
Neurobiological and Pharmacological Perspectives of D3 Receptors in Parkinson’s Disease




Abstract
:1. Introduction
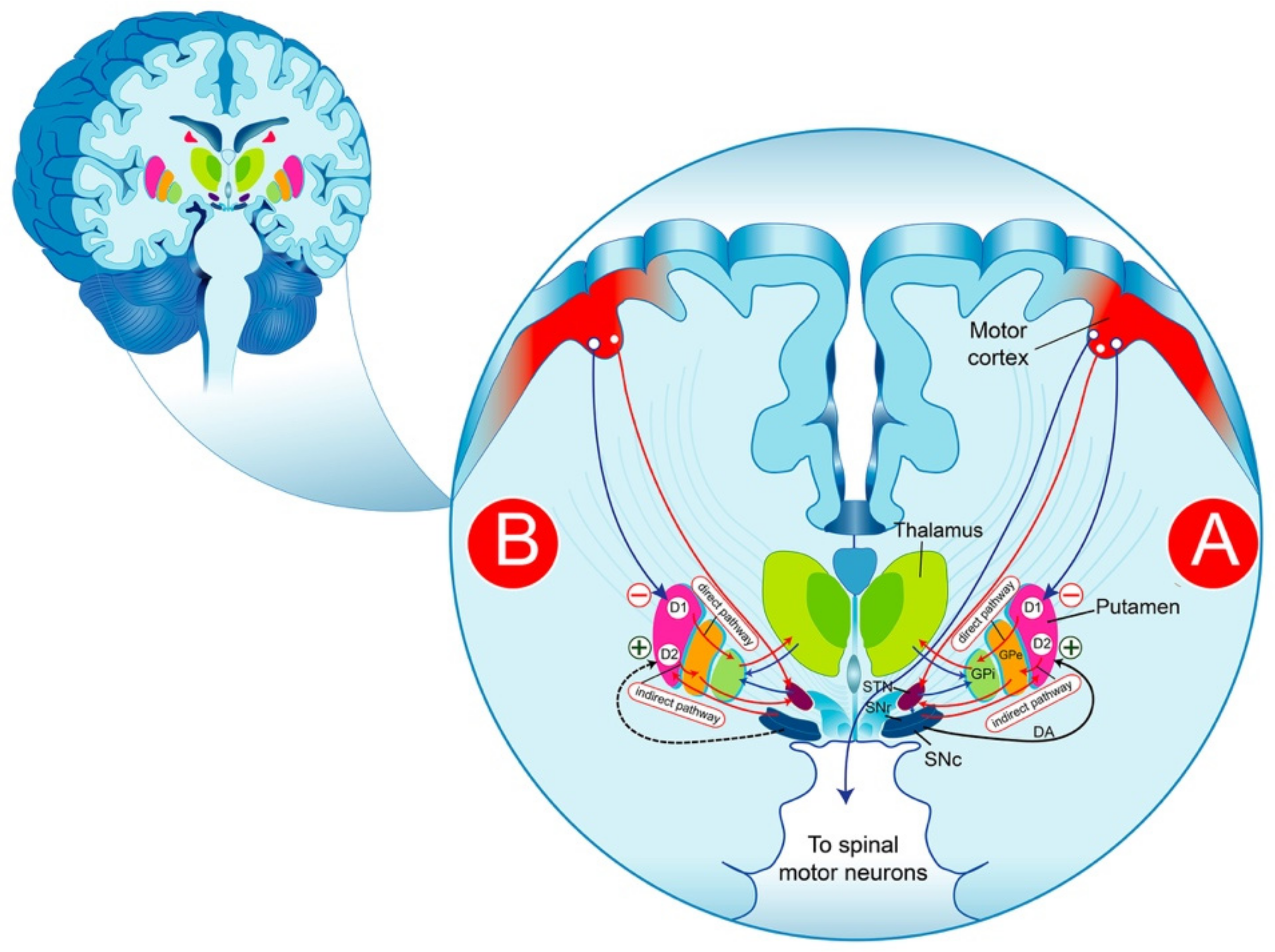
2. Functional Anatomy of the Basal Ganglia in Parkinson’s Disease
3. The Complexity of the Dopaminergic Neuron Network
4. D3 Dopamine Receptor Expression in Parkinson’s Disease
5. The Repercussions of DA Depletion on the Responsiveness of the Dopaminergic Signaling Pathways
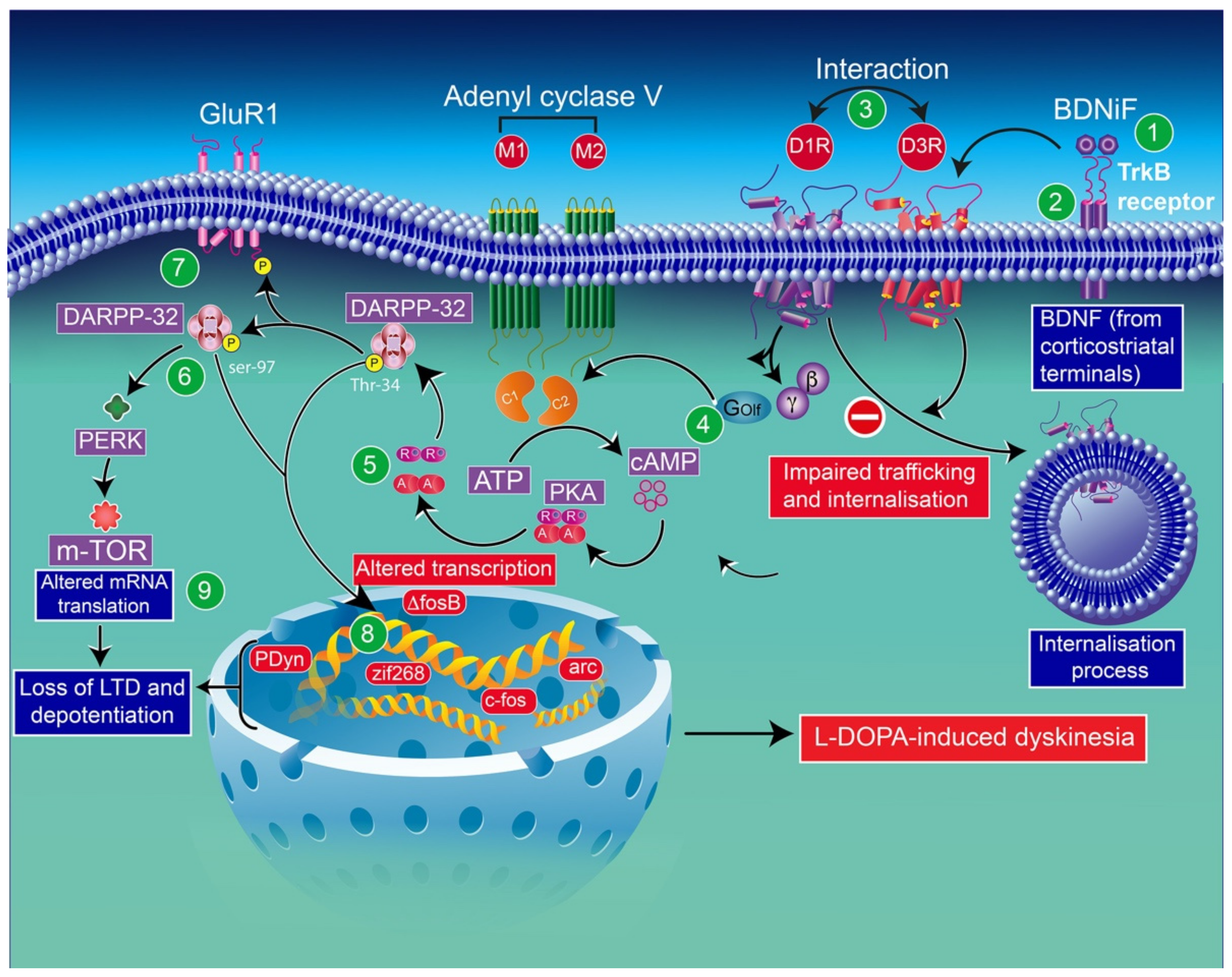
6. Molecular Consequences of l-DOPA Administration in the Context of Dopamine Depletion
6.1. Implication of D3 Receptors in l-DOPA-Induced Dyskinesias
6.2. Role for D3R in the Control of LID
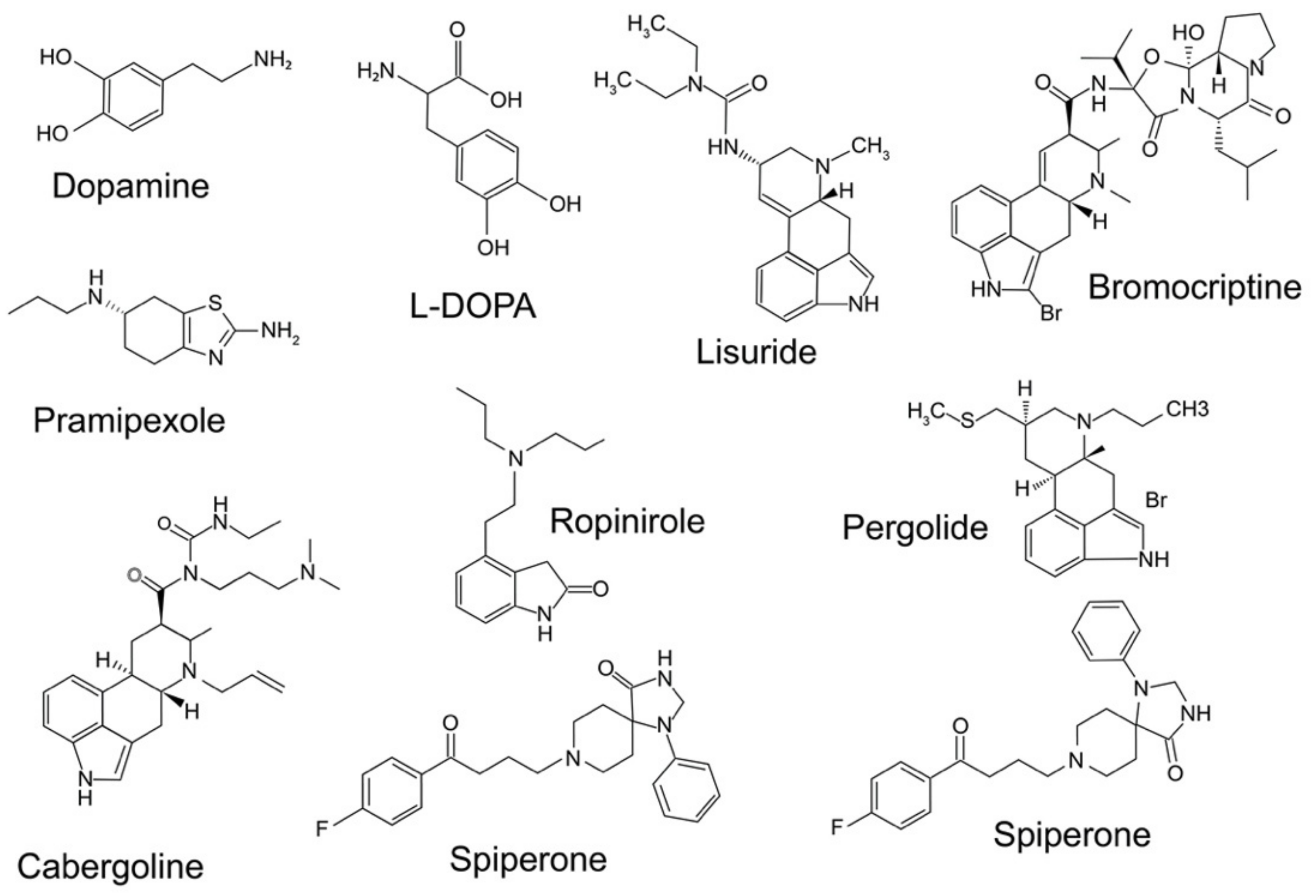
7. Neuroprotective Effects of D3 Receptor-Preferring Agonists
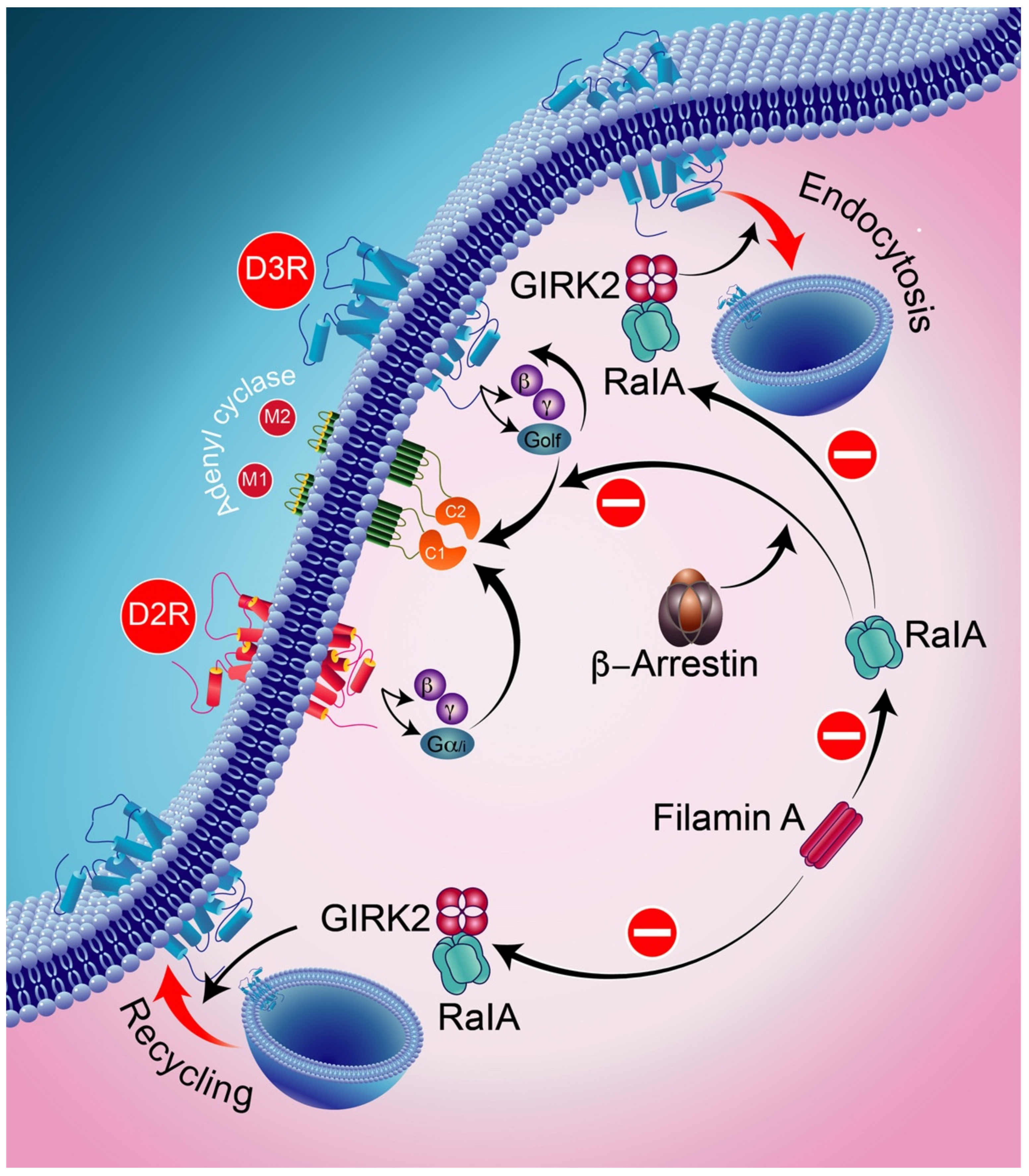
8. Desensitization Process of Dopamine D3 Receptors
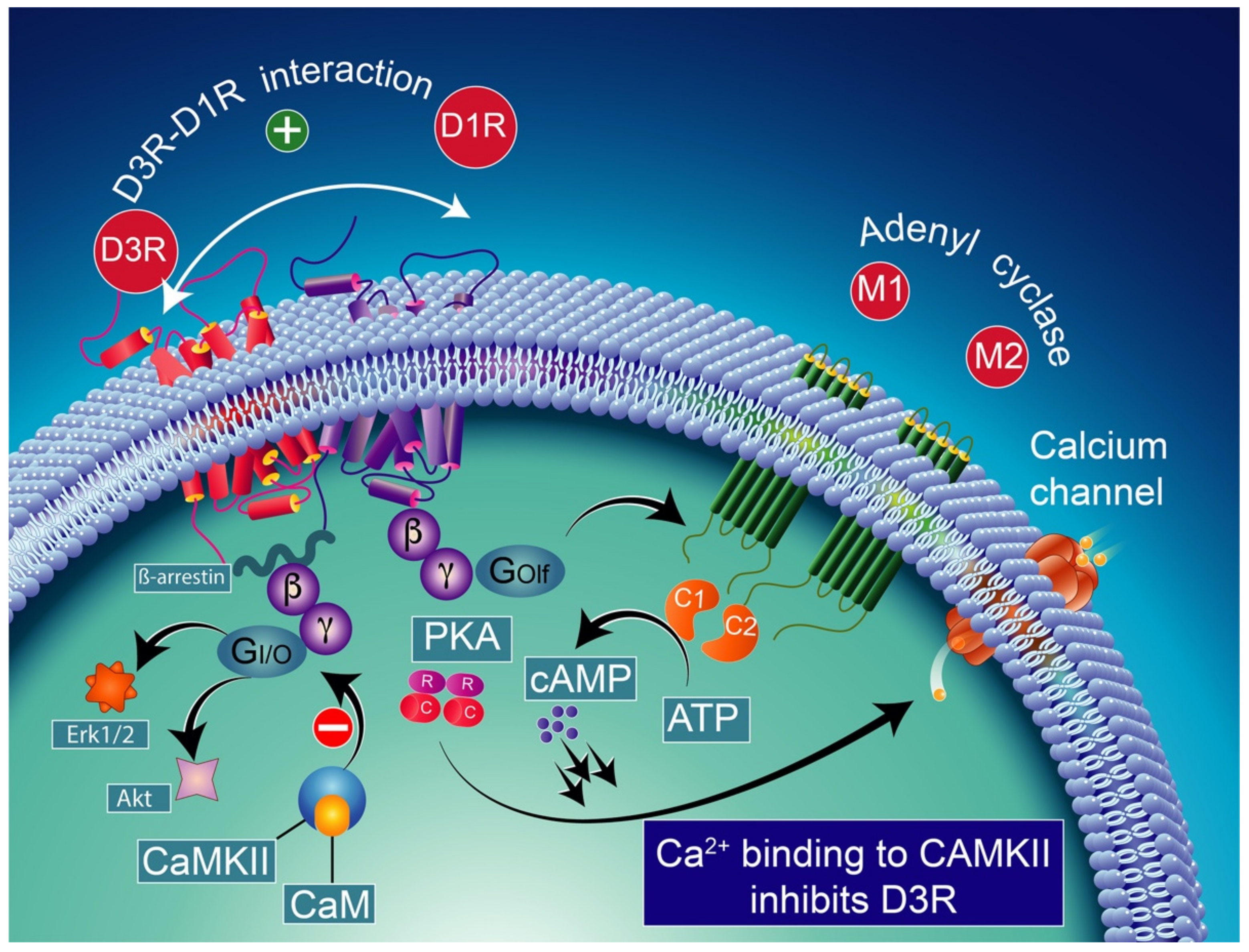
9. D3-D1 Receptors Interactions
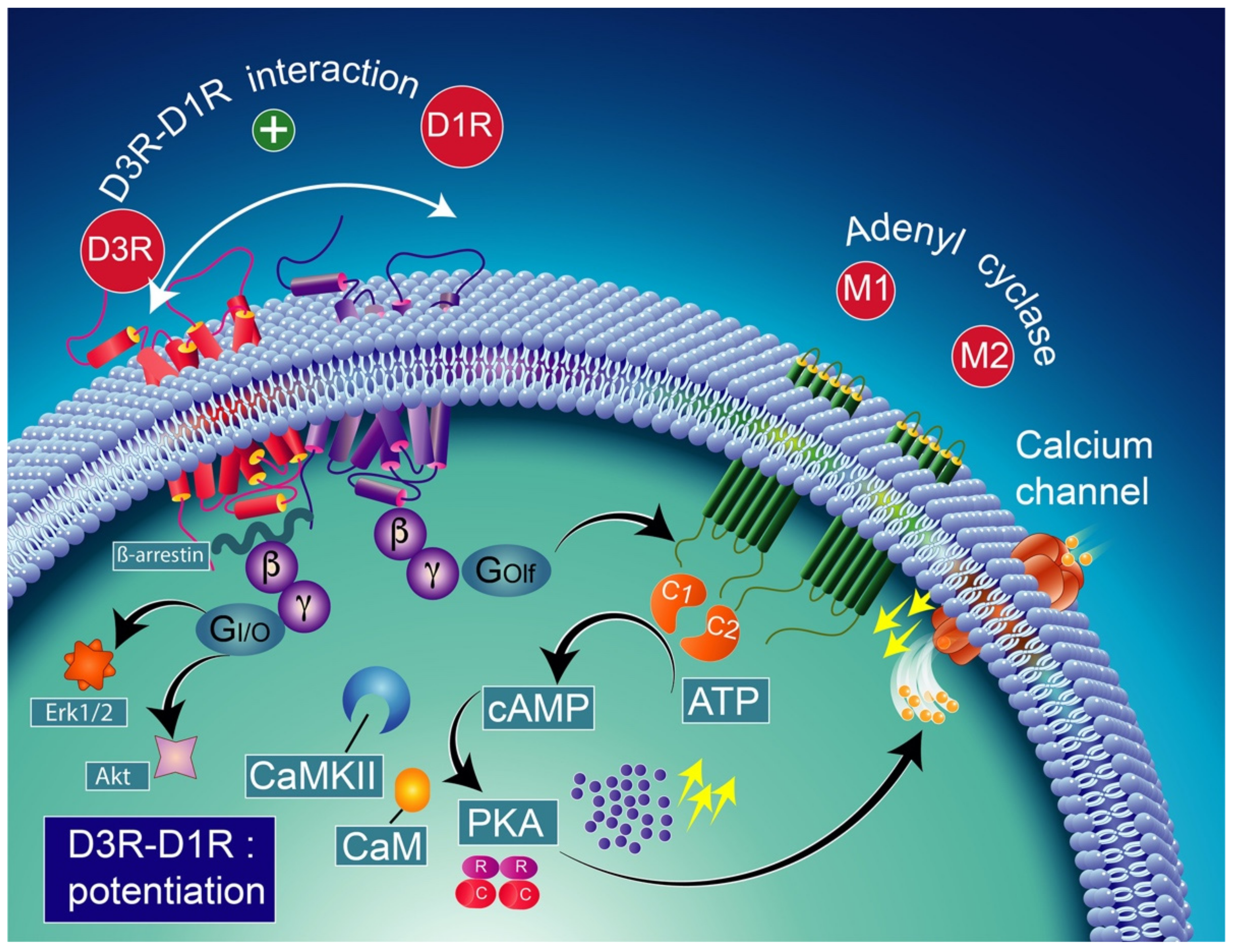
9.1. Neurobiology of Dopamine D3-D1 Receptors Interactions
9.2. Neurobiology of Functional Heteromeric Complexes of D1-D3 Receptors
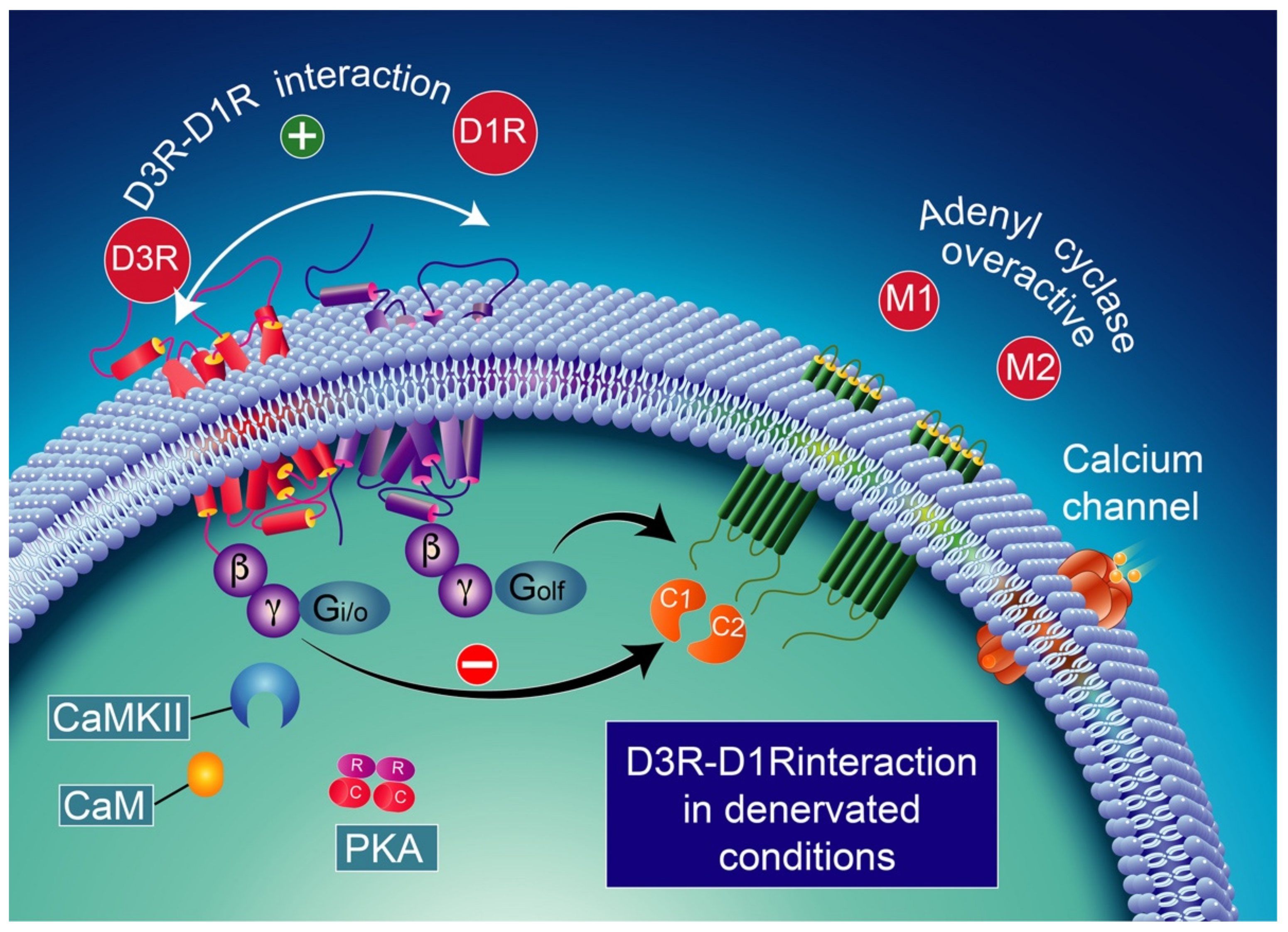
9.3. Pharmacological Significance of D3–D1 Receptor Interaction
10. D3 Receptors, l-DOPA, and DA
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sokoloff, P.; Giros, B.; Martres, M.P.; Bouthenet, M.L.; Schwartz, J.C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 1990, 347, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Bezard, E.; Ferry, S.; Mach, U.; Stark, H.; Leriche, L.; Boraud, T.; Gross, C.; Sokoloff, P. Attenuation of levodopa-induced dyskinesia by normalizing dopamine D3 receptor function. Nat. Med. 2003, 9, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Sokoloff, P.; Diaz, J.; Le Foll, B.; Guillin, O.; Leriche, L.; Bezard, E.; Gross, C. The dopamine D3 receptor: A therapeutic target for the treatment of neuropsychiatric disorders. CNS Neurol. Disord. Drug Targets 2006, 5, 25–43. [Google Scholar] [PubMed]
- Kiss, B.; Laszlovszky, I.; Krámos, B.; Visegrády, A.; Bobok, A.; Lévay, G.; Lendvai, B.; Román, V. Neuronal Dopamine D3 Receptors: Translational Implications for Preclinical Research and CNS Disorders. Biomolecules 2021, 11, 104. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, D.; Marsh, L.; Schrag, A. Neuropsychiatric symptoms in Parkinson’s disease. Mov. Disord. 2009, 24, 2175–2186. [Google Scholar] [CrossRef]
- Albin, R.L.; Young, A.B.; Penney, J.B. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989, 12, 366–375. [Google Scholar] [CrossRef]
- Albin, R.L.; Young, A.B.; Penney, J.B. The functional anatomy of disorders of the basal ganglia. Trends Neurosci. 1995, 18, 63–64. [Google Scholar] [CrossRef]
- Obeso, J.A.; Marin, C.; Rodriguez-Oroz, C.; Blesa, J.; Benitez-Temino, B.; Mena-Segovia, J.; Rodriguez, M.; Olanow, C.W. The basal ganglia in Parkinson’s disease: Current concepts and unexplained observations. Ann. Neurol. 2008, 64, S30–S46. [Google Scholar] [CrossRef]
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Rodriguez, M.; Lanciego, J.L.; Artieda, J.; Gonzalo, N.; Olanow, C.W. Pathophysiology of the basal ganglia in Parkinson’s disease. Trends Neurosci. 2000, 23, S8–S19. [Google Scholar] [CrossRef]
- Burke, R.E.; O’Malley, K. Axon degeneration in Parkinson’s disease. Exp. Neurol. 2013, 246, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Benarroch, E.E. Pedunculopontine nucleus: Functional organization and clinical implications. Neurology 2013, 80, 1148–1155. [Google Scholar] [CrossRef]
- Hortnagl, H.; Pifl, C.; Hortnagl, E.; Reiner, A.; Sperk, G. Distinct gradients of various neurotransmitter markers in caudate nucleus and putamen of the human brain. J. Neurochem. 2020, 152, 650–662. [Google Scholar] [CrossRef]
- Karachi, C.; Grabli, D.; Bernard, F.A.; Tande, D.; Wattiez, N.; Belaid, H.; Bardinet, E.; Prigent, A.; Nothacker, H.P.; Hunot, S.; et al. Cholinergic mesencephalic neurons are involved in gait and postural disorders in Parkinson disease. J. Clin. Investig. 2010, 120, 2745–2754. [Google Scholar] [CrossRef] [Green Version]
- Beaudoin-Gobert, M.; Metereau, E.; Duperrier, S.; Thobois, S.; Tremblay, L.; Sgambato, V. Pathophysiology of levodopa-induced dyskinesia: Insights from multimodal imaging and immunohistochemistry in non-human primates. Neuroimage 2018, 183, 132–141. [Google Scholar] [CrossRef]
- Carta, M.; Carlsson, T.; Kirik, D.; Bjorklund, A. Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain 2007, 130, 1819–1833. [Google Scholar] [CrossRef] [Green Version]
- De Deurwaerdere, P.; Di Giovanni, G.; Millan, M.J. Expanding the repertoire of L-DOPA’s actions: A comprehensive review of its functional neurochemistry. Prog. Neurobiol. 2017, 151, 57–100. [Google Scholar] [CrossRef]
- Navailles, S.; De Deurwaerdere, P. Contribution of serotonergic transmission to the motor and cognitive effects of high-frequency stimulation of the subthalamic nucleus or levodopa in Parkinson’s disease. Mol. Neurobiol. 2012, 45, 173–185. [Google Scholar] [CrossRef]
- Politis, M.; Wu, K.; Loane, C.; Kiferle, L.; Molloy, S.; Brooks, D.J.; Piccini, P. Staging of serotonergic dysfunction in Parkinson’s disease: An in vivo 11C-DASB PET study. Neurobiol. Dis. 2010, 40, 216–221. [Google Scholar] [CrossRef]
- Politis, M.; Wu, K.; Loane, C.; Quinn, N.P.; Brooks, D.J.; Oertel, W.H.; Bjorklund, A.; Lindvall, O.; Piccini, P. Serotonin neuron loss and nonmotor symptoms continue in Parkinson’s patients treated with dopamine grafts. Sci. Transl. Med. 2012, 4, 128ra141. [Google Scholar] [CrossRef]
- Paredes-Rodriguez, E.; Vegas-Suarez, S.; Morera-Herreras, T.; De Deurwaerdere, P.; Miguelez, C. The Noradrenergic System in Parkinson’s Disease. Front. Pharmacol. 2020, 11, 435. [Google Scholar] [CrossRef] [Green Version]
- Eskow Jaunarajs, K.L.; Angoa-Perez, M.; Kuhn, D.M.; Bishop, C. Potential mechanisms underlying anxiety and depression in Parkinson’s disease: Consequences of l-DOPA treatment. Neurosci. Biobehav. Rev. 2011, 35, 556–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanza, K.; Bishop, C. Serotonergic targets for the treatment of L-DOPA-induced dyskinesia. J. Neural Transm. 2018, 125, 1203–1216. [Google Scholar] [CrossRef] [PubMed]
- Parent, A.; Hazrati, L.N. Functional anatomy of the basal ganglia. I. The cortico-basal ganglia-thalamo-cortical loop. Brain Res. Brain Res. Rev. 1995, 20, 91–127. [Google Scholar] [CrossRef]
- Alexander, G.E.; DeLong, M.R.; Strick, P.L. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu. Rev. Neurosci. 1986, 9, 357–381. [Google Scholar] [CrossRef]
- Hoover, J.E.; Strick, P.L. Multiple output channels in the basal ganglia. Science 1993, 259, 819–821. [Google Scholar] [CrossRef] [Green Version]
- Hoover, J.E.; Strick, P.L. The organization of cerebellar and basal ganglia outputs to primary motor cortex as revealed by retrograde transneuronal transport of herpes simplex virus type 1. J. Neurosci. 1999, 19, 1446–1463. [Google Scholar] [CrossRef]
- Percheron, G.; Filion, M. Parallel processing in the basal ganglia: Up to a point. Trends Neurosci. 1991, 14, 55–59. [Google Scholar] [CrossRef]
- Castle, M.; Aymerich, M.S.; Sanchez-Escobar, C.; Gonzalo, N.; Obeso, J.A.; Lanciego, J.L. Thalamic innervation of the direct and indirect basal ganglia pathways in the rat: Ipsi- and contralateral projections. J. Comp. Neurol. 2005, 483, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Peterson, J.D.; Surmeier, D.J. Corticostriatal and thalamostriatal synapses have distinctive properties. J. Neurosci. 2008, 28, 6483–6492. [Google Scholar] [CrossRef]
- Garcia-Munoz, M.; Carrillo-Reid, L.; Arbuthnott, G.W. Functional anatomy: Dynamic States in Basal Ganglia circuits. Front. Neuroanat. 2010, 4, 144. [Google Scholar] [CrossRef] [Green Version]
- Wickens, J.R.; Begg, A.J.; Arbuthnott, G.W. Dopamine reverses the depression of rat corticostriatal synapses which normally follows high-frequency stimulation of cortex in vitro. Neuroscience 1996, 70, 1–5. [Google Scholar] [CrossRef]
- Tecuapetla, F.; Carrillo-Reid, L.; Bargas, J.; Galarraga, E. Dopaminergic modulation of short-term synaptic plasticity at striatal inhibitory synapses. Proc. Natl. Acad. Sci. USA 2007, 104, 10258–10263. [Google Scholar] [CrossRef] [Green Version]
- Ingham, C.A.; Hood, S.H.; Arbuthnott, G.W. Spine density on neostriatal neurones changes with 6-hydroxydopamine lesions and with age. Brain Res. 1989, 503, 334–338. [Google Scholar] [CrossRef]
- Smith, Y.; Raju, D.V.; Pare, J.F.; Sidibe, M. The thalamostriatal system: A highly specific network of the basal ganglia circuitry. Trends Neurosci. 2004, 27, 520–527. [Google Scholar] [CrossRef]
- Parent, A.; Smith, Y. Differential dopaminergic innervation of the two pallidal segments in the squirrel monkey (Saimiri sciureus). Brain Res. 1987, 426, 397–400. [Google Scholar] [CrossRef]
- Cossette, M.; Levesque, M.; Parent, A. Extrastriatal dopaminergic innervation of human basal ganglia. Neurosci. Res. 1999, 34, 51–54. [Google Scholar] [CrossRef]
- Lavoie, B.; Smith, Y.; Parent, A. Dopaminergic innervation of the basal ganglia in the squirrel monkey as revealed by tyrosine hydroxylase immunohistochemistry. J. Comp. Neurol. 1989, 289, 36–52. [Google Scholar] [CrossRef]
- Marcusson, J.; Eriksson, K. [3H]GBR-12935 binding to dopamine uptake sites in the human brain. Brain Res. 1988, 457, 122–129. [Google Scholar] [CrossRef]
- Prensa, L.; Cossette, M.; Parent, A. Dopaminergic innervation of human basal ganglia. J. Chem. Neuroanat. 2000, 20, 207–213. [Google Scholar] [CrossRef]
