
The Pathophysiology of H2S in Renal Glomerular Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
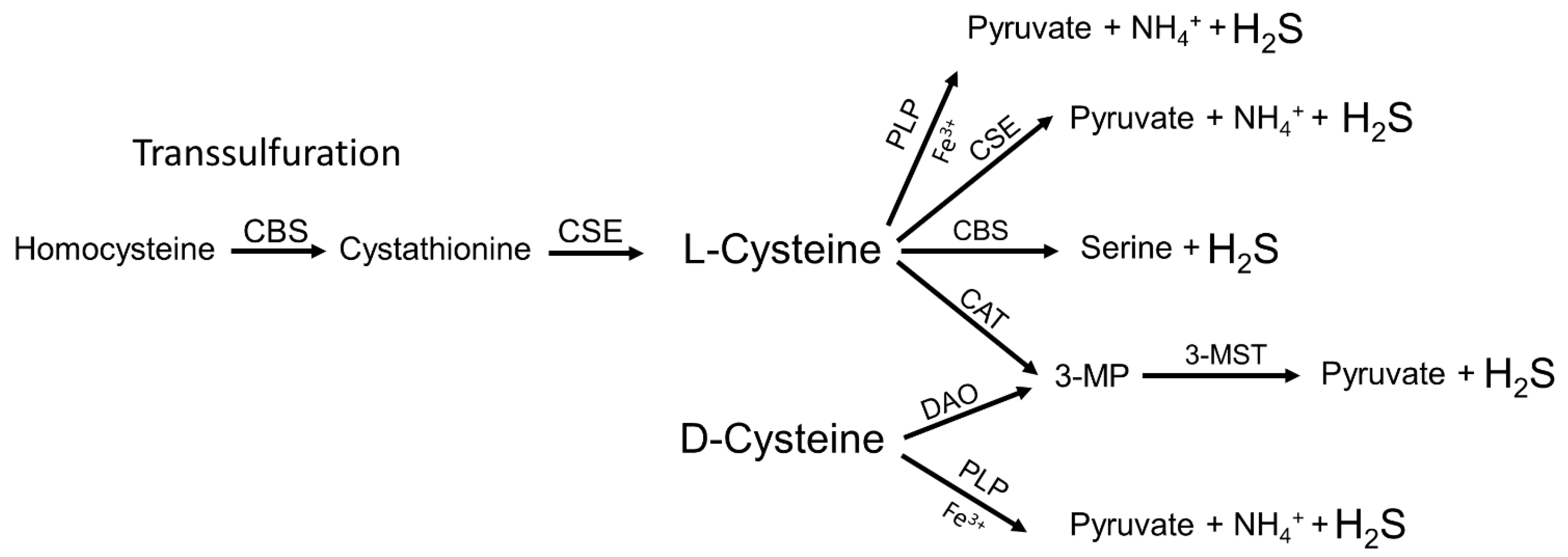
2. Endogenous Synthesis of H2S
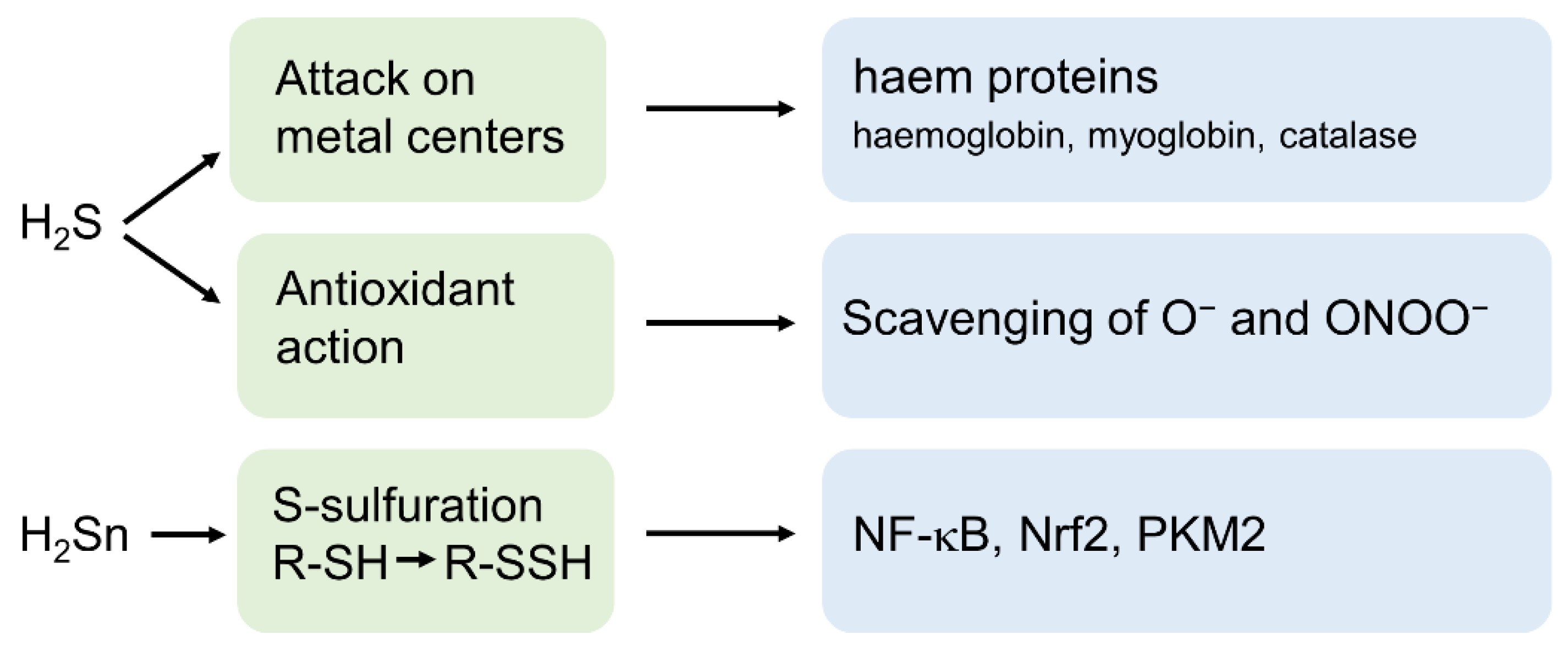
3. H2S and Its Derivatives as Important Signalling Molecules
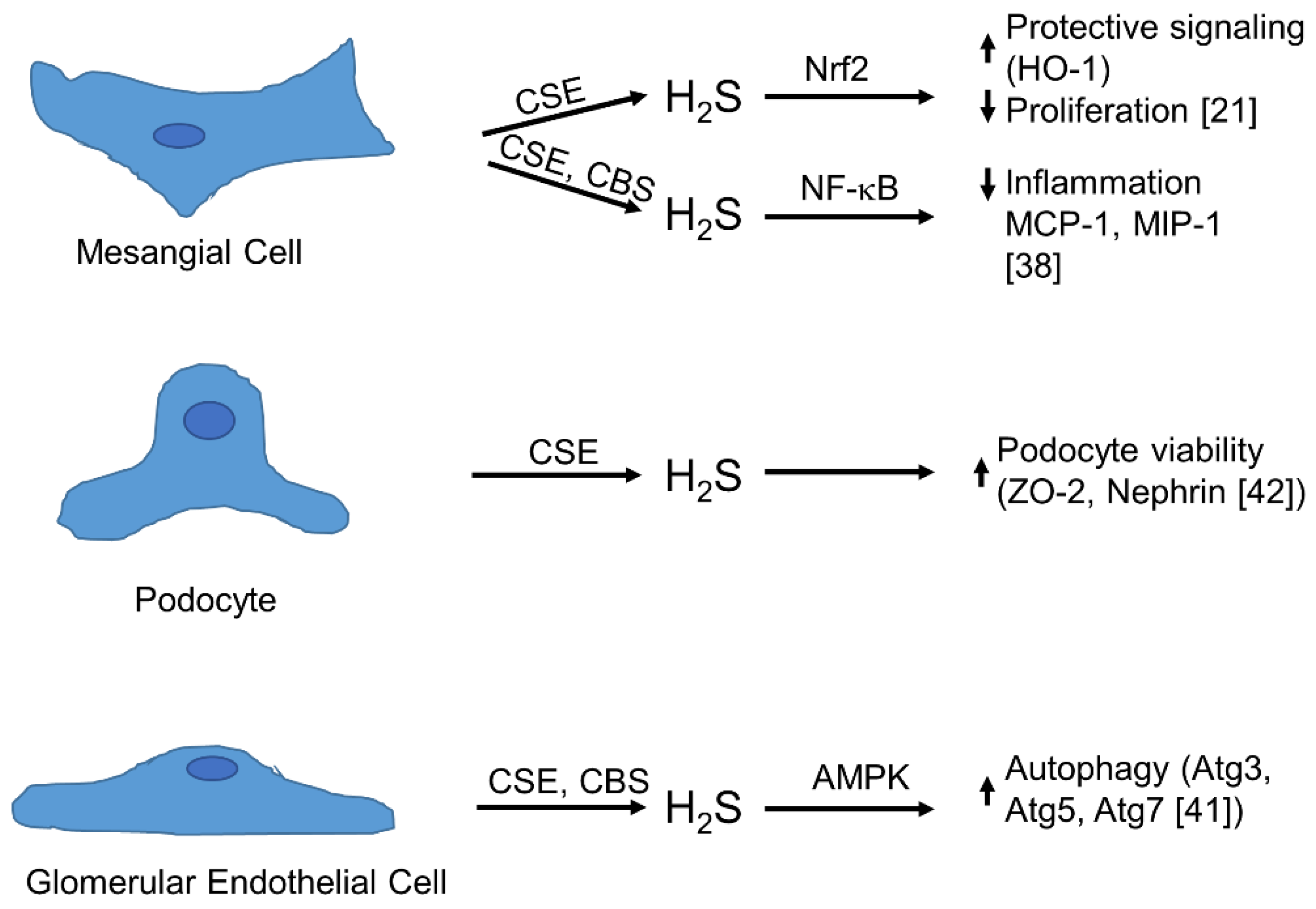
4. Physiology of Synthesis and Action of H2S in the Glomerulus
5. The Role of H2S in Glomerular Pathophysiology and Disease
5.1. Diabetic Nephropathy
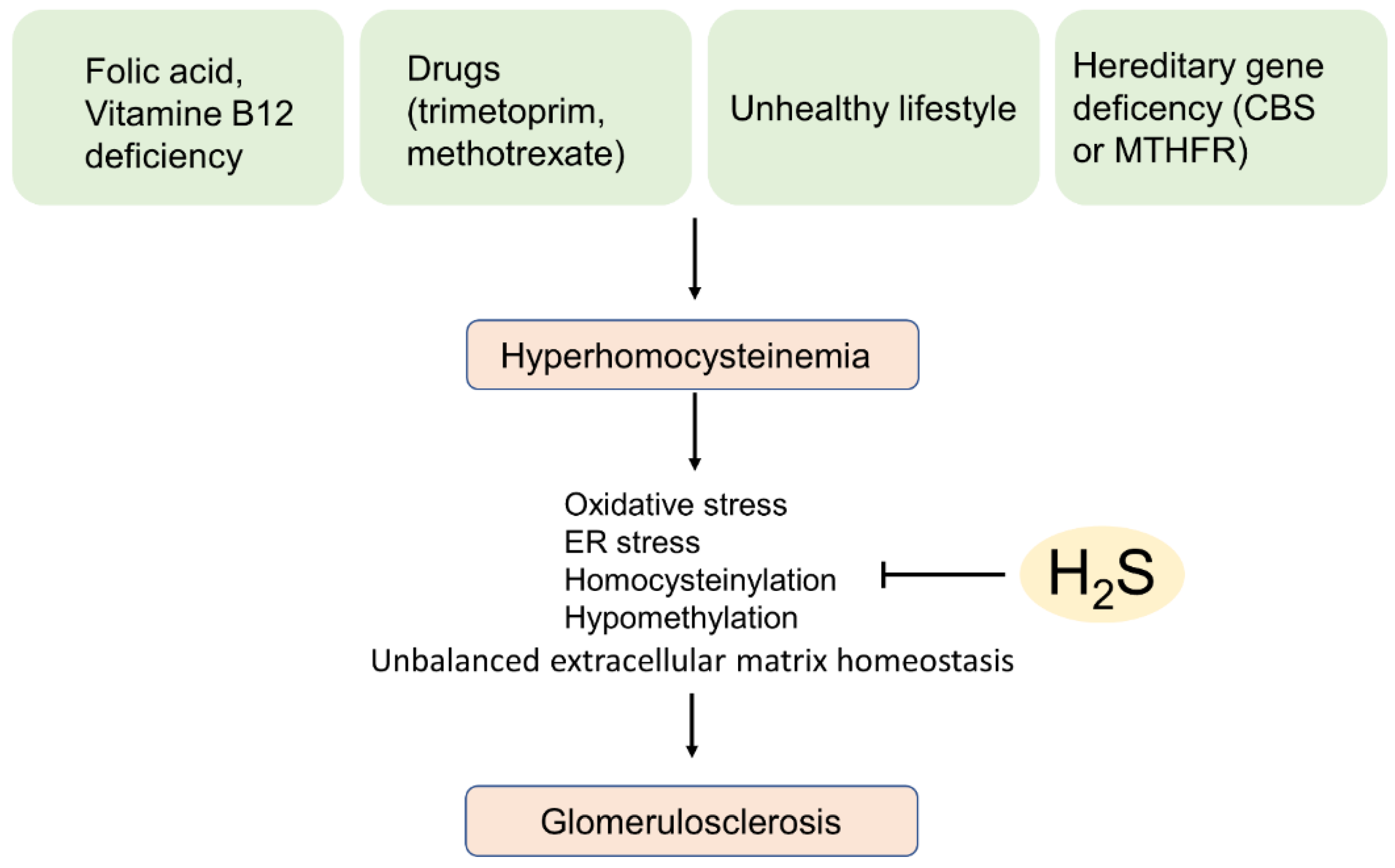
5.2. Hyperhomocysteinemia-Induced Glomerular Sclerosis
5.3. Acute Kidney Injury
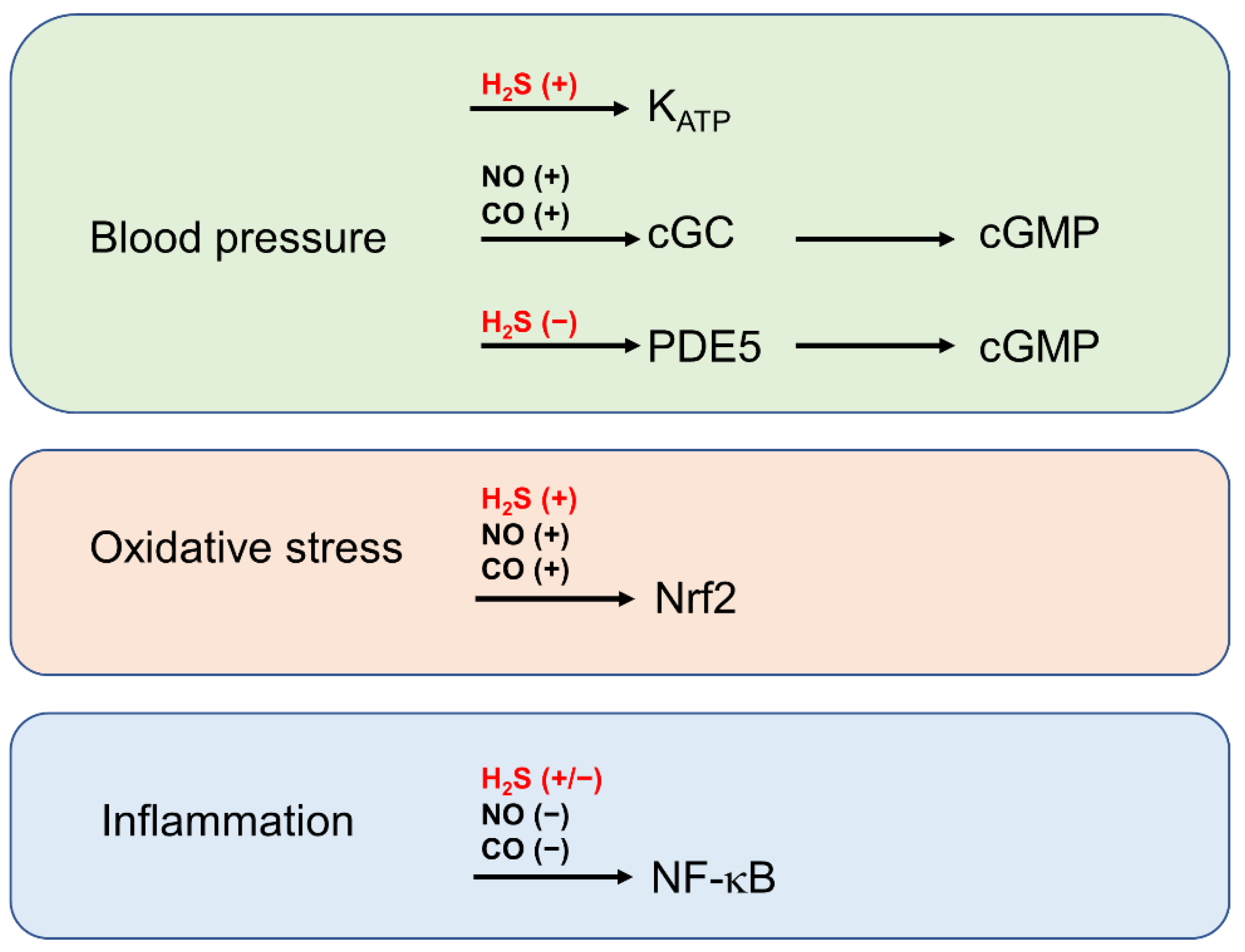
6. The Role of H2S in the Context of Gasotransmitter Signalling by NO and CO
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abe, K.; Kimura, H. The Possible Role of Hydrogen Sulfide as an Endogenous Neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R. Two’s Company, Three’s a Crowd: Can H2S Be the Third Endogenous Gaseous Transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlöndorff, D.; Banas, B. The Mesangial Cell Revisited: No Cell Is an Island. J. Am. Soc. Nephrol. 2009, 20, 1179–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeilschifter, J. Cross-Talk between Transmembrane Signalling Systems: A Prerequisite for the Delicate Regulation of Glomerular Haemodynamics by Mesangial Cells. Eur. J. Clin. Invest. 1989, 19, 347–361. [Google Scholar] [CrossRef]
- Schlöndorff, D. Putting the Glomerulus Back Together: Per Aspera Ad Astra (“a Rough Road Leads to the Stars”). Kidney Int. 2014, 85, 991–998. [Google Scholar] [CrossRef] [Green Version]
- Beck, K.-F.; Pfeilschifter, J. Gasotransmitter Synthesis and Signalling in the Renal Glomerulus. Implications for Glomerular Diseases. Cell. Signal. 2021, 77, 109823. [Google Scholar] [CrossRef]
- Ngowi, E.E.; Sarfraz, M.; Afzal, A.; Khan, N.H.; Khattak, S.; Zhang, X.; Li, T.; Duan, S.-F.; Ji, X.-Y.; Wu, D.-D. Roles of Hydrogen Sulfide Donors in Common Kidney Diseases. Front. Pharmacol. 2020, 11, 564281. [Google Scholar] [CrossRef]
- Szabo, C. A Timeline of Hydrogen Sulfide (H2S) Research: From Environmental Toxin to Biological Mediator. Biochem. Pharmacol. 2018, 149, 5–19. [Google Scholar] [CrossRef]
- Stipanuk, M.H.; Beck, P.W. Characterization of the Enzymic Capacity for Cysteine Desulphhydration in Liver and Kidney of the Rat. Biochem. J. 1982, 206, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Paget, M.S.B.; Buttner, M.J. Thiol-Based Regulatory Switches. Annu. Rev. Genet. 2003, 37, 91–121. [Google Scholar] [CrossRef] [Green Version]
