
Channelopathy Genes in Pulmonary Arterial Hypertension


Abstract
:1. Introduction
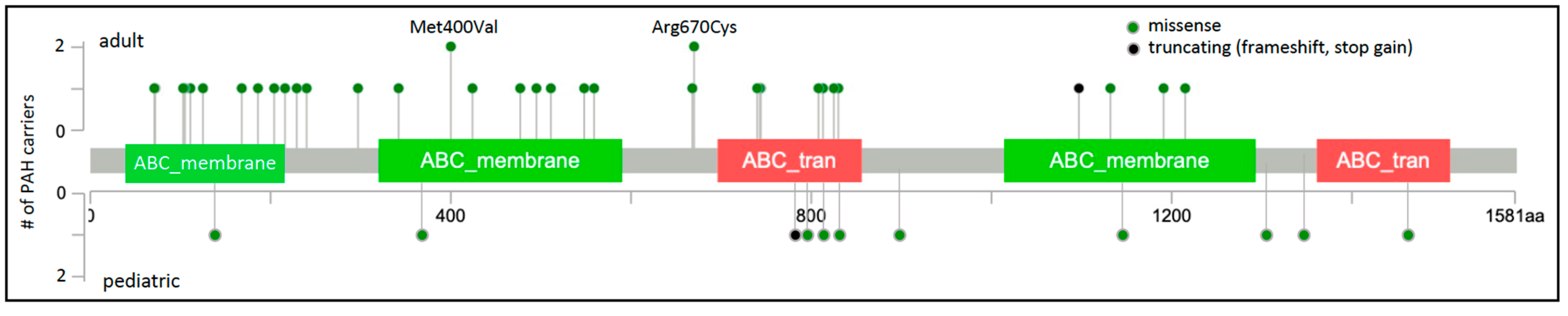
2. ABCC8 (OMIM *600509)
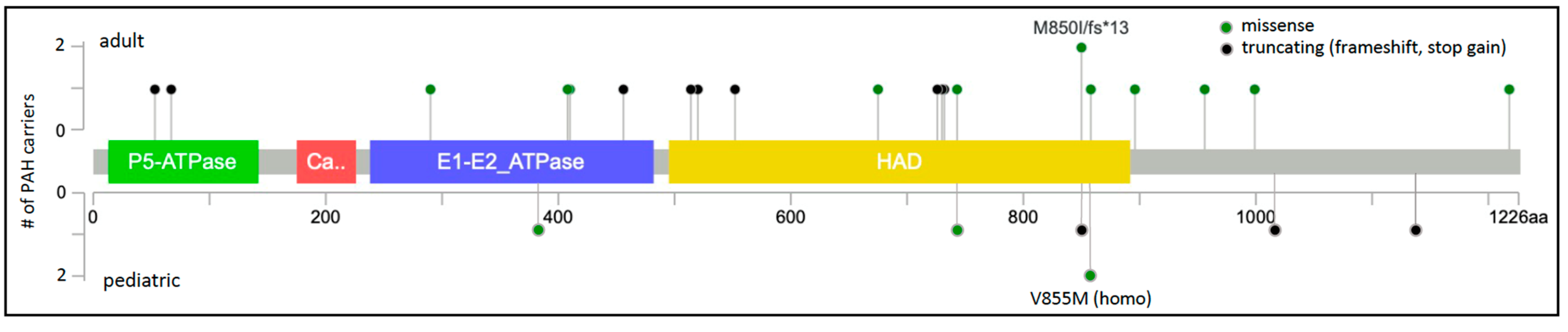
3. ATP13A3 (OMIM *610232)
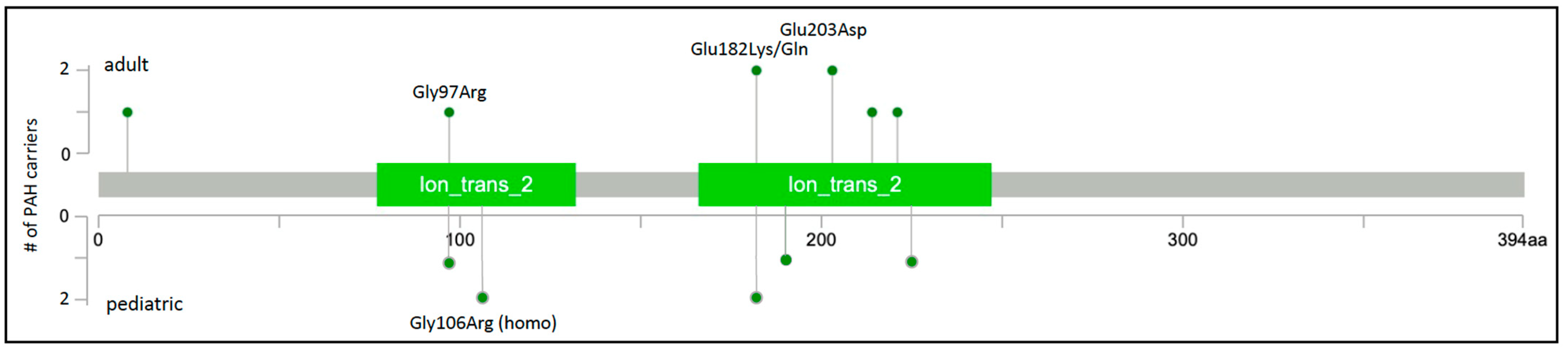
4. KCNK3 (OMIM *603220)
5. Other Channel Genes Associated with PAH
6. Therapeutic Implications
7. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vonk-Noordegraaf, A.; Haddad, F.; Chin, K.M.; Forfia, P.R.; Kawut, S.M.; Lumens, J.; Naeije, R.; Newman, J.; Oudiz, R.J.; Provencher, S.; et al. Right heart adaptation to pulmonary arterial hypertension: Physiology and pathobiology. J. Am. Coll. Cardiol. 2013, 62 (Suppl. 25), D22–D33. [Google Scholar] [CrossRef]
- Ryan, J.J.; Archer, S.L. The right ventricle in pulmonary arterial hypertension: Disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ. Res. 2014, 115, 176–188. [Google Scholar] [CrossRef] [Green Version]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmuller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Jick, S.; Breitenstein, S.; Hernandez, G.; Michel, A.; Vizcaya, D. Pulmonary arterial hypertension in the USA: An epidemiological study in a large insured pediatric population. Pulm. Circ. 2017, 7, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Swinnen, K.; Quarck, R.; Godinas, L.; Belge, C.; Delcroix, M. Learning from registries in pulmonary arterial hypertension: Pitfalls and recommendations. Eur. Respir. Rev. 2019, 28, 190050. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Pauciulo, M.W.; Welch, C.L.; Lutz, K.A.; Coleman, A.W.; Gonzaga-Jauregui, C.; Wang, J.; Grimes, J.M.; Martin, L.J.; He, H.; et al. Novel risk genes and mechanisms implicated by exome sequencing of 2572 individuals with pulmonary arterial hypertension. Genome Med. 2019, 11, 69. [Google Scholar] [CrossRef] [Green Version]
- Hoeper, M.M.; Huscher, D.; Ghofrani, H.A.; Delcroix, M.; Distler, O.; Schweiger, C.; Grunig, E.; Staehler, G.; Rosenkranz, S.; Halank, M.; et al. Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: Results from the COMPERA registry. Int. J. Cardiol. 2013, 168, 871–880. [Google Scholar] [CrossRef]
- Graf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M.; et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 2018, 9, 1416. [Google Scholar] [CrossRef] [Green Version]
- Welch, C.L.; Chung, W.K. Genetics and Genomics of Pediatric Pulmonary Arterial Hypertension. Genes 2020, 11, 1213. [Google Scholar] [CrossRef]
- Bohnen, M.S.; Ma, L.; Zhu, N.; Qi, H.; McClenaghan, C.; Gonzaga-Jauregui, C.; Dewey, F.E.; Overton, J.D.; Reid, J.G.; Shuldiner, A.R.; et al. Loss-of-Function ABCC8 Mutations in Pulmonary Arterial Hypertension. Circ. Genom. Precis. Med. 2018, 11, e002087. [Google Scholar] [CrossRef] [Green Version]