- Rommelfanger, K.S.; Wichmann, T. Extrastriatal dopaminergic circuits of the Basal Ganglia. Front. Neuroanat. 2010, 4, 139. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Muros, I.; Afonso-Oramas, D.; Abreu, P.; Barroso-Chinea, P.; Rodriguez, M.; Gonzalez, M.C.; Hernandez, T.G. Aging of the rat mesostriatal system: Differences between the nigrostriatal and the mesolimbic compartments. Exp. Neurol. 2007, 204, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Muros, I.; Afonso-Oramas, D.; Abreu, P.; Rodriguez, M.; Gonzalez, M.C.; Gonzalez-Hernandez, T. Deglycosylation and subcellular redistribution of VMAT2 in the mesostriatal system during normal aging. Neurobiol. Aging 2008, 29, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Barroso-Chinea, P.; Abdala, P.; Obeso, J.; Gonzalez-Hernandez, T. Dopamine cell degeneration induced by intraventricular administration of 6-hydroxydopamine in the rat: Similarities with cell loss in parkinson’s disease. Exp. Neurol. 2001, 169, 163–181. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Hernandez, T.; Rodriguez, M. Compartmental organization and chemical profile of dopaminergic and GABAergic neurons in the substantia nigra of the rat. J. Comp. Neurol. 2000, 421, 107–135. [Google Scholar] [CrossRef]
- Hirsch, E.; Graybiel, A.M.; Agid, Y.A. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature 1988, 334, 345–348. [Google Scholar] [CrossRef]
- Newcomer, T.A.; Palmer, A.M.; Rosenberg, P.A.; Aizenman, E. Nonenzymatic conversion of 3,4-dihydroxyphenylalanine to 2,4,5-trihydroxyphenylalanine and 2,4,5-trihydroxyphenylalanine quinone in physiological solutions. J. Neurochem. 1993, 61, 911–920. [Google Scholar] [CrossRef]
- Rescigno, A.; Rinaldi, A.C.; Sanjust, E. Some aspects of tyrosine secondary metabolism. Biochem. Pharm. 1998, 56, 1089–1096. [Google Scholar] [CrossRef]
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The substantia nigra of the human brain. I. Nigrosomes and the nigral matrix, a compartmental organization based on calbindin D(28K) immunohistochemistry. Brain 1999, 122, 1421–1436. [Google Scholar] [CrossRef] [Green Version]
- Schultz, W. Predictive reward signal of dopamine neurons. J. Neurophysiol. 1998, 80, 1–27. [Google Scholar] [CrossRef]
- Rodriguez, M.; Morales, I.; Gomez, I.; Gonzalez, S.; Gonzalez-Hernandez, T.; Gonzalez-Mora, J.L. Heterogeneous dopamine neurochemistry in the striatum: The fountain-drain matrix. J. Pharm. Exp. Ther. 2006, 319, 31–43. [Google Scholar] [CrossRef] [Green Version]
- Gonon, F.G. Nonlinear relationship between impulse flow and dopamine released by rat midbrain dopaminergic neurons as studied by in vivo electrochemistry. Neuroscience 1988, 24, 19–28. [Google Scholar] [CrossRef]
- Wightman, R.M.; Zimmerman, J.B. Control of dopamine extracellular concentration in rat striatum by impulse flow and uptake. Brain Res. Brain Res. Rev. 1990, 15, 135–144. [Google Scholar] [CrossRef]
- Braak, H.; Bohl, J.R.; Muller, C.M.; Rub, U.; de Vos, R.A.; Del Tredici, K. Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov. Disord. 2006, 21, 2042–2051. [Google Scholar] [CrossRef]
- Sun, J.; Xu, J.; Cairns, N.J.; Perlmutter, J.S.; Mach, R.H. Dopamine D1, D2, D3 receptors, vesicular monoamine transporter type-2 (VMAT2) and dopamine transporter (DAT) densities in aged human brain. PLoS ONE 2012, 7, e49483. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, E.V.; Joyce, J.N. Distribution of dopamine D3 receptor expressing neurons in the human forebrain: Comparison with D2 receptor expressing neurons. Neuropsychopharmacology 1999, 20, 60–80. [Google Scholar] [CrossRef] [Green Version]
- Bouthenet, M.L.; Souil, E.; Martres, M.P.; Sokoloff, P.; Giros, B.; Schwartz, J.C. Localization of dopamine D3 receptor mRNA in the rat brain using in situ hybridization histochemistry: Comparison with dopamine D2 receptor mRNA. Brain Res. 1991, 564, 203–219. [Google Scholar] [CrossRef]
- Meador-Woodruff, J.H.; Damask, S.P.; Wang, J.; Haroutunian, V.; Davis, K.L.; Watson, S.J. Dopamine receptor mRNA expression in human striatum and neocortex. Neuropsychopharmacology 1996, 15, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Murray, A.M.; Ryoo, H.L.; Gurevich, E.; Joyce, J.N. Localization of dopamine D3 receptors to mesolimbic and D2 receptors to mesostriatal regions of human forebrain. Proc. Natl. Acad. Sci. USA 1994, 91, 11271–11275. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Hurd, Y.L.; Sokoloff, P.; Schwartz, J.C.; Sedvall, G. D3 dopamine receptor mRNA is widely expressed in the human brain. Brain Res. 1998, 779, 58–74. [Google Scholar] [CrossRef]
- Beaulieu, J.M.; Gainetdinov, R.R. The physiology, signaling, and pharmacology of dopamine receptors. Pharm. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef] [Green Version]
- Celver, J.; Sharma, M.; Kovoor, A. RGS9-2 mediates specific inhibition of agonist-induced internalization of D2-dopamine receptors. J. Neurochem. 2010, 114, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.; Kiss, D.J.; Keserű, G.M.; Stark, H. Binding kinetics of cariprazine and aripiprazole at the dopamine D(3) receptor. Sci. Rep. 2018, 8, 12509. [Google Scholar] [CrossRef] [PubMed]
- De Deurwaerdère, P. Cariprazine: New dopamine biased agonist for neuropsychiatric disorders. Drugs Today 2016, 52, 97–110. [Google Scholar]
- Butini, S.; Nikolic, K.; Kassel, S.; Brückmann, H.; Filipic, S.; Agbaba, D.; Gemma, S.; Brogi, S.; Brindisi, M.; Campiani, G.; et al. Polypharmacology of dopamine receptor ligands. Prog. Neurobiol. 2016, 142, 68–103. [Google Scholar] [CrossRef]
- Bychkov, E.R.; Gurevich, V.V.; Joyce, J.N.; Benovic, J.L.; Gurevich, E.V. Arrestins and two receptor kinases are upregulated in Parkinson’s disease with dementia. Neurobiol. Aging 2008, 29, 379–396. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.N.; Ryoo, H.L.; Beach, T.B.; Caviness, J.N.; Stacy, M.; Gurevich, E.V.; Reiser, M.; Adler, C.H. Loss of response to levodopa in Parkinson’s disease and co-occurrence with dementia: Role of D3 and not D2 receptors. Brain Res. 2002, 955, 138–152. [Google Scholar] [CrossRef]
- Rassu, M.; Del Giudice, M.G.; Sanna, S.; Taymans, J.M.; Morari, M.; Brugnoli, A.; Frassineti, M.; Masala, A.; Esposito, S.; Galioto, M.; et al. Role of LRRK2 in the regulation of dopamine receptor trafficking. PLoS ONE 2017, 12, e0179082. [Google Scholar] [CrossRef]
- Migheli, R.; Del Giudice, M.G.; Spissu, Y.; Sanna, G.; Xiong, Y.; Dawson, T.M.; Dawson, V.L.; Galioto, M.; Rocchitta, G.; Biosa, A.; et al. LRRK2 affects vesicle trafficking, neurotransmitter extracellular level and membrane receptor localization. PLoS ONE 2013, 8, e77198. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Jiang, W.; Liu, L.; Wang, X.; Ding, C.; Tian, Z.; Zhou, R. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 2015, 160, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Tong, Y.; Pisani, A.; Martella, G.; Karouani, M.; Yamaguchi, H.; Pothos, E.N.; Shen, J. R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 14622–14627. [Google Scholar] [CrossRef] [Green Version]
- Quik, M.; Police, S.; He, L.; Di Monte, D.A.; Langston, J.W. Expression of D(3) receptor messenger RNA and binding sites in monkey striatum and substantia nigra after nigrostriatal degeneration: Effect of levodopa treatment. Neuroscience 2000, 98, 263–273. [Google Scholar] [CrossRef]
- Boraud, T.; Bezard, E.; Bioulac, B.; Gross, C.E. Dopamine agonist-induced dyskinesias are correlated to both firing pattern and frequency alterations of pallidal neurones in the MPTP-treated monkey. Brain 2001, 124, 546–557. [Google Scholar] [CrossRef]
- Herrero, M.T.; Augood, S.J.; Asensi, H.; Hirsch, E.C.; Agid, Y.; Obeso, J.A.; Emson, P.C. Effects of L-DOPA-therapy on dopamine D2 receptor mRNA expression in the striatum of MPTP-intoxicated parkinsonian monkeys. Brain Res. Mol. Brain Res. 1996, 42, 149–155. [Google Scholar] [CrossRef]
- Levesque, D.; Martres, M.P.; Diaz, J.; Griffon, N.; Lammers, C.H.; Sokoloff, P.; Schwartz, J.C. A paradoxical regulation of the dopamine D3 receptor expression suggests the involvement of an anterograde factor from dopamine neurons. Proc. Natl. Acad. Sci. USA 1995, 92, 1719–1723. [Google Scholar] [CrossRef] [Green Version]
- Bordet, R.; Ridray, S.; Carboni, S.; Diaz, J.; Sokoloff, P.; Schwartz, J.C. Induction of dopamine D3 receptor expression as a mechanism of behavioral sensitization to levodopa. Proc. Natl. Acad. Sci. USA 1997, 94, 3363–3367. [Google Scholar] [CrossRef] [Green Version]
- Bordet, R.; Ridray, S.; Schwartz, J.C.; Sokoloff, P. Involvement of the direct striatonigral pathway in levodopa-induced sensitization in 6-hydroxydopamine-lesioned rats. Eur. J. Neurosci. 2000, 12, 2117–2123. [Google Scholar] [CrossRef]
- Morissette, M.; Goulet, M.; Grondin, R.; Blanchet, P.; Bédard, P.J.; Di Paolo, T.; Lévesque, D. Associative and limbic regions of monkey striatum express high levels of dopamine D3 receptors: Effects of MPTP and dopamine agonist replacement therapies. Eur. J. Neurosci. 1998, 10, 2565–2573. [Google Scholar] [CrossRef]
- Hurley, M.J.; Stubbs, C.M.; Jenner, P.; Marsden, C.D. D3 receptor expression within the basal ganglia is not affected by Parkinson’s disease. Neurosci. Lett. 1996, 214, 75–78. [Google Scholar] [CrossRef]
- Joyce, J.N. Dopamine D3 receptor as a therapeutic target for antipsychotic and antiparkinsonian drugs. Pharm. Ther. 2001, 90, 231–259. [Google Scholar] [CrossRef]
- Van den Buuse, M. Effects of 7-hydroxy-N,N-di-n-propylaminotetralin on behaviour and blood pressure of spontaneously hypertensive rats. Eur. J. Pharm. 1993, 243, 169–177. [Google Scholar] [CrossRef]
- Ryoo, H.L.; Pierrotti, D.; Joyce, J.N. Dopamine D3 receptor is decreased and D2 receptor is elevated in the striatum of Parkinson’s disease. Mov. Disord. 1998, 13, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.N.; Ryoo, H.; Gurevich, E.V.; Adler, C.; Beach, T. Ventral striatal D(3) receptors and Parkinson’s Disease. Parkinsonism Relat. Disord. 2001, 7, 225–230. [Google Scholar] [CrossRef]