- Longen, S.; Beck, K.-F.; Pfeilschifter, J. H2S-Induced Thiol-Based Redox Switches: Biochemistry and Functional Relevance for Inflammatory Diseases. Pharmacol. Res. 2016, 111, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Carroll, K.S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef] [PubMed]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Regulators of the Transsulfuration Pathway. Br. J. Pharmacol. 2019, 176, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, N.; Tanaka, M.; Yoshida, M.; Ogasawara, Y.; Togawa, T.; Ishii, K.; Kimura, H. 3-Mercaptopyruvate Sulfurtransferase Produces Hydrogen Sulfide and Bound Sulfane Sulfur in the Brain. Antioxid. Redox. Signal. 2009, 11, 703–714. [Google Scholar] [CrossRef]
- Shibuya, N.; Koike, S.; Tanaka, M.; Ishigami-Yuasa, M.; Kimura, Y.; Ogasawara, Y.; Fukui, K.; Nagahara, N.; Kimura, H. A Novel Pathway for the Production of Hydrogen Sulfide from D-Cysteine in Mammalian Cells. Nat. Commun. 2013, 4, 1366. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Minkler, P.; Grove, D.; Wang, R.; Willard, B.; Dweik, R.; Hine, C. Non-Enzymatic Hydrogen Sulfide Production from Cysteine in Blood Is Catalyzed by Iron and Vitamin B6. Commun. Biol. 2019, 2, 194. [Google Scholar] [CrossRef] [Green Version]
- Taoka, S.; Banerjee, R. Characterization of NO Binding to Human Cystathionine Beta-Synthase: Possible Implications of the Effects of CO and NO Binding to the Human Enzyme. J. Inorg. Biochem. 2001, 87, 245–251. [Google Scholar] [CrossRef]
- Majtan, T.; Pey, A.L.; Kraus, J.P. Kinetic Stability of Cystathionine Beta-Synthase Can Be Modulated by Structural Analogs of S-Adenosylmethionine: Potential Approach to Pharmacological Chaperone Therapy for Homocystinuria. Biochimie 2016, 126, 6–13. [Google Scholar] [CrossRef]
- Yang, G.; Pei, Y.; Teng, H.; Cao, Q.; Wang, R. Specificity Protein-1 as a Critical Regulator of Human Cystathionine Gamma-Lyase in Smooth Muscle Cells. J. Biol. Chem. 2011, 286, 26450–26460. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Guo, Z.; Wang, S. The Binding Site for the Transcription Factor, NF-ΚB, on the Cystathionine γ-Lyase Promoter Is Critical for LPS-Induced Cystathionine γ-Lyase Expression. Int. J. Mol. Med. 2014, 34, 639–645. [Google Scholar] [CrossRef]
- Hassan, M.I.; Boosen, M.; Schaefer, L.; Kozlowska, J.; Eisel, F.; von Knethen, A.; Beck, M.; Hemeida, R.A.M.; El-Moselhy, M.A.M.; Hamada, F.M.A.; et al. Platelet-Derived Growth Factor-BB Induces Cystathionine γ-Lyase Expression in Rat Mesangial Cells via a Redox-Dependent Mechanism. Br. J. Pharmacol. 2012, 166, 2231–2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, P.; Kim, J.K. Sulphide as an Inhibitor and Electron Donor for the Cytochrome c Oxidase System. Can. J. Biochem. 