- Castano, J.A.T.; Hernandez-Gonzalez, I.; Gallego, N.; Perez-Olivares, C.; Ochoa Parra, N.; Arias, P.; Granda, E.; Acebo, G.G.; Lago-Docampo, M.; Palomino-Doza, J.; et al. Customized Massive Parallel Sequencing Panel for Diagnosis of Pulmonary Arterial Hypertension. Genes 2020, 11, 1158. [Google Scholar] [CrossRef] [PubMed]
- Gelinas, S.M.; Benson, C.E.; Khan, M.A.; Berger, R.M.F.; Trembath, R.C.; Machado, R.D.; Southgate, L. Whole Exome Sequence Analysis Provides Novel Insights into the Genetic Framework of Childhood-Onset Pulmonary Arterial Hypertension. Genes 2020, 11, 1328. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Mei, M.; Chen, X.; Lu, Y.; Dong, X.; Hu, L.; Hu, X.; Cheng, G.; Cao, Y.; Yang, L.; et al. Identification of genetic factors underlying persistent pulmonary hypertension of newborns in a cohort of Chinese neonates. Respir. Res. 2019, 20, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.J.; Lian, T.Y.; Jiang, X.; Liu, S.F.; Li, S.Q.; Jiang, R.; Wu, W.H.; Ye, J.; Cheng, C.Y.; Du, Y.; et al. Germline BMP9 mutation causes idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801609. [Google Scholar] [CrossRef]
- Lerche, M.; Eichstaedt, C.A.; Hinderhofer, K.; Grunig, E.; Tausche, K.; Ziemssen, T.; Halank, M.; Wirtz, H.; Seyfarth, H.J. Mutually reinforcing effects of genetic variants and interferon-beta 1a therapy for pulmonary arterial hypertension development in multiple sclerosis patients. Pulm. Circ. 2019, 9, 2045894019872192. [Google Scholar] [CrossRef] [Green Version]
- Machado, R.; Welch, C.L.; Haimel, M.; Bleda, M.; Colglazier, E.; Coulson, J.D.; Debeljak, M.; Ekstein, J.; Fineman, J.R.; Golden, W.C.; et al. Biallelic variants of ATP13A3 cause dose-dependent childhood-onset pulmonary arterial hypertension characterised by extreme morbidity and mortality. J. Med. Genet. 2021. [Google Scholar] [CrossRef]
- Ma, L.; Roman-Campos, D.; Austin, E.D.; Eyries, M.; Sampson, K.S.; Soubrier, F.; Germain, M.; Tregouet, D.A.; Borczuk, A.; Rosenzweig, E.B.; et al. A novel channelopathy in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Navas Tejedor, P.; Tenorio Castano, J.; Palomino Doza, J.; Arias Lajara, P.; Gordo Trujillo, G.; Lopez Meseguer, M.; Roman Broto, A.; Lapunzina Abadia, P.; Escribano Subia, P. An homozygous mutation in KCNK3 is associated with an aggressive form of hereditary pulmonary arterial hypertension. Clin. Genet. 2017, 91, 453–457. [Google Scholar] [CrossRef]
- Higasa, K.; Ogawa, A.; Terao, C.; Shimizu, M.; Kosugi, S.; Yamada, R.; Date, H.; Matsubara, H.; Matsuda, F. A burden of rare variants in BMPR2 and KCNK3 contributes to a risk of familial pulmonary arterial hypertension. BMC Pulm. Med. 2017, 17, 57. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.S.; Liu, Q.; Piao, C.M.; Zhu, Y.; Li, Q.Q.; Du, J.; Gu, H. Genotypes and Phenotypes of Chinese Pediatric Patients with Idiopathic and Heritable Pulmonary Arterial Hypertension-A Single-Center Study. Can. J. Cardiol. 2019, 35, 1851–1856. [Google Scholar] [CrossRef]
