidative phosphorylation in microglia and increases phagocytosis of β-amyloid. J. Neuroinflamm. 2018, 15, 247. [Google Scholar] [CrossRef] [Green Version]
- Daria, A.; Colombo, A.; Llovera, G.; Hampel, H.; Willem, M.; Liesz, A.; Haass, C.; Tahirovic, S. Young microglia restore amyloid plaque clearance of aged microglia. EMBO J. 2017, 36, 583–603. [Google Scholar] [CrossRef] [Green Version]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507.e496. [Google Scholar] [CrossRef]
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef]
- Ziegler-Waldkirch, S.; Meyer-Luehmann, M. The Role of Glial Cells and Synapse Loss in Mouse Models of Alzheimer’s Disease. Front. Cell Neurosci. 2018, 12, 473. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Kitazawa, M.; Tseng, B.P.; LaFerla, F.M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef]
- Balducci, C.; Forloni, G. Novel targets in Alzheimer’s disease: A special focus on microglia. Pharm. Res. 2018, 130, 402–413. [Google Scholar] [CrossRef]
- Shen, Y.; Lue, L.; Yang, L.; Roher, A.; Kuo, Y.; Strohmeyer, R.; Goux, W.J.; Lee, V.; Johnson, G.V.; Webster, S.D.; et al. Complement activation by neurofibrillary tangles in Alzheimer’s disease. Neurosci. Lett. 2001, 305, 165–168. [Google Scholar] [CrossRef]
- Xin, Y.R.; Jiang, J.X.; Hu, Y.; Pan, J.P.; Mi, X.N.; Gao, Q.; Xiao, F.; Zhang, W.; Luo, H.M. The Immune System Drives Synapse Loss During Lipopolysaccharide-Induced Learning and Memory Impairment in Mice. Front. Aging Neurosci. 2019, 11, 279. [Google Scholar] [CrossRef]
- Aramideh, J.A.; Vidal-Itriago, A.; Morsch, M.; Graeber, M.B. Cytokine Signalling at the Microglial Penta-Partite Synapse. Int. J. Mol. Sci. 2021, 22, 13186. [Google Scholar] [CrossRef]
- Krukowski, K.; Nolan, A.; Becker, M.; Picard, K.; Vernoux, N.; Frias, E.S.; Feng, X.; Tremblay, M.E.; Rosi, S. Novel microglia-mediated mechanisms underlying synaptic loss and cognitive impairment after traumatic brain injury. Brain Behav. Immun. 2021, 98, 122–135. [Google Scholar] [CrossRef]
- Wilton, D.K.; Dissing-Olesen, L.; Stevens, B. Neuron-Glia Signaling in Synapse Elimination. Annu. Rev. Neurosci. 2019, 42, 107–127. [Google Scholar] [CrossRef]
- Shi, X.; Luo, L.; Wang, J.; Shen, H.; Li, Y.; Mamtilahun, M.; Liu, C.; Shi, R.; Lee, J.H.; Tian, H.; et al. Stroke subtype-dependent synapse elimination by reactive gliosis in mice. Nat. Commun. 2021, 12, 6943. [Google Scholar] [CrossRef]
- Sivanesan, S.; Tan, A.; Rajadas, J. Pathogenesis of Abeta oligomers in synaptic failure. Curr. Alzheimer Res. 2013, 10, 316–323. [Google Scholar] [CrossRef]
- Lue, L.F.; Kuo, Y.M.; Roher, A.E.; Brachova, L.; Shen, Y.; Sue, L.; Beach, T.; Kurth, J.H.; Rydel, R.E.; Rogers, J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 1999, 155, 853–862. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Cellular Receptors of Amyloid β Oligomers (AβOs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 1884. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Zhao, J.; Zhang, X.; Wang, S.; Viola, K.L.; Chow, F.E.; Zhang, Y.; Lippa, C.; Klein, W.L.; Gong, Y. Amyloid Beta Oligomers Target to Extracellular and Intracellular Neuronal Synaptic Proteins in Alzheimer’s Disease. Front. Neurol. 2019, 10, 1140. [Google Scholar] [CrossRef]
- Jang, Y.N.; Jang, H.; Kim, G.H.; Noh, J.E.; Chang, K.A.; Lee, K.J. RAPGEF2 mediates oligomeric Aβ-induced synaptic loss and cognitive dysfunction in the 3xTg-AD mouse model of Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2021, 47, 625–639. [Google Scholar] [CrossRef]