1982, 60, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Ríos-González, B.B.; Román-Morales, E.M.; Pietri, R.; López-Garriga, J. Hydrogen Sulfide Activation in Hemeproteins: The Sulfheme Scenario. J. Inorg. Biochem. 2014, 133, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietri, R.; Román-Morales, E.; López-Garriga, J. Hydrogen Sulfide and Hemeproteins: Knowledge and Mysteries. Antioxid. Redox Signal. 2011, 15, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Al-Magableh, M.R.; Kemp-Harper, B.K.; Ng, H.H.; Miller, A.A.; Hart, J.L. Hydrogen Sulfide Protects Endothelial Nitric Oxide Function under Conditions of Acute Oxidative Stress in Vitro. Naunyn Schmiedebergs Arch. Pharmacol. 2014, 387, 67–74. [Google Scholar] [CrossRef]
- Whiteman, M.; Armstrong, J.S.; Chu, S.H.; Jia-Ling, S.; Wong, B.-S.; Cheung, N.S.; Halliwell, B.; Moore, P.K. The Novel Neuromodulator Hydrogen Sulfide: An Endogenous Peroxynitrite “Scavenger”? J. Neurochem. 2004, 90, 765–768. [Google Scholar] [CrossRef]
- Koike, S.; Ogasawara, Y.; Shibuya, N.; Kimura, H.; Ishii, K. Polysulfide Exerts a Protective Effect against Cytotoxicity Caused by T-Buthylhydroperoxide through Nrf2 Signaling in Neuroblastoma Cells. FEBS Lett. 2013, 587, 3548–3555. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S Signals through Protein S-Sulfhydration. Sci. Signal. 2009, 2, ra72. [Google Scholar] [CrossRef] [Green Version]
- Kimura, H. Hydrogen Sulfide (H2S) and Polysulfide (H2Sn) Signaling: The First 25 Years. Biomolecules 2021, 11, 896. [Google Scholar] [CrossRef]
- Benchoam, D.; Cuevasanta, E.; Möller, M.N.; Alvarez, B. Hydrogen Sulfide and Persulfides Oxidation by Biologically Relevant Oxidizing Species. Antioxidants 2019, 8, 48. [Google Scholar] [CrossRef] [Green Version]
- Cortese-Krott, M.M.; Kuhnle, G.G.C.; Dyson, A.; Fernandez, B.O.; Grman, M.; DuMond, J.F.; Barrow, M.P.; McLeod, G.; Nakagawa, H.; Ondrias, K.; et al. Key Bioactive Reaction Products of the NO/H2S Interaction Are S/N-Hybrid Species, Polysulfides, and Nitroxyl. Proc. Natl. Acad. Sci. USA 2015, 112, E4651–E4660. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, R.; Koike, S.; Takano, Y.; Shibuya, N.; Kimura, Y.; Hanaoka, K.; Urano, Y.; Ogasawara, Y.; Kimura, H. Polysulfides (H2Sn) Produced from the Interaction of Hydrogen Sulfide (H2S) and Nitric Oxide (NO) Activate TRPA1 Channels. Sci. Rep. 2017, 7, 45995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baud, L.; Ardaillou, R. Reactive Oxygen Species: Production and Role in the Kidney. Am. J. Physiol. 1986, 251, F765–F776. [Google Scholar] [CrossRef] [PubMed]