- Zhu, N.; Gonzaga-Jauregui, C.; Welch, C.L.; Ma, L.; Qi, H.; King, A.K.; Krishnan, U.; Rosenzweig, E.B.; Ivy, D.D.; Austin, E.D.; et al. Exome Sequencing in Children with Pulmonary Arterial Hypertension Demonstrates Differences Compared with Adults. Circ. Genom. Precis Med. 2018, 11, e001887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turro, E.; Astle, W.J.; Megy, K.; Graf, S.; Greene, D.; Shamardina, O.; Allen, H.L.; Sanchis-Juan, A.; Frontini, M.; Thys, C.; et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature 2020, 583, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Bellanne-Chantelot, C.; Saint-Martin, C.; Ribeiro, M.J.; Vaury, C.; Verkarre, V.; Arnoux, J.B.; Valayannopoulos, V.; Gobrecht, S.; Sempoux, C.; Rahier, J.; et al. ABCC8 and KCNJ11 molecular spectrum of 109 patients with diazoxide-unresponsive congenital hyperinsulinism. J. Med. Genet. 2010, 47, 752–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Garin, I.; Castano, L.; Argente, J.; Munoz-Calvo, M.T.; Perez de Nanclares, G.; Shyng, S.L. Neonatal diabetes caused by mutations in sulfonylurea receptor 1: Interplay between expression and Mg-nucleotide gating defects of ATP-sensitive potassium channels. J. Clin. Endocrinol. Metab. 2010, 95, E473–E478. [Google Scholar] [CrossRef] [Green Version]
- Lago-Docampo, M.; Tenorio, J.; Hernandez-Gonzalez, I.; Perez-Olivares, C.; Escribano-Subias, P.; Pousada, G.; Baloira, A.; Arenas, M.; Lapunzina, P.; Valverde, D. Characterization of rare ABCC8 variants identified in Spanish pulmonary arterial hypertension patients. Sci. Rep. 2020, 10, 15135. [Google Scholar] [CrossRef]
- Lee, K.P.K.; Chen, J.; MacKinnon, R. Molecular structure of human KATP in complex with ATP and ADP. eLife 2017, 6, e32481. [Google Scholar] [CrossRef]
- Badesch, D.B.; Raskob, G.E.; Elliott, C.G.; Krichman, A.M.; Farber, H.W.; Frost, A.E.; Barst, R.J.; Benza, R.L.; Liou, T.G.; Turner, M.; et al. Pulmonary arterial hypertension: Baseline characteristics from the REVEAL Registry. Chest 2010, 137, 376–387. [Google Scholar] [CrossRef] [Green Version]
- Le Ribeuz, H.; Capuano, V.; Girerd, B.; Humbert, M.; Montani, D.; Antigny, F. Implication of Potassium Channels in the Pathophysiology of Pulmonary Arterial Hypertension. Biomolecules 2020, 10, 1261. [Google Scholar] [CrossRef]
- Woo, S.K.; Kwon, M.S.; Geng, Z.; Chen, Z.; Ivanov, A.; Bhatta, S.; Gerzanich, V.; Simard, J.M. Sequential activation of hypoxia-inducible factor 1 and specificity protein 1 is required for hypoxia-induced transcriptional stimulation of Abcc8. J. Cereb. Blood Flow Metab. 2012, 32, 525–536. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Al-Mehdi, A.B.; Levitan, I.; Stevens, T.; Fisher, A.B. Shear stress increases expression of a KATP channel in rat and bovine pulmonary vascular endothelial cells. Am. J. Physiol. Cell Physiol. 2003, 285, C959–C967. [Google Scholar] [CrossRef] [Green Version]