- Conde, C.; Caceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar] [CrossRef]
- Chidambaram, H.; Das, R.; Chinnathambi, S. Interaction of Tau with the chemokine receptor, CX3CR1 and its effect on microglial activation, migration and proliferation. Cell Biosci. 2020, 10, 109. [Google Scholar] [CrossRef]
- Gao, Y.; Tan, L.; Yu, J.T.; Tan, L. Tau in Alzheimer’s Disease: Mechanisms and Therapeutic Strategies. Curr. Alzheimer Res. 2018, 15, 283–300. [Google Scholar] [CrossRef]
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17, e12840. [Google Scholar] [CrossRef]
- Ying, Z.; Wang, H.; Wang, G. The ubiquitin proteasome system as a potential target for the treatment of neurodegenerative diseases. Curr. Pharm. Des. 2013, 19, 3305–3314. [Google Scholar] [CrossRef]
- Laurent, C.; Buee, L.; Blum, D. Tau and neuroinflammation: What impact for Alzheimer’s Disease and Tauopathies? Biomed. J. 2018, 41, 21–33. [Google Scholar] [CrossRef]
- Alonso, A.D.; Cohen, L.S. Our Tau Tales from Normal to Pathological Behavior. J. Alzheimer’s Dis. JAD 2018, 64, S507–S516. [Google Scholar] [CrossRef]
- Saha, P.; Sen, N. Tauopathy: A common mechanism for neurodegeneration and brain aging. Mech. Ageing Dev. 2019, 178, 72–79. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, M.; Yin, X.; Chen, K.; Hu, Z.; Zhou, Q.; Cao, X.; Chen, Z.; Liu, D. The role of pathological tau in synaptic dysfunction in Alzheimer’s diseases. Transl. Neurodegener. 2021, 10, 45. [Google Scholar] [CrossRef]
- Mondragon-Rodriguez, S.; Trillaud-Doppia, E.; Dudilot, A.; Bourgeois, C.; Lauzon, M.; Leclerc, N.; Boehm, J. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J. Biol. Chem. 2012, 287, 32040–32053. [Google Scholar] [CrossRef] [Green Version]
- Dinda, B.; Dinda, M.; Kulsi, G.; Chakraborty, A.; Dinda, S. Therapeutic potentials of plant iridoids in Alzheimer’s and Parkinson’s diseases: A review. Eur J. Med. Chem. 2019, 169, 185–199. [Google Scholar] [CrossRef]
- Pallas-Bazarra, N.; Draffin, J.; Cuadros, R.; Antonio Esteban, J.; Avila, J. Tau is required for the function of extrasynaptic NMDA receptors. Sci. Rep. 2019, 9, 9116. [Google Scholar] [CrossRef] [Green Version]
- Jay, T.R.; Miller, C.M.; Cheng, P.J.; Graham, L.C.; Bemiller, S.; Broihier, M.L.; Xu, G.; Margevicius, D.; Karlo, J.C.; Sousa, G.L.; et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 2015, 212, 287–295. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Bhaskar, K.; Konerth, M.; Kokiko-Cochran, O.N.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T. Regulation of tau pathology by the microglial fractalkine receptor. Neuron 2010, 68, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Lodder, C.; Scheyltjens, I.; Stancu, I.C.; Botella Lucena, P.; Gutiérrez de Ravé, M.; Vanherle, S.; Vanmierlo, T.; Cremers, N.; Vanrusselt, H.; Brône, B.; et al. CSF1R inhibition rescues tau pathology and neurodegeneration in an A/T/N model with combined AD pathologies, while preserving plaque associated microglia. Acta Neuropathol. Commun. 2021, 9, 108. [Google Scholar] [CrossRef]
- Jolivel, V.; Bicker, F.; Binamé, F.; Ploen, R.; Keller, S.; Gollan, R.; Jurek, B.; Birkenstock, J.; Poisa-Beiro, L.; Bruttger, J.; et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. 2015, 129, 279–295. [Google Scholar] [CrossRef]