- Pfeilschifter, J.; Schwarzenbach, H. Interleukin 1 and Tumor Necrosis Factor Stimulate CGMP Formation in Rat Renal Mesangial Cells. FEBS Lett. 1990, 273, 185–187. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.-N.; Tain, Y.-L. Gasotransmitters for the Therapeutic Prevention of Hypertension and Kidney Disease. Int. J. Mol. Sci. 2021, 22, 7808. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, H.; Guo, N. Protective Effect of Hydrogen Sulfide on the Kidney (Review). Mol. Med. Rep. 2021, 24, 696. [Google Scholar] [CrossRef]
- Fan, Q.-L.; Yang, G.; Liu, X.-D.; Ma, J.-F.; Feng, J.-M.; Jiang, Y.; Wang, L.-N. Effect of Losartan on the Glomerular Protein Expression Profile of Type 2 Diabetic KKAy Mice. J. Nephrol. 2013, 26, 517–526. [Google Scholar] [CrossRef]
- Sen, U.; Givvimani, S.; Abe, O.A.; Lederer, E.D.; Tyagi, S.C. Cystathionine β-Synthase and Cystathionine γ-Lyase Double Gene Transfer Ameliorate Homocysteine-Mediated Mesangial Inflammation through Hydrogen Sulfide Generation. Am. J. Physiol. Cell. Physiol. 2011, 300, C155–C163. [Google Scholar] [CrossRef] [Green Version]
- Yuan, P.; Xue, H.; Zhou, L.; Qu, L.; Li, C.; Wang, Z.; Ni, J.; Yu, C.; Yao, T.; Huang, Y.; et al. Rescue of Mesangial Cells from High Glucose-Induced over-Proliferation and Extracellular Matrix Secretion by Hydrogen Sulfide. Nephrol. Dial. Transplant. 2011, 26, 2119–2126. [Google Scholar] [CrossRef] [Green Version]
- Xue, H.; Yuan, P.; Ni, J.; Li, C.; Shao, D.; Liu, J.; Shen, Y.; Wang, Z.; Zhou, L.; Zhang, W.; et al. H2S Inhibits Hyperglycemia-Induced Intrarenal Renin-Angiotensin System Activation via Attenuation of Reactive Oxygen Species Generation. PLoS ONE 2013, 8, e74366. [Google Scholar] [CrossRef]
- Kundu, S.; Pushpakumar, S.; Khundmiri, S.J.; Sen, U. Hydrogen Sulfide Mitigates Hyperglycemic Remodeling via Liver Kinase B1-Adenosine Monophosphate-Activated Protein Kinase Signaling. Biochim. Biophys. Acta 2014, 1843, 2816–2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhao, H.; Qiang, Y.; Qian, G.; Lu, S.; Chen, J.; Wang, X.; Guan, Q.; Liu, Y.; Fu, Y. Effects of Hydrogen Sulfide on High Glucose-Induced Glomerular Podocyte Injury in Mice. Int. J. Clin. Exp. Pathol. 2015, 8, 6814–6820. [Google Scholar] [PubMed]
- Lee, H.J.; Feliers, D.; Mariappan, M.M.; Sataranatarajan, K.; Choudhury, G.G.; Gorin, Y.; Kasinath, B.S. Tadalafil Integrates Nitric Oxide-Hydrogen Sulfide Signaling to Inhibit High Glucose-Induced Matrix Protein Synthesis in Podocytes. J. Biol. Chem. 2015, 290, 12014–12026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, C. Roles of Hydrogen Sulfide in the Pathogenesis of Diabetes Mellitus and Its Complications. Antioxid. Redox Signal. 2012, 17, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-J.; Wu, Z.