- Hamouda, N.N.; Van den Haute, C.; Vanhoutte, R.; Sannerud, R.; Azfar, M.; Mayer, R.; Cortes Calabuig, A.; Swinnen, J.V.; Agostinis, P.; Baekelandt, V.; et al. ATP13A3 is a major component of the enigmatic mammalian polyamine transport system. J. Biol. Chem. 2020, 1, 296. [Google Scholar] [CrossRef]
- Rhodes, C.J.; Ghataorhe, P.; Wharton, J.; Rue-Albrecht, K.C.; Hadinnapola, C.; Watson, G.; Bleda, M.; Haimel, M.; Coghlan, G.; Corris, P.A.; et al. Plasma Metabolomics Implicates Modified Transfer RNAs and Altered Bioenergetics in the Outcomes of Pulmonary Arterial Hypertension. Circulation 2017, 135, 460–475. [Google Scholar] [CrossRef]
- He, Y.Y.; Yan, Y.; Jiang, X.; Zhao, J.H.; Wang, Z.; Wu, T.; Wang, Y.; Guo, S.S.; Ye, J.; Lian, T.Y.; et al. Spermine promotes pulmonary vascular remodelling and its synthase is a therapeutic target for pulmonary arterial hypertension. Eur. Respir. J. 2020, 56, 2000522. [Google Scholar] [CrossRef] [PubMed]
- Savale, L.; Chaumais, M.C.; O’Connell, C.; Humbert, M.; Sitbon, O. Interferon-induced pulmonary hypertension: An update. Curr. Opin. Pulm. Med. 2016, 22, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.; Colglazier, E.; Parker, C.; Amin, E.K.; Nawaytou, H.; Teitel, D.; Reddy, V.M.; Welch, C.L.; Chung, W.K.; Fineman, J.R. Genetics dictating therapeutic decisions in pediatric pulmonary hypertension? A case report suggesting we are getting closer. Pulm. Circ. 2022, 12, e12033. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, D.M.; Holemans, T.; van Veen, S.; Martin, S.; Arslan, T.; Haagendahl, I.W.; Holen, H.W.; Hamouda, N.N.; Eggermont, J.; Palmgren, M.; et al. Parkinson disease related ATP13A2 evolved early in animal evolution. PLoS ONE 2018, 13, e0193228. [Google Scholar]
- Cunningham, K.P.; Holden, R.G.; Escribano-Subias, P.M.; Cogolludo, A.; Veale, E.L.; Mathie, A. Characterization and regulation of wild-type and mutant TASK-1 two pore domain potassium channels indicated in pulmonary arterial hypertension. J. Physiol. 2019, 597, 1087–1101. [Google Scholar] [CrossRef] [Green Version]
- Antigny, F.; Hautefort, A.; Meloche, J.; Belacel-Ouari, M.; Manoury, B.; Rucker-Martin, C.; Pechoux, C.; Potus, F.; Nadeau, V.; Tremblay, E.; et al. Potassium Channel Subfamily K Member 3 (KCNK3) Contributes to the Development of Pulmonary Arterial Hypertension. Circulation 2016, 133, 1371–1385. [Google Scholar] [CrossRef]
- Manoury, B.; Lamalle, C.; Oliveira, R.; Reid, J.; Gurney, A.M. Contractile and electrophysiological properties of pulmonary artery smooth muscle are not altered in TASK-1 knockout mice. J. Physiol. 2011, 589, 3231–3246. [Google Scholar] [CrossRef]