- Apátiga-Pérez, R.; Soto-Rojas, L.O.; Campa-Córdoba, B.B.; Luna-Viramontes, N.I.; Cuevas, E.; Villanueva-Fierro, I.; Ontiveros-Torres, M.A.; Bravo-Muñoz, M.; Flores-Rodríguez, P.; Garcés-Ramirez, L.; et al. Neurovascular dysfunction and vascular amyloid accumulation as early events in Alzheimer’s disease. Metab. Brain Dis. 2022, 37, 39–50. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Munoz, D.G. Nonsteroidal anti-inflammatory drug use and Alzheimer-type pathology in aging. Neurology 1998, 50, 986–990. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, E.J.; Wang, W.J.; Wang, Y.L.; Li, X.G.; Wang, X.; Li, S.H.; Zhang, S.J.; Li, M.Z.; Zhou, Q.Z.; et al. Microglia CREB-Phosphorylation Mediates Amyloid-β-Induced Neuronal Toxicity. J. Alzheimer’s Dis. JAD 2018, 66, 333–345. [Google Scholar] [CrossRef]
- Howard, R.; Zubko, O.; Bradley, R.; Harper, E.; Pank, L.; O’Brien, J.; Fox, C.; Tabet, N.; Livingston, G.; Bentham, P.; et al. Minocycline at 2 Different Dosages vs Placebo for Patients with Mild Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 164–174. [Google Scholar] [CrossRef]
- Dhapola, R.; Hota, S.S.; Sarma, P.; Bhattacharyya, A.; Medhi, B.; Reddy, D.H. Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer’s disease. Inflammopharmacology 2021, 29, 1669–1681. [Google Scholar] [CrossRef]
- Gold, M.; Alderton, C.; Zvartau-Hind, M.; Egginton, S.; Saunders, A.M.; Irizarry, M.; Craft, S.; Landreth, G.; Linnamagi, U.; Sawchak, S. Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: Results from a randomized, double-blind, placebo-controlled phase III study. Dement. Geriatr. Cogn. Disord. 2010, 30, 131–146. [Google Scholar] [CrossRef] [Green Version]
- Son, S.M.; Shin, H.J.; Byun, J.; Kook, S.Y.; Moon, M.; Chang, Y.J.; Mook-Jung, I. Metformin Facilitates Amyloid-beta Generation by beta- and gamma-Secretases via Autophagy Activation. J. Alzheimers Dis. 2016, 51, 1197–1208. [Google Scholar] [CrossRef]
- Ngandu, T.; Lehtisalo, J.; Solomon, A.; Levalahti, E.; Ahtiluoto, S.; Antikainen, R.; Backman, L.; Hanninen, T.; Jula, A.; Laatikainen, T.; et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): A randomised controlled trial. Lancet 2015, 385, 2255–2263. [Google Scholar] [CrossRef]
- Li, J.W.; Zong, Y.; Cao, X.P.; Tan, L.; Tan, L. Microglial priming in Alzheimer’s disease. Ann. Transl. Med. 2018, 6, 176. [Google Scholar] [CrossRef]
- Zhang, S.S.; Zhu, L.; Peng, Y.; Zhang, L.; Chao, F.L.; Jiang, L.; Xiao, Q.; Liang, X.; Tang, J.; Yang, H.; et al. Long-term running exercise improves cognitive function and promotes microglial glucose metabolism and morphological plasticity in the hippocampus of APP/PS1 mice. J. Neuroinflamm. 2022, 19, 34. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, N.; Deng, M.; Hu, G.; Li, N.; Yuan, H.; Zhou, Y. New Insights into Microglial Mechanisms of Memory Impairment in Alzheimer’s Disease. Biomolecules 2022, 12, 1722. https://doi.org/10.3390/biom12111722
Li N, Deng M, Hu G, Li N, Yuan H, Zhou Y. New Insights into Microglial Mechanisms of Memory Impairment in Alzheimer’s Disease. Biomolecules. 2022; 12(11):1722. https://doi.org/10.3390/biom12111722
Chicago/Turabian StyleLi, Na, Mingru Deng, Gonghui Hu, Nan Li, Haicheng Yuan, and Yu Zhou. 2022. "New Insights into Microglial Mechanisms of Memory Impairment in Alzheimer’s Disease" Biomolecules 12, no. 11: 1722. https://doi.org/10.3390/biom12111722
APA StyleLi, N., Deng, M., Hu, G., Li, N., Yuan, H., & Zhou, Y. (2022). New Insights into Microglial Mechanisms of Memory Impairment in Alzheimer’s Disease. Biomolecules, 12(11), 1722. https://doi.org/10.3390/biom12111722