-Y.; Cao, L.; Zhu, M.-Y.; Liu, T.-T.; Guo, L.; Lin, Y.; Nie, X.-W.; Bian, J.-S. Hydrogen Sulfide: Recent Progression and Perspectives for the Treatment of Diabetic Nephropathy. Molecules 2019, 24, 2857. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Feng, Y.; Zhan, Z.; Chen, J. Hydrogen Sulfide Alleviates Diabetic Nephropathy in a Streptozotocin-Induced Diabetic Rat Model. J. Biol. Chem. 2014, 289, 28827–28834. [Google Scholar] [CrossRef] [Green Version]
- John, A.M.S.P.; Kundu, S.; Pushpakumar, S.; Fordham, M.; Weber, G.; Mukhopadhyay, M.; Sen, U. GYY4137, a Hydrogen Sulfide Donor Modulates MiR194-Dependent Collagen Realignment in Diabetic Kidney. Sci. Rep. 2017, 7, 10924. [Google Scholar] [CrossRef]
- Qian, X.; Li, X.; Ma, F.; Luo, S.; Ge, R.; Zhu, Y. Novel Hydrogen Sulfide-Releasing Compound, S-Propargyl-Cysteine, Prevents STZ-Induced Diabetic Nephropathy. Biochem. Biophys. Res. Commun. 2016, 473, 931–938. [Google Scholar] [CrossRef]
- Paganelli, F.; Mottola, G.; Fromonot, J.; Marlinge, M.; Deharo, P.; Guieu, R.; Ruf, J. Hyperhomocysteinemia and Cardiovascular Disease: Is the Adenosinergic System the Missing Link? Int. J. Mol. Sci. 2021, 22, 1690. [Google Scholar] [CrossRef]
- Angelini, A.; Cappuccilli, M.L.; Magnoni, G.; Croci Chiocchini, A.L.; Aiello, V.; Napoletano, A.; Iacovella, F.; Troiano, A.; Mancini, R.; Capelli, I.; et al. The Link between Homocysteine, Folic Acid and Vitamin B12 in Chronic Kidney Disease. G. Ital. Nefrol. 2021, 38, 1–17. [Google Scholar]
- Cordaro, M.; Siracusa, R.; Fusco, R.; Cuzzocrea, S.; Di Paola, R.; Impellizzeri, D. Involvements of Hyperhomocysteinemia in Neurological Disorders. Metabolites 2021, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Kruger, W.D. Cystathionine β-Synthase Deficiency: Of Mice and Men. Mol. Genet. Metab. 2017, 121, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Frosst, P.; Blom, H.J.; Milos, R.; Goyette, P.; Sheppard, C.A.; Matthews, R.G.; Boers, G.J.; den Heijer, M.; Kluijtmans, L.A.; van den Heuvel, L.P. A Candidate Genetic Risk Factor for Vascular Disease: A Common Mutation in Methylenetetrahydrofolate Reductase. Nat. Genet. 1995, 10, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Li, P.-L. Mechanisms of Homocysteine-Induced Glomerular Injury and Sclerosis. Am. J. Nephrol. 2008, 28, 254–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, U.; Basu, P.; Abe, O.A.; Givvimani, S.; Tyagi, N.; Metreveli, N.; Shah, K.S.; Passmore, J.C.; Tyagi, S.C. Hydrogen Sulfide Ameliorates Hyperhomocysteinemia-Associated Chronic Renal Failure. Am. J. Physiol. Renal Physiol. 2009, 297, F410–F419. [Google Scholar] [CrossRef] [Green Version]