- Pandit, L.M.; Lloyd, E.E.; Reynolds, J.O.; Lawrence, W.S.; Reynolds, C.; Wehrens, X.H.; Bryan, R.M. TWIK-2 channel deficiency leads to pulmonary hypertension through a rho-kinase-mediated process. Hypertension 2014, 64, 1260–1265. [Google Scholar] [CrossRef] [Green Version]
- Lambert, M.; Capuano, V.; Boet, A.; Tesson, L.; Bertero, T.; Nakhleh, M.K.; Remy, S.; Anegon, I.; Pechoux, C.; Hautefort, A.; et al. Characterization of Kcnk3-Mutated Rat, a Novel Model of Pulmonary Hypertension. Circ. Res. 2019, 125, 678–695. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.; Mendes-Ferreira, P.; Ghigna, M.R.; LeRibeuz, H.; Adao, R.; Boet, A.; Capuano, V.; Rucker-Martin, C.; Bras-Silva, C.; Quarck, R.; et al. Kcnk3 dysfunction exaggerates the development of pulmonary hypertension induced by left ventricular pressure overload. Cardiovasc. Res. 2021, 117, 2474–2488. [Google Scholar] [CrossRef] [PubMed]
- Barros, F.; Pardo, L.A.; Dominguez, P.; Sierra, L.M.; De la Pena, P. New Structures and Gating of Voltage-Dependent Potassium (Kv) Channels and Their Relatives: A Multi-Domain and Dynamic Question. Int. J. Mol. Sci. 2019, 20, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.L.; Souil, E.; Dinh-Xuan, A.T.; Schremmer, B.; Mercier, J.C.; El Yaagoubi, A.; Nguyen-Huu, L.; Reeve, H.L.; Hampl, V. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J. Clin. Investig. 1998, 101, 2319–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, K.A.; Ivester, C.; West, J.; Carr, M.; Rodman, D.M. BMP signaling controls PASMC KV channel expression in vitro and in vivo. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L841–L848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remillard, C.V.; Tigno, D.D.; Platoshyn, O.; Burg, E.D.; Brevnova, E.E.; Conger, D.; Nicholson, A.; Rana, B.K.; Channick, R.N.; Rubin, L.J.; et al. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am. J. Physiol. Cell Physiol. 2007, 292, C1837–C1853. [Google Scholar] [CrossRef]
- Wipff, J.; Dieude, P.; Guedj, M.; Ruiz, B.; Riemekasten, G.; Cracowski, J.L.; Matucci-Cerinic, M.; Melchers, I.; Humbert, M.; Hachulla, E.; et al. Association of a KCNA5 gene polymorphism with systemic sclerosis-associated pulmonary arterial hypertension in the European Caucasian population. Arthritis Rheum. 2010, 62, 3093–3100. [Google Scholar] [CrossRef]
- Wang, G.; Knight, L.; Ji, R.; Lawrence, P.; Kanaan, U.; Li, L.; Das, A.; Cui, B.; Zou, W.; Penny, D.J.; et al. Early onset severe pulmonary arterial hypertension with ‘two-hit’ digenic mutations in both BMPR2 and KCNA5 genes. Int. J. Cardiol. 2014, 177, e167–e169. [Google Scholar] [CrossRef]