- Bychkov, E.; Ahmed, M.R.; Dalby, K.N.; Gurevich, E.V. Dopamine depletion and subsequent treatment with L-DOPA, but not the long-lived dopamine agonist pergolide, enhances activity of the Akt pathway in the rat striatum. J. Neurochem. 2007, 102, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Gerfen, C.R.; McGinty, J.F.; Young, W.S., 3rd. Dopamine differentially regulates dynorphin, substance P, and enkephalin expression in striatal neurons: In situ hybridization histochemical analysis. J. Neurosci. 1991, 11, 1016–1031. [Google Scholar] [CrossRef] [Green Version]
- Chagraoui, A.; Boulain, M.; Juvin, L.; Anouar, Y.; Barriere, G.; Deurwaerdere, P. L-DOPA in Parkinson’s Disease: Looking at the “False” Neurotransmitters and Their Meaning. Int. J. Mol. Sci. 2019, 21, 294. [Google Scholar] [CrossRef] [Green Version]
- Prieto, G.A.; Perez-Burgos, A.; Palomero-Rivero, M.; Galarraga, E.; Drucker-Colin, R.; Bargas, J. Upregulation of D2-class signaling in dopamine-denervated striatum is in part mediated by D3 receptors acting on Ca V 2.1 channels via PIP2 depletion. J. Neurophysiol. 2011, 105, 2260–2274. [Google Scholar] [CrossRef]
- Prieto, G.A.; Perez-Burgos, A.; Fiordelisio, T.; Salgado, H.; Galarraga, E.; Drucker-Colin, R.; Bargas, J. Dopamine D(2)-class receptor supersensitivity as reflected in Ca2+ current modulation in neostriatal neurons. Neuroscience 2009, 164, 345–350. [Google Scholar] [CrossRef]
- Richtand, N.M.; Liu, Y.; Ahlbrand, R.; Sullivan, J.R.; Newman, A.H.; McNamara, R.K. Dopaminergic regulation of dopamine D3 and D3nf receptor mRNA expression. Synapse 2010, 64, 634–643. [Google Scholar] [CrossRef] [Green Version]
- Schmauss, C. Enhanced cleavage of an atypical intron of dopamine D3-receptor pre-mRNA in chronic schizophrenia. J. Neurosci. 1996, 16, 7902–7909. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.N.; Bachus, S.E.; McDonald, C.G.; Smith, R.F. Role of the D3 dopamine receptor in nicotine sensitization. Behav. Brain Res. 2015, 289, 92–104. [Google Scholar] [CrossRef]
- Elmhurst, J.L.; Xie, Z.; O’Dowd, B.F.; George, S.R. The splice variant D3nf reduces ligand binding to the D3 dopamine receptor: Evidence for heterooligomerization. Brain Res. Mol. Brain Res. 2000, 80, 63–74. [Google Scholar] [CrossRef]
- Ng, G.Y.; Mouillac, B.; George, S.R.; Caron, M.; Dennis, M.; Bouvier, M.; O’Dowd, B.F. Desensitization, phosphorylation and palmitoylation of the human dopamine D1 receptor. Eur. J. Pharm. 1994, 267, 7–19. [Google Scholar] [CrossRef]
- Cai, G.; Wang, H.Y.; Friedman, E. Increased dopamine receptor signaling and dopamine receptor-G protein coupling in denervated striatum. J. Pharm. Exp. Ther. 2002, 302, 1105–1112. [Google Scholar] [CrossRef] [Green Version]
- Corvol, J.C.; Muriel, M.P.; Valjent, E.; Feger, J.; Hanoun, N.; Girault, J.A.; Hirsch, E.C.; Herve, D. Persistent increase in olfactory type G-protein alpha subunit levels may underlie D1 receptor functional hypersensitivity in Parkinson disease. J. Neurosci. 2004, 24, 7007–7014. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.K.; Gardner, E.L.; Katzman, R.; Makman, M.H. Enhancement of dopamine-stimulated adenylate cyclase activity in rat caudate after lesions in substantia nigra: Evidence for denervation supersensitivity. Proc. Natl. Acad. Sci. USA 1974, 71, 3883–3887. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.K.; Marshall, A.M.; Varmuza, S.L. Supersensitivity in rat caudate nucleus: Effects of 6-hydroxydopamine on the time course of dopamine receptor and cyclic AMP changes. Brain Res. 1980, 200, 47–57. [Google Scholar] [CrossRef]
- Pifl, C.; Nanoff, C.; Schingnitz, G.; Schutz, W.; Hornykiewicz, O. Sensitization of dopamine-stimulated adenylyl cyclase in the striatum of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated rhesus monkeys and patients with idiopathic Parkinson’s disease. J. Neurochem. 1992, 58, 1997–2004. [Google Scholar] [CrossRef]
- Pifl, C.; Reither, H.; Hornykiewicz, O. Functional sensitization of striatal dopamine D1 receptors in the 6-hydroxydopamine-lesioned rat. Brain Res. 1992, 572, 87–93. [Google Scholar] [CrossRef]
- Satoh, H.; Satoh, Y.; Notsu, Y.; Honda, F. Adenosine 3′,5′-cyclic monophosphate as a possible mediator of rotational behaviour induced by dopaminergic receptor stimulation in rats lesioned unilaterally in the substantia nigra. Eur. J. Pharm. 1976, 39, 365–377. [Google Scholar] [CrossRef]
- Tong, J.; Fitzmaurice, P.S.; Ang, L.C.; Furukawa, Y.; Guttman, M.; Kish, S.J. Brain dopamine-stimulated adenylyl cyclase activity in Parkinson’s disease, multiple system atrophy, and progressive supranuclear palsy. Ann. Neurol. 2004, 55, 125–129. [Google Scholar] [CrossRef]
- Santini, E.; Valjent, E.; Usiello, A.; Carta, M.; Borgkvist, A.; Girault, J.A.; Herve, D.; Greengard, P.; Fisone, G. Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J. Neurosci. 2007, 27, 6995–7005. [Google Scholar] [CrossRef]
- Hemmings, H.C., Jr.; Williams, K.R.; Konigsberg, W.H.; Greengard, P. DARPP-32, a dopamine- and adenosine 3′:5′-monophosphate-regulated neuronal phosphoprotein. I. Amino acid sequence around the phosphorylated threonine. J. Biol. Chem. 1984, 259, 14486–14490. [Google Scholar] [CrossRef]
- Fienberg, A.A.; Hiroi, N.; Mermelstein, P.G.; Song, W.; Snyder, G.L.; Nishi, A.; Cheramy, A.; O’Callaghan, J.P.; Miller, D.B.; Cole, D.G.; et al. DARPP-32: Regulator of the efficacy of dopaminergic neurotransmission. Science 1998, 281, 838–842. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, E.V.; Bordelon, Y.; Shapiro, R.M.; Arnold, S.E.; Gur, R.E.; Joyce, J.N. Mesolimbic dopamine D3 receptors and use of antipsychotics in patients with schizophrenia. A Postmortem Study. Arch. Gen. Psychiatry 1997, 54, 225–232. [Google Scholar] [CrossRef]
- Pivonello, R.; Ferone, D.; de Herder, W.W.; Faggiano, A.; Bodei, L.; de Krijger, R.R.; Lombardi, G.; Colao, A.; Lamberts, S.W.; Hofland, L.J. Dopamine receptor expression and function in corticotroph ectopic tumors. J. Clin. Endocrinol. Metab. 2007, 92, 65–69. [Google Scholar] [CrossRef] [Green Version]
- De Mei, C.; Ramos, M.; Iitaka, C.; Borrelli, E. Getting specialized: Presynaptic and postsynaptic dopamine D2 receptors. Curr. Opin. Pharm. 2009, 9, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Starke, K.; Reimann, W.; Zumstein, A.; Hertting, G. Effect of dopamine receptor agonists and antagonists on release of dopamine in the rabbit caudate nucleus in vitro. Naunyn Schmiedeberg’s Arch. Pharm. 1978, 305, 27–36. [Google Scholar] [CrossRef]
- Wreggett, K.A.; Seeman, P. Agonist high- and low-affinity states of the D2-dopamine receptor in calf brain. Partial conversion by guanine nucleotide. Mol. Pharm. 1984, 25, 10–17. [Google Scholar]
- Nakajima, S.; Gerretsen, P.; Takeuchi, H.; Caravaggio, F.; Chow, T.; Le Foll, B.; Mulsant, B.; Pollock, B.; Graff-Guerrero, A. The potential role of dopamine D₃ receptor neurotransmission in cognition. Eur. Neuropsychopharmacol. 2013, 23, 799–813. [Google Scholar] [CrossRef] [Green Version]
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Rodriguez, M.; DeLong, M.R.; Olanow, C.W. Pathophysiology of levodopa-induced dyskinesias in Parkinson’s disease: Problems with the current model. Ann. Neurol. 2000, 47, S22–S32; discussion S32-24. [Google Scholar] [PubMed]
- Crossman, A.R. A hypothesis on the pathophysiological mechanisms that underlie levodopa- or dopamine agonist-induced dyskinesia in Parkinson’ disease: Implications for future strategies in treatment. Mov. Disord. 1990, 5, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Porras, G.; De Deurwaerdere, P.; Li, Q.; Marti, M.; Morgenstern, R.; Sohr, R.; Bezard, E.; Morari, M.; Meissner, W.G. L-dopa-induced dyskinesia: Beyond an excessive dopamine tone in the striatum. Sci. Rep. 2014, 4, 3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezard, E.; Brotchie, J.M.; Gross, C.E. Pathophysiology of levodopa-induced dyskinesia: Potential for new therapies. Nat. Rev. 2001, 2, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, G.; Brotchie, J.M.; Grandas, F.; Nomoto, M.; Goetz, C.G. Levodopa-induced dyskinesias. Mov. Disord. 2007, 22, 1379–1389; quiz 1523. [Google Scholar] [CrossRef]
- Le Foll, B.; Diaz, J.; Sokoloff, P. Neuroadaptations to hyperdopaminergia in dopamine D3 receptor-deficient mice. Life Sci. 2005, 76, 1281–1296. [Google Scholar] [CrossRef]
- Joseph, J.D.; Wang, Y.M.; Miles, P.R.; Budygin, E.A.; Picetti, R.; Gainetdinov, R.R.; Caron, M.G.; Wightman, R.M. Dopamine autoreceptor regulation of release and uptake in mouse brain slices in the absence of D(3) receptors. Neuroscience 2002, 112, 39–49. [Google Scholar] [CrossRef]
- Asin, K.E.; Bednarz, L.; Nikkel, A.; Perner, R. Rotation and striatal c-fos expression after repeated, daily treatment with selective dopamine receptor agonists and levodopa. J. Pharm. Exp. Ther. 1995, 273, 1483–1490. [Google Scholar]
- Rascol, O.; Nutt, J.G.; Blin, O.; Goetz, C.G.; Trugman, J.M.; Soubrouillard, C.; Carter, J.H.; Currie, L.J.; Fabre, N.; Thalamas, C.; et al. Induction by dopamine D1 receptor agonist ABT-431 of dyskinesia similar to levodopa in patients with Parkinson disease. Arch. Neurol. 2001, 58, 249–254. [Google Scholar] [CrossRef]
- Blanchet, P.J.; Konitsiotis, S.; Chase, T.N. Motor response to a dopamine D3 receptor preferring agonist compared to apomorphine in levodopa-primed 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine monkeys. J. Pharm. Exp. Ther. 1997, 283, 794–799. [Google Scholar]
- Blanchet, P.J.; Grondin, R.; Bedard, P.J. Dyskinesia and wearing-off following dopamine D1 agonist treatment in drug-naive 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned primates. Mov. Disord. 1996, 11, 91–94. [Google Scholar] [CrossRef]