- Sen, U.; Munjal, C.; Qipshidze, N.; Abe, O.; Gargoum, R.; Tyagi, S.C. Hydrogen Sulfide Regulates Homocysteine-Mediated Glomerulosclerosis. Am. J. Nephrol. 2010, 31, 442–455. [Google Scholar] [CrossRef] [Green Version]
- Majumder, S.; Ren, L.; Pushpakumar, S.; Sen, U. Hydrogen Sulphide Mitigates Homocysteine-Induced Apoptosis and Matrix Remodelling in Mesangial Cells through Akt/FOXO1 Signalling Cascade. Cell. Signal. 2019, 61, 66–77. [Google Scholar] [CrossRef]
- Pushpakumar, S.; Kundu, S.; Sen, U. Hydrogen Sulfide Protects Hyperhomocysteinemia-Induced Renal Damage by Modulation of Caveolin and ENOS Interaction. Sci. Rep. 2019, 9, 2223. [Google Scholar] [CrossRef]
- Waz, S.; Heeba, G.H.; Hassanin, S.O.; Abdel-Latif, R.G. Nephroprotective Effect of Exogenous Hydrogen Sulfide Donor against Cyclophosphamide-Induced Toxicity Is Mediated by Nrf2/HO-1/NF-ΚB Signaling Pathway. Life Sci. 2021, 264, 118630. [Google Scholar] [CrossRef]
- Pieretti, J.C.; Junho, C.V.C.; Carneiro-Ramos, M.S.; Seabra, A.B. H2S- and NO-Releasing Gasotransmitter Platform: A Crosstalk Signaling Pathway in the Treatment of Acute Kidney Injury. Pharmacol. Res. 2020, 161, 105121. [Google Scholar] [CrossRef]
- Scammahorn, J.J.; Nguyen, I.T.N.; Bos, E.M.; Van Goor, H.; Joles, J.A. Fighting Oxidative Stress with Sulfur: Hydrogen Sulfide in the Renal and Cardiovascular Systems. Antioxidants 2021, 10, 373. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.R.; Botting, C.H.; Panico, M.; Morris, H.R.; Hay, R.T. Inhibition of NF-KappaB DNA Binding by Nitric Oxide. Nucleic. Acids. Res. 1996, 24, 2236–2242. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, Z.T.; Matsumoto, A.; Stamler, J.S.; Marshall, H.E. NOS2 Regulation of NF-KappaB by S-Nitrosylation of P65. J. Biol. Chem. 2007, 282, 30667–30672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen Sulfide-Linked Sulfhydration of NF-ΚB Mediates Its Antiapoptotic Actions. Mol. Cell. 2012, 45, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Huang, Y.; Yan, H.; Zhang, Q.; Zhao, M.; Zhu, M.; Liu, J.; Chen, S.X.; Bu, D.; Tang, C.; et al. Hydrogen Sulfide Suppresses Oxidized Low-Density Lipoprotein (Ox-LDL)-Stimulated Monocyte Chemoattractant Protein 1 Generation from Macrophages via the Nuclear Factor ΚB (NF-ΚB) Pathway. J. Biol. Chem. 2014, 289, 9741–9753. [Google Scholar] [CrossRef] [Green Version]
- Yeh, P.-Y.; Li, C.-Y.; Hsieh, C.-W.; Yang, Y.-C.; Yang, P.-M.; Wung, B.-S. CO-Releasing Molecules and Increased Heme Oxygenase-1 Induce Protein S-Glutathionylation to Modulate NF-ΚB Activity in Endothelial Cells. Free Radic. Biol. Med. 2014, 70, 1–13. [Google Scholar] [CrossRef]