- Bossini-Castillo, L.; Simeon, C.P.; Beretta, L.; Broen, J.; Vonk, M.C.; Callejas, J.L.; Carreira, P.; Rodriguez-Rodriguez, L.; Garcia-Portales, R.; Gonzalez-Gay, M.A.; et al. KCNA5 gene is not confirmed as a systemic sclerosis-related pulmonary arterial hypertension genetic susceptibility factor. Arthritis Res. Ther. 2012, 14, R273. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Y.R.; Wang, W.; Lei, P.C.; Jia, H.P.; Dong, J.; Gou, Y.Q.; Chen, C.L.; Cao, J.; Wang, Y.F.; Zhu, Y.K. 5-HTT, BMPR2, EDN1, ENG, KCNA5 gene polymorphisms and susceptibility to pulmonary arterial hypertension: A meta-analysis. Gene 2019, 680, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J.; Batai, K.; Bleda, M.; Haimel, M.; Southgate, L.; Germain, M.; Pauciulo, M.W.; Hadinnapola, C.; Aman, J.; Girerd, B.; et al. Genetic determinants of risk in pulmonary arterial hypertension: International genome-wide association studies and meta-analysis. Lancet Respir. Med. 2019, 7, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Stoller, D.A.; Fahrenbach, J.P.; Chalupsky, K.; Tan, B.H.; Aggarwal, N.; Metcalfe, J.; Hadhazy, M.; Shi, N.Q.; Makielski, J.C.; McNally, E.M. Cardiomyocyte sulfonylurea receptor 2-KATP channel mediates cardioprotection and ST segment elevation. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1100–H1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harakalova, M.; Van Harssel, J.J.; Terhal, P.A.; Van Lieshout, S.; Duran, K.; Renkens, I.; Amor, D.J.; Wilson, L.C.; Kirk, E.P.; Turner, C.L.; et al. Dominant missense mutations in ABCC9 cause Cantu syndrome. Nat. Genet. 2012, 44, 793–796. [Google Scholar] [CrossRef]
- Park, J.Y.; Koo, S.H.; Jung, Y.J.; Lim, Y.J.; Chung, M.L. A patient with Cantu syndrome associated with fatal bronchopulmonary dysplasia and pulmonary hypertension. Am. J. Med. Genet. A 2014, 164, 2118–2120. [Google Scholar] [CrossRef]
- Ma, A.; Gurnasinghani, S.; Kirk, E.P.; McClenaghan, C.; Singh, G.K.; Grange, D.K.; Pandit, C.; Zhu, Y.; Roscioli, T.; Elakis, G.; et al. Glibenclamide treatment in a Cantu syndrome patient with a pathogenic ABCC9 gain-of-function variant: Initial experience. Am. J. Med. Genet. A 2019, 179, 1585–1590. [Google Scholar] [CrossRef] [Green Version]
- Malczyk, M.; Erb, A.; Veith, C.; Ghofrani, H.A.; Schermuly, R.T.; Gudermann, T.; Dietrich, A.; Weissmann, N.; Sydykov, A. The Role of Transient Receptor Potential Channel 6 Channels in the Pulmonary Vasculature. Front. Immunol. 2017, 8, 707. [Google Scholar] [CrossRef] [Green Version]
- Winn, M.P.; Conlon, P.J.; Lynn, K.L.; Farrington, M.K.; Creazzo, T.; Hawkins, A.F.; Daskalakis, N.; Kwan, S.Y.; Ebersviller, S.; Burchette, J.L.; et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 2005, 308, 1801–1804. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Keller, S.H.; Remillard, C.V.; Safrina, O.; Nicholson, A.; Zhang, S.L.; Jiang, W.; Vangala, N.; Landsberg, J.W.; Wang, J.Y.; et al. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 2009, 119, 2313–2322. [Google Scholar] [CrossRef] [Green Version]