- Pearce, R.K.; Jackson, M.; Britton, D.R.; Shiosaki, K.; Jenner, P.; Marsden, C.D. Actions of the D1 agonists A-77636 and A-86929 on locomotion and dyskinesia in MPTP-treated L-dopa-primed common marmosets. Psychopharmacology 1999, 142, 51–60. [Google Scholar] [CrossRef]
- Rascol, O.; Blin, O.; Thalamas, C.; Descombes, S.; Soubrouillard, C.; Azulay, P.; Fabre, N.; Viallet, F.; Lafnitzegger, K.; Wright, S.; et al. ABT-431, a D1 receptor agonist prodrug, has efficacy in Parkinson’s disease. Ann. Neurol. 1999, 45, 736–741. [Google Scholar] [CrossRef]
- Rinne, U.K.; Laihinen, A.; Rinne, J.O.; Nagren, K.; Bergman, J.; Ruotsalainen, U. Positron emission tomography demonstrates dopamine D2 receptor supersensitivity in the striatum of patients with early Parkinson’s disease. Mov. Disord. 1990, 5, 55–59. [Google Scholar] [CrossRef]
- Antonini, A.; Schwarz, J.; Oertel, W.H.; Pogarell, O.; Leenders, K.L. Long-term changes of striatal dopamine D2 receptors in patients with Parkinson’s disease: A study with positron emission tomography and [11C]raclopride. Mov. Disord. 1997, 12, 33–38. [Google Scholar] [CrossRef]
- Turjanski, N.; Lees, A.J.; Brooks, D.J. In vivo studies on striatal dopamine D1 and D2 site binding in L-dopa-treated Parkinson’s disease patients with and without dyskinesias. Neurology 1997, 49, 717–723. [Google Scholar] [CrossRef]
- Thobois, S.; Vingerhoets, F.; Fraix, V.; Xie-Brustolin, J.; Mollion, H.; Costes, N.; Mertens, P.; Benabid, A.L.; Pollak, P.; Broussolle, E. Role of dopaminergic treatment in dopamine receptor down-regulation in advanced Parkinson disease: A positron emission tomographic study. Arch. Neurol. 2004, 61, 1705–1709. [Google Scholar] [CrossRef]
- Morissette, M.; Goulet, M.; Calon, F.; Falardeau, P.; Blanchet, P.J.; Bedard, P.J.; Di Paolo, T. Changes of D1 and D2 dopamine receptor mRNA in the brains of monkeys lesioned with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: Correction with chronic administration of L-3,4-dihydroxyphenylalanine. Mol. Pharmacol. 1996, 50, 1073–1079. [Google Scholar]
- Kim, D.S.; Szczypka, M.S.; Palmiter, R.D. Dopamine-deficient mice are hypersensitive to dopamine receptor agonists. J. Neurosci. 2000, 20, 4405–4413. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.S.; Palmiter, R.D.; Cummins, A.; Gerfen, C.R. Reversal of supersensitive striatal dopamine D1 receptor signaling and extracellular signal-regulated kinase activity in dopamine-deficient mice. Neuroscience 2006, 137, 1381–1388. [Google Scholar] [CrossRef]
- Bezard, E.; Gross, C.E.; Qin, L.; Gurevich, V.V.; Benovic, J.L.; Gurevich, E.V. L-DOPA reverses the MPTP-induced elevation of the arrestin2 and GRK6 expression and enhanced ERK activation in monkey brain. Neurobiol. Dis. 2005, 18, 323–335. [Google Scholar] [CrossRef]
- Brown, A.M.; Deutch, A.Y.; Colbran, R.J. Dopamine depletion alters phosphorylation of striatal proteins in a model of Parkinsonism. Eur. J. Neurosci. 2005, 22, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Picconi, B.; Gardoni, F.; Centonze, D.; Mauceri, D.; Cenci, M.A.; Bernardi, G.; Calabresi, P.; Di Luca, M. Abnormal Ca2+-calmodulin-dependent protein kinase II function mediates synaptic and motor deficits in experimental parkinsonism. J. Neurosci. 2004, 24, 5283–5291. [Google Scholar] [CrossRef]
- Berke, J.D.; Paletzki, R.F.; Aronson, G.J.; Hyman, S.E.; Gerfen, C.R. A complex program of striatal gene expression induced by dopaminergic stimulation. J. Neurosci. 1998, 18, 5301–5310. [Google Scholar] [CrossRef]
- Cenci, M.A.; Tranberg, A.; Andersson, M.; Hilbertson, A. Changes in the regional and compartmental distribution of FosB- and JunB-like immunoreactivity induced in the dopamine-denervated rat striatum by acute or chronic L-dopa treatment. Neuroscience 1999, 94, 515–527. [Google Scholar] [CrossRef]
- McClung, C.A.; Ulery, P.G.; Perrotti, L.I.; Zachariou, V.; Berton, O.; Nestler, E.J. DeltaFosB: A molecular switch for long-term adaptation in the brain. Brain Res. Mol. Brain Res. 2004, 132, 146–154. [Google Scholar] [CrossRef]
- Henry, B.; Crossman, A.R.; Brotchie, J.M. Effect of repeated L-DOPA, bromocriptine, or lisuride administration on preproenkephalin-A and preproenkephalin-B mRNA levels in the striatum of the 6-hydroxydopamine-lesioned rat. Exp. Neurol. 1999, 155, 204–220. [Google Scholar] [CrossRef]
- Morissette, M.; Goulet, M.; Soghomonian, J.J.; Blanchet, P.J.; Calon, F.; Bedard, P.J.; Di Paolo, T. Preproenkephalin mRNA expression in the caudate-putamen of MPTP monkeys after chronic treatment with the D2 agonist U91356A in continuous or intermittent mode of administration: Comparison with L-DOPA therapy. Brain Res. Mol. Brain Res. 1997, 49, 55–62. [Google Scholar] [CrossRef]
- Morissette, M.; Grondin, R.; Goulet, M.; Bedard, P.J.; Di Paolo, T. Differential regulation of striatal preproenkephalin and preprotachykinin mRNA levels in MPTP-lesioned monkeys chronically treated with dopamine D1 or D2 receptor agonists. J. Neurochem. 1999, 72, 682–692. [Google Scholar] [CrossRef] [Green Version]
- Granata, R.; Wenning, G.K.; Jolkkonen, J.; Jenner, P.; Marsden, C.D. Effect of repeated administration of dopamine agonists on striatal neuropeptide mRNA expression in rats with a unilateral nigral 6-hydroxydopamine lesion. J. Neural Transm. 1996, 103, 249–260. [Google Scholar] [CrossRef]
- Calon, F.; Birdi, S.; Rajput, A.H.; Hornykiewicz, O.; Bedard, P.J.; Di Paolo, T. Increase of preproenkephalin mRNA levels in the putamen of Parkinson disease patients with levodopa-induced dyskinesias. J. Neuropathol. Exp. Neurol. 2002, 61, 186–196. [Google Scholar] [CrossRef]
- Ahmed, M.R.; Bychkov, E.; Gurevich, V.V.; Benovic, J.L.; Gurevich, E.V. Altered expression and subcellular distribution of GRK subtypes in the dopamine-depleted rat basal ganglia is not normalized by l-DOPA treatment. J. Neurochem. 2008, 104, 1622–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Hilaire, M.; Landry, E.; Levesque, D.; Rouillard, C. Denervation and repeated L-DOPA induce a coordinate expression of the transcription factor NGFI-B in striatal projection pathways in hemi-parkinsonian rats. Neurobiol. Dis. 2003, 14, 98–109. [Google Scholar] [CrossRef]
- Gerfen, C.R.; Engber, T.M.; Mahan, L.C.; Susel, Z.; Chase, T.N.; Monsma, F.J., Jr.; Sibley, D.R. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 1990, 250, 1429–1432. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.D.; Chartisathian, K.; Ahmed, S.M.; Chase, T.N. Cyclic AMP responsive element binding protein phosphorylation and persistent expression of levodopa-induced response alterations in unilateral nigrostriatal 6-OHDA lesioned rats. J. Neurosci. Res. 2003, 72, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.M.; Tirotta, E.; Sotnikova, T.D.; Masri, B.; Salahpour, A.; Gainetdinov, R.R.; Borrelli, E.; Caron, M.G. Regulation of Akt signaling by D2 and D3 dopamine receptors in vivo. J. Neurosci. 2007, 27, 881–885. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, J.M.; Gainetdinov, R.R.; Caron, M.G. The Akt-GSK-3 signaling cascade in the actions of dopamine. Trends Pharm. Sci. 2007, 28, 166–172. [Google Scholar] [CrossRef]
- Kumar, R.; Riddle, L.R.; Griffin, S.A.; Chu, W.; Vangveravong, S.; Neisewander, J.; Mach, R.H.; Luedtke, R.R. Evaluation of D2 and D3 dopamine receptor selective compounds on L-dopa-dependent abnormal involuntary movements in rats. Neuropharmacology 2009, 56, 956–969. [Google Scholar] [CrossRef] [Green Version]
- Monville, C.; Torres, E.M.; Dunnett, S.B. Validation of the l-dopa-induced dyskinesia in the 6-OHDA model and evaluation of the effects of selective dopamine receptor agonists and antagonists. Brain Res. Bull. 2005, 68, 16–23. [Google Scholar] [CrossRef]
- Berthet, A.; Bezard, E. Dopamine receptors and L-dopa-induced dyskinesia. Parkinsonism Relat. Disord. 2009, 15 (Suppl. 4), S8–S12. [Google Scholar] [CrossRef]
- Joyce, J.N.; Millan, M.J. Dopamine D3 receptor agonists for protection and repair in Parkinson’s disease. Curr. Opin. Pharmacol. 2007, 7, 100–105. [Google Scholar] [CrossRef]
- Visanji, N.P.; Fox, S.H.; Johnston, T.; Reyes, G.; Millan, M.J.; Brotchie, J.M. Dopamine D3 receptor stimulation underlies the development of L-DOPA-induced dyskinesia in animal models of Parkinson’s disease. Neurobiol. Dis. 2009, 35, 184–192. [Google Scholar] [CrossRef]
- Solis, O.; Garcia-Montes, J.R.; Gonzalez-Granillo, A.; Xu, M.; Moratalla, R. Dopamine D3 Receptor Modulates l-DOPA-Induced Dyskinesia by Targeting D1 Receptor-Mediated Striatal Signaling. Cereb. Cortex 2015, 27, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Mela, F.; Millan, M.J.; Brocco, M.; Morari, M. The selective D(3) receptor antagonist, S33084, improves parkinsonian-like motor dysfunction but does not affect L-DOPA-induced dyskinesia in 6-hydroxydopamine hemi-lesioned rats. Neuropharmacology 2010, 58, 528–536. [Google Scholar] [CrossRef]
- Sokoloff, P.; Andrieux, M.; Besançon, R.; Pilon, C.; Martres, M.P.; Giros, B.; Schwartz, J.C. Pharmacology of human dopamine D3 receptor expressed in a mammalian cell line: Comparison with D2 receptor. Eur. J. Pharm. 1992, 225, 331–337. [Google Scholar] [CrossRef]
- Tadori, Y.; Forbes, R.A.; McQuade, R.D.; Kikuchi, T. Functional potencies of dopamine agonists and antagonists at human dopamine D₂ and D₃ receptors. Eur. J. Pharm. 2011, 666, 43–52. [Google Scholar] [CrossRef]
- Millan, M.J.; Maiofiss, L.; Cussac, D.; Audinot, V.; Boutin, J.A.; Newman-Tancredi, A. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. I. A multivariate analysis of the binding profiles of 14 drugs at 21 native and cloned human receptor subtypes. J. Pharm. Exp. Ther. 2002, 303, 791–804. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Hazeldine, S.; Ghosh, B.; Parrington, I.; Kuzhikandathil, E.; Reith, M.E.; Dutta, A.K. Bioisosteric heterocyclic versions of 7-{[2-(4-phenyl-piperazin-1-yl)ethyl]propylamino}-5,6,7,8-tetrahydronaphthalen-2-ol: Identification of highly potent and selective agonists for dopamine D3 receptor with potent in vivo activity. J. Med. Chem. 2008, 51, 3005–3019. [Google Scholar] [CrossRef]