- Itoh, K.; Igarashi, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Cloning and Characterization of a Novel Erythroid Cell-Derived CNC Family Transcription Factor Heterodimerizing with the Small Maf Family Proteins. Mol. Cell. Biol. 1995, 15, 4184–4193. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Yamamoto, M. Molecular Mechanisms Activating the Nrf2-Keap1 Pathway of Antioxidant Gene Regulation. Antioxid. Redox Signal. 2005, 7, 385–394. [Google Scholar] [CrossRef]
- Um, H.-C.; Jang, J.-H.; Kim, D.-H.; Lee, C.; Surh, Y.-J. Nitric Oxide Activates Nrf2 through S-Nitrosylation of Keap1 in PC12 Cells. Nitric. Oxide 2011, 25, 161–168. [Google Scholar] [CrossRef]
- Lee, B.-S.; Heo, J.; Kim, Y.-M.; Shim, S.M.; Pae, H.-O.; Kim, Y.-M.; Chung, H.-T. Carbon Monoxide Mediates Heme Oxygenase 1 Induction via Nrf2 Activation in Hepatoma Cells. Biochem. Biophys. Res. Commun. 2006, 343, 965–972. [Google Scholar] [CrossRef]
- D’Araio, E.; Shaw, N.; Millward, A.; Demaine, A.; Whiteman, M.; Hodgkinson, A. Hydrogen Sulfide Induces Heme Oxygenase-1 in Human Kidney Cells. Acta. Diabetol. 2014, 51, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Cao, W.; Biswal, S.; Doré, S. Carbon Monoxide-Activated Nrf2 Pathway Leads to Protection against Permanent Focal Cerebral Ischemia. Stroke 2011, 42, 2605–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilz, R.B.; Casteel, D.E. Regulation of Gene Expression by Cyclic GMP. Circ. Res. 2003, 93, 1034–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, H.; Wang, Y.; Loof, T.; Martini, S.; Kron, S.; Krämer, S.; Neumayer, H.-H. Expression and Activity of Soluble Guanylate Cyclase in Injury and Repair of Anti-Thy1 Glomerulonephritis. Kidney Int. 2004, 66, 2224–2236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Krämer, S.; Loof, T.; Martini, S.; Kron, S.; Kawachi, H.; Shimizu, F.; Neumayer, H.-H.; Peters, H. Stimulation of Soluble Guanylate Cyclase Slows Progression in Anti-Thy1-Induced Chronic Glomerulosclerosis. Kidney Int. 2005, 68, 47–61. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; D’Ambrosio, M.A.; Garvin, J.L.; Wang, H.; Carretero, O.A. Mechanism of Inhibition of Tubuloglomerular Feedback by CO and CGMP. Hypertension 2013, 62, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Morita, T.; Perrella, M.A.; Lee, M.E.; Kourembanas, S. Smooth Muscle Cell-Derived Carbon Monoxide Is a Regulator of Vascular CGMP. Proc. Natl. Acad. Sci. USA 1995, 92, 1475–1479. [Google Scholar] [CrossRef] [Green Version]