- Nebesio, T.D.; Hoover, W.C.; Caldwell, R.L.; Nitu, M.E.; Eugster, E.A. Development of pulmonary hypertension in an infant treated with diazoxide. J. Pediatr. Endocrinol. Metab. 2007, 20, 939–944. [Google Scholar] [CrossRef]
- Timlin, M.R.; Black, A.B.; Delaney, H.M.; Matos, R.I.; Percival, C.S. Development of Pulmonary Hypertension During Treatment with Diazoxide: A Case Series and Literature Review. Pediatr. Cardiol. 2017, 38, 1247–1250. [Google Scholar] [CrossRef] [PubMed]
- Kylat, R.I. Pulmonary hypertension occurring with diazoxide use in a preterm infant with hypoglycemia. Drug Healthc. Patient Saf. 2019, 11, 7–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.C.; Dastamani, A.; Pintus, D.; Yau, D.; Aftab, S.; Bath, L.; Swinburne, C.; Hunter, L.; Giardini, A.; Christov, G.; et al. Diazoxide-induced pulmonary hypertension in hyperinsulinaemic hypoglycaemia: Recommendations from a multicentre study in the United Kingdom. Clin. Endocrinol. 2019, 91, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Pozeg, Z.I.; Michelakis, E.D.; McMurtry, M.S.; Thebaud, B.; Wu, X.C.; Dyck, J.R.; Hashimoto, K.; Wang, S.; Moudgil, R.; Harry, G.; et al. In vivo gene transfer of the O2-sensitive potassium channel Kv1.5 reduces pulmonary hypertension and restores hypoxic pulmonary vasoconstriction in chronically hypoxic rats. Circulation 2003, 107, 2037–2044. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Name | Number of Cases (%) | PAH Subclass 1 | Variant Type 2 | Mode of Inheritance 3 |
|---|---|---|---|---|---|
| ABCC8 | ATP-binding cassette subfamily C member 8 | 49/3521 (1.4%) [6,10,11,12,13] | H/I/APAH APAH-CTD APAH-CHD APAH-HIV PPHN | missense, LGD | AD |
| ATP13A3 | ATPase 13A3 | 27/4012 (0.7%) [6,8,12,14,15,16] | H/IPAH APAH-CTD APAH-CHD APAH-MS/IFNβ-1a | LGD, missense | Semi-dominant |
| KCNK3 | Potassium two-pore domain channel subfamily K member 3 | 14/4682 (0.3%) [6,17,18,19,20,21,22] | H/IPAH | missense | AD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Welch, C.L.; Chung, W.K. Channelopathy Genes in Pulmonary Arterial Hypertension. Biomolecules 2022, 12, 265. https://doi.org/10.3390/biom12020265
Welch CL, Chung WK. Channelopathy Genes in Pulmonary Arterial Hypertension. Biomolecules. 2022; 12(2):265. https://doi.org/10.3390/biom12020265
Chicago/Turabian StyleWelch, Carrie L., and Wendy K. Chung. 2022. "Channelopathy Genes in Pulmonary Arterial Hypertension" Biomolecules 12, no. 2: 265. https://doi.org/10.3390/biom12020265
APA StyleWelch, C. L., & Chung, W. K. (2022). Channelopathy Genes in Pulmonary Arterial Hypertension. Biomolecules, 12(2), 265. https://doi.org/10.3390/biom12020265