- Johnson, M.; Antonio, T.; Reith, M.E.; Dutta, A.K. Structure-activity relationship study of N⁶-(2-(4-(1H-Indol-5-yl)piperazin-1-yl)ethyl)-N⁶-propyl-4,5,6,7-tetrahydrobenzo[d]thiazole-2,6-diamine analogues: Development of highly selective D3 dopamine receptor agonists along with a highly potent D2/D3 agonist and their pharmacological characterization. J. Med. Chem. 2012, 55, 5826–5840. [Google Scholar] [CrossRef] [Green Version]
- Boeckler, F.; Gmeiner, P. The structural evolution of dopamine D3 receptor ligands: Structure-activity relationships and selected neuropharmacological aspects. Pharm. Ther. 2006, 112, 281–333. [Google Scholar] [CrossRef]
- Pilon, C.; Lévesque, D.; Dimitriadou, V.; Griffon, N.; Martres, M.P.; Schwartz, J.C.; Sokoloff, P. Functional coupling of the human dopamine D3 receptor in a transfected NG 108-15 neuroblastoma-glioma hybrid cell line. Eur. J. Pharm. 1994, 268, 129–139. [Google Scholar] [CrossRef]
- Perachon, S.; Schwartz, J.C.; Sokoloff, P. Functional potencies of new antiparkinsonian drugs at recombinant human dopamine D1, D2 and D3 receptors. Eur. J. Pharm. 1999, 366, 293–300. [Google Scholar] [CrossRef]
- Mierau, J.; Schneider, F.J.; Ensinger, H.A.; Chio, C.L.; Lajiness, M.E.; Huff, R.M. Pramipexole binding and activation of cloned and expressed dopamine D2, D3 and D4 receptors. Eur. J. Pharm. 1995, 290, 29–36. [Google Scholar] [CrossRef]
- Sautel, F.; Griffon, N.; Lévesque, D.; Pilon, C.; Schwartz, J.C.; Sokoloff, P. A functional test identifies dopamine agonists selective for D3 versus D2 receptors. Neuroreport 1995, 6, 329–332. [Google Scholar] [CrossRef]
- Van Vliet, L.A.; Tepper, P.G.; Dijkstra, D.; Damsma, G.; Wikström, H.; Pugsley, T.A.; Akunne, H.C.; Heffner, T.G.; Glase, S.A.; Wise, L.D. Affinity for dopamine D2, D3, and D4 receptors of 2-aminotetralins. Relevance of D2 agonist binding for determination of receptor subtype selectivity. J. Med. Chem. 1996, 39, 4233–4237. [Google Scholar] [CrossRef]
- De Keyser, J.; De Backer, J.P.; Wilczak, N.; Herroelen, L. Dopamine agonists used in the treatment of Parkinson’s disease and their selectivity for the D1, D2, and D3 dopamine receptors in human striatum. Prog. Neuropsychopharmacol. Biol. Psychiatry 1995, 19, 1147–1154. [Google Scholar] [CrossRef]
- Gille, G.; Rausch, W.D.; Hung, S.T.; Moldzio, R.; Ngyuen, A.; Janetzky, B.; Engfer, A.; Reichmann, H. Protection of dopaminergic neurons in primary culture by lisuride. J. Neural. Transm. 2002, 109, 157–169. [Google Scholar] [CrossRef]
- Bettinetti, L.; Schlotter, K.; Hübner, H.; Gmeiner, P. Interactive SAR studies: Rational discovery of super-potent and highly selective dopamine D3 receptor antagonists and partial agonists. J. Med. Chem. 2002, 45, 4594–4597. [Google Scholar] [CrossRef]
- Pilla, M.; Perachon, S.; Sautel, F.; Garrido, F.; Mann, A.; Wermuth, C.G.; Schwartz, J.C.; Everitt, B.J.; Sokoloff, P. Selective inhibition of cocaine-seeking behaviour by a partial dopamine D3 receptor agonist. Nature 1999, 400, 371–375. [Google Scholar] [CrossRef]
- Wicke, K.; Garcia-Ladona, J. The dopamine D3 receptor partial agonist, BP 897, is an antagonist at human dopamine D3 receptors and at rat somatodendritic dopamine D3 receptors. Eur. J. Pharm. 2001, 424, 85–90. [Google Scholar] [CrossRef]
- Van Vliet, L.A.; Rodenhuis, N.; Wikström, H.; Pugsley, T.A.; Serpa, K.A.; Meltzer, L.T.; Heffner, T.G.; Wise, L.D.; Lajiness, M.E.; Huff, R.M.; et al. Thiazoloindans and thiazolobenzopyrans: A novel class of orally active central dopamine (partial) agonists. J. Med. Chem. 2000, 43, 3549–3557. [Google Scholar] [CrossRef]
- Murray, P.J.; Helden, R.M.; Johnson, M.R.; Robertson, G.M.; Scopes, D.I.C.; Stokes, M.; Wadman, S.; Whitehead, J.W.F.; Hayes, A.G.; Kilpatrick, G.J.; et al. Novel 6-substituted 2-aminotetralins with potent and selective affinity for the dopamine D3 receptor. Bioorg. Med. Chem. Lett. 1996, 6, 403–408. [Google Scholar] [CrossRef]
- Missale, C.; Nash, S.R.; Robinson, S.W.; Jaber, M.; Caron, M.G. Dopamine receptors: From structure to function. Physiol. Rev. 1998, 78, 189–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Chen, X.; Brodbeck, R.; Primus, R.; Braun, J.; Wasley, J.W.; Thurkauf, A. NGB 2904 and NGB 2849: Two highly selective dopamine D3 receptor antagonists. Bioorg. Med. Chem. Lett. 1998, 8, 2715–2718. [Google Scholar] [CrossRef]
- Bancroft, G.N.; Morgan, K.A.; Flietstra, R.J.; Levant, B. Binding of [3H]PD 128907, a putatively selective ligand for the D3 dopamine receptor, in rat brain: A receptor binding and quantitative autoradiographic study. Neuropsychopharmacology 1998, 18, 305–316. [Google Scholar] [CrossRef]
- Sautel, F.; Griffon, N.; Sokoloff, P.; Schwartz, J.C.; Launay, C.; Simon, P.; Costentin, J.; Schoenfelder, A.; Garrido, F.; Mann, A.; et al. Nafadotride, a potent preferential dopamine D3 receptor antagonist, activates locomotion in rodents. J. Pharm. Exp. Ther. 1995, 275, 1239–1246. [Google Scholar]
- Millan, M.J.; Audinot, V.; Rivet, J.M.; Gobert, A.; Vian, J.; Prost, J.F.; Spedding, M.; Peglion, J.L. S 14297, a novel selective ligand at cloned human dopamine D3 receptors, blocks 7-OH-DPAT-induced hypothermia in rats. Eur. J. Pharm. 1994, 260, R3–R5. [Google Scholar] [CrossRef]
- Rivet, J.M.; Audinot, V.; Gobert, A.; Peglion, J.L.; Millan, M.J. Modulation of mesolimbic dopamine release by the selective dopamine D3 receptor antagonist, (+)-S 14297. Eur. J. Pharmacol. 1994, 265, 175–177. [Google Scholar] [CrossRef]
- Millan, M.J.; Gobert, A.; Newman-Tancredi, A.; Lejeune, F.; Cussac, D.; Rivet, J.M.; Audinot, V.; Dubuffet, T.; Lavielle, G. S33084, a novel, potent, selective, and competitive antagonist at dopamine D(3)-receptors: I. Receptorial, electrophysiological and neurochemical profile compared with GR218,231 and L741,626. J. Pharmacol. Exp. Ther. 2000, 293, 1048–1062. [Google Scholar]
- Dubuffet, T.; Newman-Tancredi, A.; Cussac, D.; Audinot, V.; Loutz, A.; Millan, M.J.; Lavielle, G. Novel benzopyrano[3,4-c]pyrrole derivatives as potent and selective dopamine D3 receptor antagonist. Bioorg. Med. Chem. Lett. 1999, 9, 2059–2064. [Google Scholar] [CrossRef]
- Stemp, G.; Ashmeade, T.; Branch, C.L.; Hadley, M.S.; Hunter, A.J.; Johnson, C.N.; Nash, D.J.; Thewlis, K.M.; Vong, A.K.; Austin, N.E.; et al. Design and synthesis of trans-N-[4-[2-(6-cyano-1,2,3, 4-tetrahydroisoquinolin-2-yl)ethyl]cyclohexyl]-4-quinolinecarboxamide (SB-277011): A potent and selective dopamine D(3) receptor antagonist with high oral bioavailability and CNS penetration in the rat. J. Med. Chem. 2000, 43, 1878–1885. [Google Scholar] [CrossRef]
- Mach, U.R.; Hackling, A.E.; Perachon, S.; Ferry, S.; Wermuth, C.G.; Schwartz, J.C.; Sokoloff, P.; Stark, H. Development of novel 1,2,3,4-tetrahydroisoquinoline derivatives and closely related compounds as potent and selective dopamine D3 receptor ligands. Chembiochem 2004, 5, 508–518. [Google Scholar] [CrossRef]
- Haadsma-Svensson, S.R.; Cleek, K.A.; Dinh, D.M.; Duncan, J.N.; Haber, C.L.; Huff, R.M.; Lajiness, M.E.; Nichols, N.F.; Smith, M.W.; Svensson, K.A.; et al. Dopamine D(3) receptor antagonists. 1. Synthesis and structure-activity relationships of 5,6-dimethoxy-N-alkyl- and N-alkylaryl-substituted 2-aminoindans. J. Med. Chem. 2001, 44, 4716–4732. [Google Scholar] [CrossRef]
- Guillin, O.; Diaz, J.; Carroll, P.; Griffon, N.; Schwartz, J.C.; Sokoloff, P. BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature 2001, 411, 86–89. [Google Scholar] [CrossRef]
- Berendse, H.W.; Galis-de Graaf, Y.; Groenewegen, H.J. Topographical organization and relationship with ventral striatal compartments of prefrontal corticostriatal projections in the rat. J. Comp. Neurol. 1992, 316, 314–347. [Google Scholar] [CrossRef]
- Cotzias, G.C.; Papavasiliou, P.S.; Gellene, R. Modification of Parkinsonism--chronic treatment with L-dopa. N. Engl. J. Med. 1969, 280, 337–345. [Google Scholar] [CrossRef]
- Fiorentini, C.; Savoia, P.; Savoldi, D.; Bono, F.; Busi, C.; Barbon, A.; Missale, C. Shp-2 knockdown prevents l-dopa-induced dyskinesia in a rat model of Parkinson’s disease. Mov. Disord. 2016, 31, 512–520. [Google Scholar] [CrossRef]
- Farré, D.; Muñoz, A.; Moreno, E.; Reyes-Resina, I.; Canet-Pons, J.; Dopeso-Reyes, I.G.; Rico, A.J.; Lluís, C.; Mallol, J.; Navarro, G.; et al. Stronger Dopamine D1 Receptor-Mediated Neurotransmission in Dyskinesia. Mol. Neurobiol. 2015, 52, 1408–1420. [Google Scholar] [CrossRef]
- Cote, S.R.; Kuzhikandathil, E.V. Chronic levodopa treatment alters expression and function of dopamine D3 receptor in the MPTP/p mouse model of Parkinson’s disease. Neurosci. Lett. 2015, 585, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Pernaute, R.; Jenkins, B.G.; Choi, J.K.; Iris Chen, Y.C.; Isacson, O. In vivo evidence of D3 dopamine receptor sensitization in parkinsonian primates and rodents with l-DOPA-induced dyskinesias. Neurobiol. Dis. 2007, 27, 220–227. [Google Scholar] [CrossRef] [Green Version]
- Khaled, M.A.T.M.; Farid Araki, K.; Li, B.; Coen, K.M.; Marinelli, P.W.; Varga, J.; Gaál, J.; Le Foll, B. The selective dopamine D3 receptor antagonist SB 277011-A, but not the partial agonist BP 897, blocks cue-induced reinstatement of nicotine-seeking. Int. J. Neuropsychopharmacol. 2010, 13, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Silverdale, M.A.; Nicholson, S.L.; Ravenscroft, P.; Crossman, A.R.; Millan, M.J.; Brotchie, J.M. Selective blockade of D(3) dopamine receptors enhances the anti-parkinsonian properties of ropinirole and levodopa in the MPTP-lesioned primate. Exp. Neurol. 2004, 188, 128–138. [Google Scholar] [CrossRef]