- Bucci, M.; Papapetropoulos, A.; Vellecco, V.; Zhou, Z.; Pyriochou, A.; Roussos, C.; Roviezzo, F.; Brancaleone, V.; Cirino, G. Hydrogen Sulfide Is an Endogenous Inhibitor of Phosphodiesterase Activity. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1998–2004. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Martin, E.; Sharina, I.; Esposito, I.; Szabo, C.; Bucci, M.; Cirino, G.; Papapetropoulos, A. Regulation of Soluble Guanylyl Cyclase Redox State by Hydrogen Sulfide. Pharmacol. Res. 2016, 111, 556–562. [Google Scholar] [CrossRef] [Green Version]
- Eberhardt, W.; Kunz, D.; Hummel, R.; Pfeilschifter, J. Molecular Cloning of the Rat Inducible Nitric Oxide Synthase Gene Promoter. Biochem. Biophys. Res. Commun. 1996, 223, 752–756. [Google Scholar] [CrossRef]
- Beck, K.F.; Sterzel, R.B. Cloning and Sequencing of the Proximal Promoter of the Rat INOS Gene: Activation of NFkappaB Is Not Sufficient for Transcription of the INOS Gene in Rat Mesangial Cells. FEBS Lett. 1996, 394, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Gao, Y.; Ding, Y.; Cao, Y.; Chen, J.; Lv, G.; Lu, J.; Yu, B.; Peng, M.; Xu, H.; et al. Catalpol Ameliorates Advanced Glycation End Product-Induced Dysfunction of Glomerular Endothelial Cells via Regulating Nitric Oxide Synthesis by Inducible Nitric Oxide Synthase and Endothelial Nitric Oxide Synthase. IUBMB Life 2019, 71, 1268–1283. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Signaling Pathways for the Vascular Effects of Hydrogen Sulfide. Curr. Opin. Nephrol. Hypertens. 2011, 20, 107–112. [Google Scholar] [CrossRef]
- Schmidlin, C.J.; Rojo de la Vega, M.; Perer, J.; Zhang, D.D.; Wondrak, G.T. Activation of NRF2 by Topical Apocarotenoid Treatment Mitigates Radiation-Induced Dermatitis. Redox Biol. 2020, 37, 101714. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Yamawaki, K. Bardoxolone Methyl: Drug Development for Diabetic Kidney Disease. Clin. Exp. Nephrol. 2020, 24, 857–864. [Google Scholar] [CrossRef]
- Yang, W.; Yang, G.; Jia, X.; Wu, L.; Wang, R. Activation of KATP channels by H2S in rat insulin-secreting cells and the underlying mechanisms. J. Physiol. 2005, 569, 519–531. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beck, K.-F.; Pfeilschifter, J. The Pathophysiology of H2S in Renal Glomerular Diseases. Biomolecules 2022, 12, 207. https://doi.org/10.3390/biom12020207
Beck K-F, Pfeilschifter J. The Pathophysiology of H2S in Renal Glomerular Diseases. Biomolecules. 2022; 12(2):207. https://doi.org/10.3390/biom12020207
Chicago/Turabian StyleBeck, Karl-Friedrich, and Josef Pfeilschifter. 2022. "The Pathophysiology of H2S in Renal Glomerular Diseases" Biomolecules 12, no. 2: 207. https://doi.org/10.3390/biom12020207
APA StyleBeck, K. -F., & Pfeilschifter, J. (2022). The Pathophysiology of H2S in Renal Glomerular Diseases. Biomolecules, 12(2), 207. https://doi.org/10.3390/biom12020207