- Hurley, M.J.; Jolkkonen, J.; Stubbs, C.M.; Jenner, P.; Marsden, C.D. Dopamine D3 receptors in the basal ganglia of the common marmoset and following MPTP and L-DOPA treatment. Brain Res. 1996, 709, 259–264. [Google Scholar] [CrossRef]
- Huot, P.; Johnston, T.H.; Koprich, J.B.; Aman, A.; Fox, S.H.; Brotchie, J.M. L-745,870 reduces L-DOPA-induced dyskinesia in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned macaque model of Parkinson’s disease. J. Pharm. Exp. Ther. 2012, 342, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Reavill, C.; Taylor, S.G.; Wood, M.D.; Ashmeade, T.; Austin, N.E.; Avenell, K.Y.; Boyfield, I.; Branch, C.L.; Cilia, J.; Coldwell, M.C.; et al. Pharmacological actions of a novel, high-affinity, and selective human dopamine D(3) receptor antagonist, SB-277011-A. J. Pharmacol. Exp. Ther. 2000, 294, 1154–1165. [Google Scholar]
- Accili, D.; Fishburn, C.S.; Drago, J.; Steiner, H.; Lachowicz, J.E.; Park, B.H.; Gauda, E.B.; Lee, E.J.; Cool, M.H.; Sibley, D.R.; et al. A targeted mutation of the D3 dopamine receptor gene is associated with hyperactivity in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 1945–1949. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Riddle, L.; Griffin, S.A.; Grundt, P.; Newman, A.H.; Luedtke, R.R. Evaluation of the D3 dopamine receptor selective antagonist PG01037 on L-dopa-dependent abnormal involuntary movements in rats. Neuropharmacology 2009, 56, 944–955. [Google Scholar] [CrossRef] [Green Version]
- Riddle, L.R.; Kumar, R.; Griffin, S.A.; Grundt, P.; Newman, A.H.; Luedtke, R.R. Evaluation of the D3 dopamine receptor selective agonist/partial agonist PG01042 on L-dopa dependent animal involuntary movements in rats. Neuropharmacology 2011, 60, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Cote, S.R.; Chitravanshi, V.C.; Bleickardt, C.; Sapru, H.N.; Kuzhikandathil, E.V. Overexpression of the dopamine D3 receptor in the rat dorsal striatum induces dyskinetic behaviors. Behav. Brain Res. 2014, 263, 46–50. [Google Scholar] [CrossRef]
- Lanza, K.; Centner, A.; Coyle, M.; Del Priore, I.; Manfredsson, F.P.; Bishop, C. Genetic suppression of the dopamine D3 receptor in striatal D1 cells reduces the development of L-DOPA-induced dyskinesia. Exp. Neurol. 2021, 336, 113534. [Google Scholar] [CrossRef]
- Lanza, K.; Chemakin, K.; Lefkowitz, S.; Saito, C.; Chambers, N.; Bishop, C. Reciprocal cross-sensitization of D1 and D3 receptors following pharmacological stimulation in the h.hemiparkinsonian rat. Psychopharmacology 2020, 237, 155–165. [Google Scholar] [CrossRef]
- Nomoto, M.; Jenner, P.; Marsden, C.D. The dopamine D2 agonist LY 141865, but not the D1 agonist SKF 38393, reverses parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in the common marmoset. Neurosci. Lett. 1985, 57, 37–41. [Google Scholar] [CrossRef]
- Löschmann, P.A.; Smith, L.A.; Lange, K.W.; Jähnig, P.; Jenner, P.; Marsden, C.D. Motor activity following the administration of selective D-1 and D-2 dopaminergic drugs to MPTP-treated common marmosets. Psychopharmacology 1992, 109, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.N.; Presgraves, S.; Renish, L.; Borwege, S.; Osredkar, T.; Hagner, D.; Replogle, M.; PazSoldan, M.; Millan, M.J. Neuroprotective effects of the novel D3/D2 receptor agonist and antiparkinson agent, S32504, in vitro against 1-methyl-4-phenylpyridinium (MPP+) and in vivo against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): A comparison to ropinirole. Exp. Neurol. 2003, 184, 393–407. [Google Scholar] [CrossRef]
- Joyce, J.N.; Woolsey, C.; Ryoo, H.; Borwege, S.; Hagner, D. Low dose pramipexole is neuroprotective in the MPTP mouse model of Parkinson’s disease, and downregulates the dopamine transporter via the D3 receptor. BMC Biol. 2004, 2, 22. [Google Scholar] [CrossRef] [Green Version]
- Iravani, M.M.; Haddon, C.O.; Cooper, J.M.; Jenner, P.; Schapira, A.H. Pramipexole protects against MPTP toxicity in non-human primates. J. Neurochem. 2006, 96, 1315–1321. [Google Scholar] [CrossRef]
- Vu, T.Q.; Ling, Z.D.; Ma, S.Y.; Robie, H.C.; Tong, C.W.; Chen, E.Y.; Lipton, J.W.; Carvey, P.M. Pramipexole attenuates the dopaminergic cell loss induced by intraventricular 6-hydroxydopamine. J. Neural Transm. 2000, 107, 159–176. [Google Scholar] [CrossRef]
- Clarke, C.E.; Guttman, M. Dopamine agonist monotherapy in Parkinson’s disease. Lancet 2002, 360, 1767–1769. [Google Scholar] [CrossRef]
- Kitamura, Y.; Kohno, Y.; Nakazawa, M.; Nomura, Y. Inhibitory effects of talipexole and pramipexole on MPTP-induced dopamine reduction in the striatum of C57BL/6N mice. Jpn. J. Pharm. 1997, 74, 51–57. [Google Scholar] [CrossRef]
- Zou, L.; Xu, J.; Jankovic, J.; He, Y.; Appel, S.H.; Le, W. Pramipexole inhibits lipid peroxidation and reduces injury in the substantia nigra induced by the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in C57BL/6 mice. Neurosci. Lett. 2000, 281, 167–170. [Google Scholar] [CrossRef]
- Takata, K.; Kitamura, Y.; Kakimura, J.; Kohno, Y.; Taniguchi, T. Increase of bcl-2 protein in neuronal dendritic processes of cerebral cortex and hippocampus by the antiparkinsonian drugs, talipexole and pramipexole. Brain Res. 2000, 872, 236–241. [Google Scholar] [CrossRef]
- Ling, Z.D.; Tong, C.W.; Carvey, P.M. Partial purification of a pramipexole-induced trophic activity directed at dopamine neurons in ventral mesencephalic cultures. Brain Res. 1998, 791, 137–145. [Google Scholar] [CrossRef]
- Ramirez, A.D.; Wong, S.K.; Menniti, F.S. Pramipexole inhibits MPTP toxicity in mice by dopamine D3 receptor dependent and independent mechanisms. Eur. J. Pharm. 2003, 475, 29–35. [Google Scholar] [CrossRef]
- Anderson, D.W.; Neavin, T.; Smith, J.A.; Schneider, J.S. Neuroprotective effects of pramipexole in young and aged MPTP-treated mice. Brain Res. 2001, 905, 44–53. [Google Scholar] [CrossRef]
- Le, W.D.; Jankovic, J.; Xie, W.; Appel, S.H. Antioxidant property of pramipexole independent of dopamine receptor activation in neuroprotection. J. Neural Transm. 2000, 107, 1165–1173. [Google Scholar] [CrossRef]
- Kim, M.K.; Park, H.S.; Cho, J.H.; Kim, G.S.; Won, C. Pramipexole protects dopaminergic neurons through paraplegin against 6-hydroxydopamine. Neuroreport 2015, 26, 74–80. [Google Scholar] [CrossRef]
- Shah, M.; Rajagopalan, S.; Xu, L.; Voshavar, C.; Shurubor, Y.; Beal, F.; Andersen, J.K.; Dutta, A.K. The high-affinity D2/D3 agonist D512 protects PC12 cells from 6-OHDA-induced apoptotic cell death and rescues dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. J. Neurochem. 2014, 131, 74–85. [Google Scholar] [CrossRef]
- Biswas, S.; Zhang, S.; Fernandez, F.; Ghosh, B.; Zhen, J.; Kuzhikandathil, E.; Reith, M.E.; Dutta, A.K. Further structure-activity relationships study of hybrid 7-{[2-(4-phenylpiperazin-1-yl)ethyl]propylamino}-5,6,7,8-tetrahydronaphthalen-2-o l analogues: Identification of a high-affinity D3-preferring agonist with potent in vivo activity with long duration of action. J. Med. Chem. 2008, 51, 101–117. [Google Scholar] [CrossRef]
- Horowski, R.; Wachtel, H. Direct dopaminergic action of lisuride hydrogen maleate, an ergot derivative, in mice. Eur. J. Pharm. 1976, 36, 373–383. [Google Scholar] [CrossRef]
- Schechter, M.D. Evidence for a direct dopaminergic effect of lisuride. Pharm. Biochem. Behav. 1984, 21, 185–189. [Google Scholar] [CrossRef]
- Kim, M.; Lee, S.; Cho, J.; Kim, G.; Won, C. Dopamine D3 receptor-modulated neuroprotective effects of lisuride. Neuropharmacology 2017, 117, 14–20. [Google Scholar] [CrossRef]
- Carvey, P.M.; Pieri, S.; Ling, Z.D. Attenuation of levodopa-induced toxicity in mesencephalic cultures by pramipexole. J. Neural Trans. 1997, 104, 209–228. [Google Scholar] [CrossRef]
- Sethy, V.H.; Wu, H.; Oostveen, J.A.; Hall, E.D. Neuroprotective effects of the dopamine agonists pramipexole and bromocriptine in 3-acetylpyridine-treated rats. Brain Res. 1997, 754, 181–186. [Google Scholar] [CrossRef]
- Inden, M.; Kitamura, Y.; Tamaki, A.; Yanagida, T.; Shibaike, T.; Yamamoto, A.; Takata, K.; Yasui, H.; Taira, T.; Ariga, H.; et al. Neuroprotective effect of the antiparkinsonian drug pramipexole against nigrostriatal dopaminergic degeneration in rotenone-treated mice. Neurochem. Int. 2009, 55, 760–767. [Google Scholar] [CrossRef]
- Oster, S.; Radad, K.; Scheller, D.; Hesse, M.; Balanzew, W.; Reichmann, H.; Gille, G. Rotigotine protects against glutamate toxicity in primary dopaminergic cell culture. Eur. J. Pharm. 2014, 724, 31–42. [Google Scholar] [CrossRef]
- Matsuo, T.; Izumi, Y.; Kume, T.; Takada-Takatori, Y.; Sawada, H.; Akaike, A. Protective effect of aripiprazole against glutamate cytotoxicity in dopaminergic neurons of rat mesencephalic cultures. Neurosci. Lett. 2010, 481, 78–81. [Google Scholar] [CrossRef] [Green Version]
- Westrich, L.; Gil-Mast, S.; Kortagere, S.; Kuzhikandathil, E.V. Development of tolerance in D3 dopamine receptor signaling is accompanied by distinct changes in receptor conformation. Biochem. Pharm. 2010, 79, 897–907. [Google Scholar] [CrossRef]
- Gil-Mast, S.; Kortagere, S.; Kota, K.; Kuzhikandathil, E.V. An amino acid residue in the second extracellular loop determines the agonist-dependent tolerance property of the human D3 dopamine receptor. ACS Chem. Neurosci. 2013, 4, 940–951. [Google Scholar] [CrossRef] [Green Version]
- Ng, G.Y.; Varghese, G.; Chung, H.T.; Trogadis, J.; Seeman, P.; O’Dowd, B.F.; George, S.R. Resistance of the dopamine D2L receptor to desensitization accompanies the up-regulation of receptors on to the surface of Sf9 cells. Endocrinology 1997, 138, 4199–4206. [Google Scholar] [CrossRef]
- Zhang, L.J.; Lachowicz, J.E.; Sibley, D.R. The D2S and D2L dopamine receptor isoforms are differentially regulated in Chinese hamster ovary cells. Mol. Pharm. 1994, 45, 878–889. [Google Scholar]
- Cho, E.Y.; Cho, D.I.; Park, J.H.; Kurose, H.; Caron, M.G.; Kim, K.M. Roles of protein kinase C and actin-binding protein 280 in the regulation of intracellular trafficking of dopamine D3 receptor. Mol. Endocrinol. 2007, 21, 2242–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, O.J.; Gardner, B.R.; Williams, D.B.; Marinec, P.S.; Cabrera, D.M.; Peters, J.D.; Mak, C.C.; Kim, K.M.; Sibley, D.R. The role of phosphorylation in D1 dopamine receptor desensitization: Evidence for a novel mechanism of arrestin association. J. Biol. Chem. 2004, 279, 7999–8010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.M.; Valenzano, K.J.; Robinson, S.R.; Yao, W.D.; Barak, L.S.; Caron, M.G. Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinases and beta-arrestins. J. Biol. Chem. 2001, 276, 37409–37414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.M.; Gainetdinov, R.R.; Laporte, S.A.; Caron, M.G.; Barak, L.S. G protein-coupled receptor kinase regulates dopamine D3 receptor signaling by modulating the stability of a receptor-filamin-beta-arrestin complex. A case of autoreceptor regulation. J. Biol. Chem. 2005, 280, 12774–12780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simms, S.L.; Huettner, D.P.; Kortagere, S. In vivo characterization of a novel dopamine D3 receptor agonist to treat motor symptoms of Parkinson’s disease. Neuropharmacology 2016, 100, 106–115. [Google Scholar] [CrossRef]
- Bonifati, V.; Fabrizio, E.; Cipriani, R.; Vanacore, N.; Meco, G. Buspirone in levodopa-induced dyskinesias. Clin. Neuropharmacol. 1994, 17, 73–82. [Google Scholar] [CrossRef]
- Schwartz, J.C.; Diaz, J.; Bordet, R.; Griffon, N.; Perachon, S.; Pilon, C.; Ridray, S.; Sokoloff, P. Functional implications of multiple dopamine receptor subtypes: The D1/D3 receptor coexistence. Brain Res. Brain Res. Rev. 1998, 26, 236–242. [Google Scholar] [CrossRef]
- Fiorentini, C.; Busi, C.; Gorruso, E.; Gotti, C.; Spano, P.; Missale, C. Reciprocal regulation of dopamine D1 and D3 receptor function and trafficking by heterodimerization. Mol. Pharm. 2008, 74, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Marcellino, D.; Ferre, S.; Casado, V.; Cortes, A.; Le Foll, B.; Mazzola, C.; Drago, F.; Saur, O.; Stark, H.; Soriano, A.; et al. Identification of dopamine D1-D3 receptor heteromers. Indications for a role of synergistic D1-D3 receptor interactions in the striatum. J. Biol. Chem. 2008, 283, 26016–26025. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Trujillo, R.; Avalos-Fuentes, A.; Rangel-Barajas, C.; Paz-Bermudez, F.; Sierra, A.; Escartin-Perez, E.; Aceves, J.; Erlij, D.; Floran, B. D3 dopamine receptors interact with dopamine D1 but not D4 receptors in the GABAergic terminals of the SNr of the rat. Neuropharmacology 2013, 67, 370–378. [Google Scholar] [CrossRef]
- Guitart, X.; Moreno, E.; Rea, W.; Sánchez-Soto, M.; Cai, N.S.; Quiroz, C.; Kumar, V.; Bourque, L.; Cortés, A.; Canela, E.I.; et al. Biased G Protein-Independent Signaling of Dopamine D(1)-D(3) Receptor Heteromers in the Nucleus Accumbens. Mol. Neurobiol. 2019, 56, 6756–6769. [Google Scholar] [CrossRef]
- Avalos-Fuentes, A.; Loya-López, S.; Flores-Pérez, A.; Recillas-Morales, S.; Cortés, H.; Paz-Bermúdez, F.; Aceves, J.; Erlij, D.; Florán, B. Presynaptic CaMKIIα modulates dopamine D3 receptor activation in striatonigral terminals of the rat brain in a Ca²⁺ dependent manner. Neuropharmacology 2013, 71, 273–281. [Google Scholar] [CrossRef]
- Zapata, A.; Kivell, B.; Han, Y.; Javitch, J.A.; Bolan, E.A.; Kuraguntla, D.; Jaligam, V.; Oz, M.; Jayanthi, L.D.; Samuvel, D.J.; et al. Regulation of dopamine transporter function and cell surface expression by D3 dopamine receptors. J. Biol. Chem. 2007, 282, 35842–35854. [Google Scholar] [CrossRef] [Green Version]
- Collo, G.; Bono, F.; Cavalleri, L.; Plebani, L.; Merlo Pich, E.; Millan, M.J.; Spano, P.F.; Missale, C. Pre-synaptic dopamine D(3) receptor mediates cocaine-induced structural plasticity in mesencephalic dopaminergic neurons via ERK and Akt pathways. J. Neurochem. 2012, 120, 765–778. [Google Scholar] [CrossRef]
- Jaworski, J.; Spangler, S.; Seeburg, D.P.; Hoogenraad, C.C.; Sheng, M. Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J. Neurosci. 2005, 25, 11300–11312. [Google Scholar] [CrossRef]
- Kumar, V.; Zhang, M.X.; Swank, M.W.; Kunz, J.; Wu, G.Y. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J. Neurosci. 2005, 25, 11288–11299. [Google Scholar] [CrossRef] [Green Version]
- Avalos-Fuentes, A.; Albarran-Bravo, S.; Loya-Lopez, S.; Cortes, H.; Recillas-Morales, S.; Magana, J.J.; Paz-Bermudez, F.; Rangel-Barajas, C.; Aceves, J.; Erlij, D.; et al. Dopaminergic denervation switches dopamine D3 receptor signaling and disrupts its Ca(2+) dependent modulation by CaMKII and calmodulin in striatonigral projections of the rat. Neurobiol. Dis. 2015, 74, 336–346. [Google Scholar] [CrossRef]
- Liu, X.Y.; Mao, L.M.; Zhang, G.C.; Papasian, C.J.; Fibuch, E.E.; Lan, H.X.; Zhou, H.F.; Xu, M.; Wang, J.Q. Activity-dependent modulation of limbic dopamine D3 receptors by CaMKII. Neuron 2009, 61, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.L.; Liu, X.Y.; Mao, L.M.; Wang, J.Q. Regulation of dopamine D3 receptors by protein-protein interactions. Neurosci. Bull. 2010, 26, 163–167. [Google Scholar] [CrossRef]
- Moriguchi, S.; Yabuki, Y.; Fukunaga, K. Reduced calcium/calmodulin-dependent protein kinase II activity in the hippocampus is associated with impaired cognitive function in MPTP-treated mice. J. Neurochem. 2012, 120, 541–551. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Song, W.J.; Yan, Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J. Neurosci. 1996, 16, 6579–6591. [Google Scholar] [CrossRef] [Green Version]
- Fiorentini, C.; Busi, C.; Spano, P.; Missale, C. Dimerization of dopamine D1 and D3 receptors in the regulation of striatal function. Curr. Opin. Pharm. 2010, 10, 87–92. [Google Scholar] [CrossRef]
- Bono, F.; Mutti, V.; Fiorentini, C.; Missale, C. Dopamine D3 Receptor Heteromerization: Implications for Neuroplasticity and Neuroprotection. Biomolecules 2020, 10, 1016. [Google Scholar] [CrossRef]
- Koschatzky, S.; Gmeiner, P. Selective agonists for dopamine/neurotensin receptor heterodimers. ChemMedChem 2012, 7, 509–514. [Google Scholar] [CrossRef]
- Zeng, C.; Asico, L.D.; Yu, C.; Villar, V.A.; Shi, W.; Luo, Y.; Wang, Z.; He, D.; Liu, Y.; Huang, L.; et al. Renal D3 dopamine receptor stimulation induces natriuresis by endothelin B receptor interactions. Kidney Int. 2008, 74, 750–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjaergaard, S.K.; Pedersen, O.F.; Taudorf, E.; Mølhave, L. Assessment of changes in eye redness by a photographic method and the relation to sensory eye irritation. Int. Arch. Occup. Environ. Health 1990, 62, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, M.; Novi, F.; Schallmach, E.; Lin, R.; Baragli, A.; Colzi, A.; Griffon, N.; Corsini, G.U.; Sokoloff, P.; Levenson, R.; et al. D2/D3 dopamine receptor heterodimers exhibit unique functional properties. J. Biol. Chem. 2001, 276, 30308–30314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dam, E.M.; Robinson, P.J. Ral: Mediator of membrane trafficking. Int. J. Biochem. Cell Biol. 2006, 38, 1841–1847. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.; Zheng, M.; Min, C.; Ma, L.; Kurose, H.; Park, J.H.; Kim, K.M. Agonist-induced endocytosis and receptor phosphorylation mediate resensitization of dopamine D(2) receptors. Mol. Endocrinol. 2010, 24, 574–586. [Google Scholar] [CrossRef] [Green Version]
- Fiorentini, C.; Savoia, P.; Savoldi, D.; Missale, C. Receptor heteromers in Parkinson’s disease and L-DOPA-induced dyskinesia. CNS Neurol. Disord. Drug Targets 2013, 12, 1101–1113. [Google Scholar]
- Aubert, I.; Guigoni, C.; Hakansson, K.; Li, Q.; Dovero, S.; Barthe, N.; Bioulac, B.H.; Gross, C.E.; Fisone, G.; Bloch, B.; et al. Increased D1 dopamine receptor signaling in levodopa-induced dyskinesia. Ann. Neurol. 2005, 57, 17–26. [Google Scholar] [CrossRef]
- Mahmoudi, S.; Levesque, D.; Blanchet, P.J. Upregulation of dopamine D3, not D2, receptors correlates with tardive dyskinesia in a primate model. Mov. Disord. 2014, 29, 1125–1133. [Google Scholar] [CrossRef] [Green Version]
- Van Kampen, J.M.; Stoessl, A.J. Effects of oligonucleotide antisense to dopamine D3 receptor mRNA in a rodent model of behavioural sensitization to levodopa. Neuroscience 2003, 116, 307–314. [Google Scholar] [CrossRef]
- Ferre, S.; Lluis, C.; Lanciego, J.L.; Franco, R. Prime time for G-protein-coupled receptor heteromers as therapeutic targets for CNS disorders: The dopamine D(1)-D(3) receptor heteromer. CNS Neurol. Disord. Drug Targets 2010, 9, 596–600. [Google Scholar] [CrossRef]
- Ferre, S.; Casado, V.; Devi, L.A.; Filizola, M.; Jockers, R.; Lohse, M.J.; Milligan, G.; Pin, J.P.; Guitart, X. G protein-coupled receptor oligomerization revisited: Functional and pharmacological perspectives. Pharm. Rev. 2014, 66, 413–434. [Google Scholar] [CrossRef] [Green Version]
- Darmopil, S.; Martin, A.B.; De Diego, I.R.; Ares, S.; Moratalla, R. Genetic inactivation of dopamine D1 but not D2 receptors inhibits L-DOPA-induced dyskinesia and histone activation. Biol. Psychiatry 2009, 66, 603–613. [Google Scholar] [CrossRef]
- Murer, M.G.; Moratalla, R. Striatal Signaling in L-DOPA-Induced Dyskinesia: Common Mechanisms with Drug Abuse and Long Term Memory Involving D1 Dopamine Receptor Stimulation. Front. Neuroanat. 2011, 5, 51. [Google Scholar] [CrossRef] [Green Version]
- Berthet, A.; Porras, G.; Doudnikoff, E.; Stark, H.; Cador, M.; Bezard, E.; Bloch, B. Pharmacological analysis demonstrates dramatic alteration of D1 dopamine receptor neuronal distribution in the rat analog of L-DOPA-induced dyskinesia. J. Neurosci. 2009, 29, 4829–4835. [Google Scholar] [CrossRef]
- Staley, J.K.; Mash, D.C. Adaptive increase in D3 dopamine receptors in the brain reward circuits of human cocaine fatalities. J. Neurosci. 1996, 16, 6100–6106. [Google Scholar] [CrossRef] [Green Version]
- Fuxe, K.; Ferre, S.; Woods, A. Novel Strategies for the Treatment of Parkinson’s Disease. Focus on Receptor-Receptor Interactions in the Basal Ganglia; Kehr, J., Fuxe, K., Ungerstedt, U., Svensson, T., Eds.; Monitoring Molecules in Neuroscience; Karolinska University Press: Stockholm, Sweden, 2003. [Google Scholar]
- Guigoni, C.; Doudnikoff, E.; Li, Q.; Bloch, B.; Bezard, E. Altered D(1) dopamine receptor trafficking in parkinsonian and dyskinetic non-human primates. Neurobiol. Dis. 2007, 26, 452–463. [Google Scholar] [CrossRef]
- Gross, C.E.; Ravenscroft, P.; Dovero, S.; Jaber, M.; Bioulac, B.; Bezard, E. Pattern of levodopa-induced striatal changes is different in normal and MPTP-lesioned mice. J. Neurochem. 2003, 84, 1246–1255. [Google Scholar] [CrossRef] [Green Version]
- Aristieta, A.; Azkona, G.; Sagarduy, A.; Miguelez, C.; Ruiz-Ortega, J.A.; Sanchez-Pernaute, R.; Ugedo, L. The role of the subthalamic nucleus in L-DOPA induced dyskinesia in 6-hydroxydopamine lesioned rats. PLoS ONE 2012, 7, e42652. [Google Scholar] [CrossRef] [PubMed]
- Bagetta, V.; Sgobio, C.; Pendolino, V.; Del Papa, G.; Tozzi, A.; Ghiglieri, V.; Giampa, C.; Zianni, E.; Gardoni, F.; Calabresi, P.; et al. Rebalance of striatal NMDA/AMPA receptor ratio underlies the reduced emergence of dyskinesia during D2-like dopamine agonist treatment in experimental Parkinson’s disease. J. Neurosci. 2012, 32, 17921–17931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindenbach, D.; Conti, M.M.; Ostock, C.Y.; George, J.A.; Goldenberg, A.A.; Melikhov-Sosin, M.; Nuss, E.E.; Bishop, C. The Role of Primary Motor Cortex (M1) Glutamate and GABA Signaling in l-DOPA-Induced Dyskinesia in Parkinsonian Rats. J. Neurosci. 2016, 36, 9873–9887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavón, N.; Martín, A.B.; Mendialdua, A.; Moratalla, R. ERK phosphorylation and FosB expression are associated with L-DOPA-induced dyskinesia in hemiparkinsonian mice. Biol. Psychiatry 2006, 59, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Lanza, K.; Meadows, S.M.; Chambers, N.E.; Nuss, E.; Deak, M.M.; Ferré, S.; Bishop, C. Behavioral and cellular dopamine D1 and D3 receptor-mediated synergy: Implications for L-DOPA-induced dyskinesia. Neuropharmacology 2018, 138, 304–314. [Google Scholar] [CrossRef]
- Bunney, B.S.; Aghajanian, G.K.; Roth, R.H. Comparison of effects of L-dopa, amphetamine and apomorphine on firing rate of rat dopaminergic neurones. Nat. New Biol. 1973, 245, 123–125. [Google Scholar] [CrossRef]
- Harden, D.G.; Grace, A.A. Activation of dopamine cell firing by repeated L-DOPA administration to dopamine-depleted rats: Its potential role in mediating the therapeutic response to L-DOPA treatment. J. Neurosci. 1995, 15, 6157–6166. [Google Scholar] [CrossRef]
- Zetterstrom, T.; Herrera-Marschitz, M.; Ungerstedt, U. Simultaneous measurement of dopamine release and rotational behaviour in 6-hydroxydopamine denervated rats using intracerebral dialysis. Brain Res. 1986, 376, 1–7. [Google Scholar] [CrossRef]
- Navailles, S.; Benazzouz, A.; Bioulac, B.; Gross, C.; De Deurwaerdere, P. High-frequency stimulation of the subthalamic nucleus and L-3,4-dihydroxyphenylalanine inhibit in vivo serotonin release in the prefrontal cortex and hippocampus in a rat model of Parkinson’s disease. J. Neurosci. 2010, 30, 2356–2364. [Google Scholar] [CrossRef] [Green Version]
- Navailles, S.; Bioulac, B.; Gross, C.; De Deurwaerdere, P. Serotonergic neurons mediate ectopic release of dopamine induced by L-DOPA in a rat model of Parkinson’s disease. Neurobiol. Dis. 2010, 38, 136–143. [Google Scholar] [CrossRef]
- Tanaka, H.; Kannari, K.; Maeda, T.; Tomiyama, M.; Suda, T.; Matsunaga, M. Role of serotonergic neurons in L-DOPA-derived extracellular dopamine in the striatum of 6-OHDA-lesioned rats. Neuroreport 1999, 10, 631–634. [Google Scholar] [CrossRef]
- Carta, M.; Carlsson, T.; Munoz, A.; Kirik, D.; Bjorklund, A. Role of serotonin neurons in the induction of levodopa- and graft-induced dyskinesias in Parkinson’s disease. Mov. Disord. 2010, 25 (Suppl. 1), S174–S179. [Google Scholar] [CrossRef]
- Bastide, M.F.; Meissner, W.G.; Picconi, B.; Fasano, S.; Fernagut, P.O.; Feyder, M.; Francardo, V.; Alcacer, C.; Ding, Y.; Brambilla, R.; et al. Pathophysiology of L-dopa-induced motor and non-motor complications in Parkinson’s disease. Prog. Neurobiol. 2015, 132, 96–168. [Google Scholar] [CrossRef]
- Navailles, S.; Bioulac, B.; Gross, C.; De Deurwaerdere, P. Chronic L-DOPA therapy alters central serotonergic function and L-DOPA-induced dopamine release in a region-dependent manner in a rat model of Parkinson’s disease. Neurobiol. Dis. 2011, 41, 585–590. [Google Scholar] [CrossRef]
- Navailles, S.; De Deurwaerdere, P. Imbalanced Dopaminergic Transmission Mediated by Serotonergic Neurons in L-DOPA-Induced Dyskinesia. Park. Dis. 2012, 2012, 323686. [Google Scholar] [CrossRef]
- Nevalainen, N.; Af Bjerken, S.; Lundblad, M.; Gerhardt, G.A.; Stromberg, I. Dopamine release from serotonergic nerve fibers is reduced in L-DOPA-induced dyskinesia. J. Neurochem. 2011, 118, 12–23. [Google Scholar] [CrossRef]
- Di Giovanni, G.; Chagraoui, A.; Puginier, E.; Galati, S.; De Deurwaerdere, P. Reciprocal interaction between monoaminergic systems and the pedunculopontine nucleus: Implication in the mechanism of L-DOPA. Neurobiol. Dis. 2019, 128, 9–18. [Google Scholar] [CrossRef]
- Millan, M.J. From the cell to the clinic: A comparative review of the partial D(2)/D(3)receptor agonist and alpha2-adrenoceptor antagonist, piribedil, in the treatment of Parkinson’s disease. Pharm. Ther. 2010, 128, 229–273. [Google Scholar] [CrossRef]
- Millan, M.J.; Dekeyne, A.; Gobert, A.; Brocco, M.; Mannoury la Cour, C.; Ortuno, J.C.; Watson, D.; Fone, K.C.F. Dual-acting agents for improving cognition and real-world function in Alzheimer’s disease: Focus on 5-HT6 and D3 receptors as hubs. Neuropharmacology 2020, 177, 108099. [Google Scholar] [CrossRef]
- Beom, S.; Cheong, D.; Torres, G.; Caron, M.G.; Kim, K.M. Comparative studies of molecular mechanisms of dopamine D2 and D3 receptors for the activation of extracellular signal-regulated kinase. J. Biol. Chem. 2004, 279, 28304–28314. [Google Scholar] [CrossRef] [Green Version]
- Levant, B.; Ling, Z.D.; Carvey, P.M. Dopamine D3 Receptors. CNS Drugs 1999, 12, 391–402. [Google Scholar] [CrossRef]
- Boulay, D.; Depoortere, R.; Rostene, W.; Perrault, G.; Sanger, D.J. Dopamine D3 receptor agonists produce similar decreases in body temperature and locomotor activity in D3 knock-out and wild-type mice. Neuropharmacology 1999, 38, 555–565. [Google Scholar] [CrossRef]
- Millan, M.J.; Dekeyne, A.; Rivet, J.M.; Dubuffet, T.; Lavielle, G.; Brocco, M. S33084, a novel, potent, selective, and competitive antagonist at dopamine D(3)-receptors: II. Functional and behavioral profile compared with GR218,231 and L741,626. J. Pharmacol. Exp. Ther. 2000, 293, 1063–1073. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D3 Receptor Agonists | |||
|---|---|---|---|
| Ligand | Ki D3R | Selectivity over D2 Receptors | References |
| DA | 3.9 nM | DR2/DR3 = 0.4 | [154] |
| Bromocriptine | 0.5–5 nM | [155] | |
| Cabergoline | <0.5 nM | [156] | |
| CJ-1037 | |||
| D-264 | 0.92 nM | DR2/DR3 = 253 | [157] |
| D-440 | DR2/DR3 = 583.2 | [158] | |
| Quinpirole | 0.96 nM | DR2/DR3 = 133 | [159,160] |
| Pergolide | 0.5–5 nM | [155] | |
| Pramipexole | 0.5–8.5 nM | DR2/DR3 = 253 | [161,162,163] |
| R-7-OH-DPAT | DR2/DR3 = 26 | [164] | |
| Ropinirole | 69 nM | DR2/DR3 = 8.3 | |
| Lisuride | 1.08 nM | DR2/DR3 = 2.5 | [165,166] |
| D3 Receptor Antagonists | |||
| Ligand | Ki D3R | Selectivity over D2 Receptors | References |
| FAUC 365 | 0.50 nM | DR2/DR3 = 200 | [167] |
| BP 897 | 0.92 nM | DR2/D3R = 66 | [168,169] |
| GMC1111 | 1.4 nM | DR2/DR3 = 19 | [170] |
| GR103691 | |||
| GR218231 | 1.3 nM | DR2/DR3 = 380 | [159,171] |
| Nemonapride | <0.5 nM | [172] | |
| NGB 2849 | 0.9 nM | DR2/DR3 = 290 | [173] |
| PD128907 | 5–50 nM | [174] | |
| Raclopride | 0.5–5 nM | [172] | |
| S-5-OH-DPAT | DR2/DR3 = 60 | [164] | |
| S-nafadotride | 0.3 nM | DR2/DR3 = 10 | [175] |
| S14297 | 13 nM | DR2/DR3 = 23 | [176,177] |
| S33084 | 0.32 nM | DR2/DR3 = 100 | [159,178,179] |
| SB 277011 | 10 nM | DR2/DR3 = 100 | [180] |
| Spiperone | 0.5–5 nM | [172] | |
| ST 198 | 12 nM | DR2/DR3 = 65 | [2,181] |
| U99194A | 31 nM | DR2/DR3 = 32 | [182] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chagraoui, A.; Di Giovanni, G.; De Deurwaerdère, P. Neurobiological and Pharmacological Perspectives of D3 Receptors in Parkinson’s Disease. Biomolecules 2022, 12, 243. https://doi.org/10.3390/biom12020243
Chagraoui A, Di Giovanni G, De Deurwaerdère P. Neurobiological and Pharmacological Perspectives of D3 Receptors in Parkinson’s Disease. Biomolecules. 2022; 12(2):243. https://doi.org/10.3390/biom12020243
Chicago/Turabian StyleChagraoui, Abdeslam, Giuseppe Di Giovanni, and Philippe De Deurwaerdère. 2022. "Neurobiological and Pharmacological Perspectives of D3 Receptors in Parkinson’s Disease" Biomolecules 12, no. 2: 243. https://doi.org/10.3390/biom12020243
APA StyleChagraoui, A., Di Giovanni, G., & De Deurwaerdère, P. (2022). Neurobiological and Pharmacological Perspectives of D3 Receptors in Parkinson’s Disease. Biomolecules, 12(2), 243. https://doi.org/10.3390/biom12020243