
DNA Methylation Malleability and Dysregulation in Cancer Progression: Understanding the Role of PARP1



Abstract
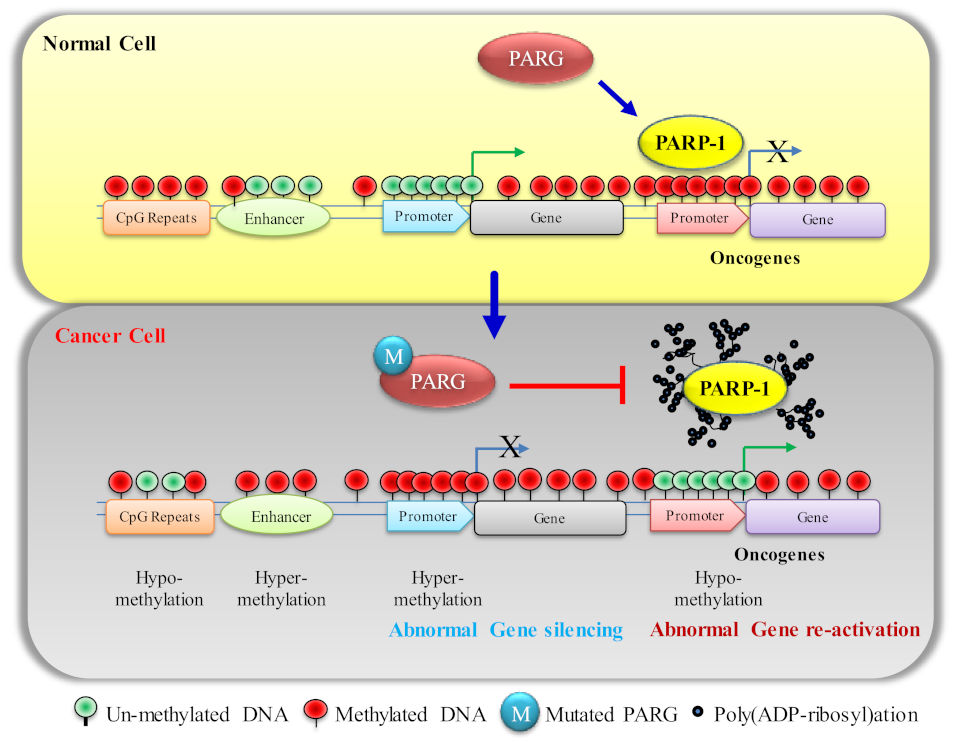
:1. Introduction
2. Dynamic Behavior of DNA Methylation and Demethylation
3. DNA Methylation and Demethylation in Cancer Progression
4. Poly(ADP-ry)lation of DNMT1 Determines DNA Methylation
5. PARP1 in DNA Hypermethylation and Its Effect on Cancer Progression
5.1. Effect of DNA Hypermethylation on TSG (P53 and NF-κB) Expression
- P53, one of the major tumor suppressor proteins, and its loss of function by mutations or loss of expression cause more than 50% of human cancers. P53 also plays a key role in a multitude of DNA-damage response pathways [86]. It has been reported in several papers that P53 and PARP1 interact at multiple levels [108]. P53 is not only a covalent poly(ADP-ribosyl)ation target [109,110], but it also possesses a high-affinity non-covalent association with poly(ADP-ribosyl) [111]. Dysregulated poly(ribosyl)ation activity in cancer cells could be one of the possibilities to downregulate P53 expression via DNA hypermethylation on its gene region.
- NF-κB, the master regulator, mediates the crosstalk between cancer and inflammation at multiple levels. Enhanced NF-κB function can cause pro-inflammatory cytokine production in tumor tissues, which significantly contributes to the pro-tumorigenic microenvironment [112].
5.2. Control of DNA Hypermethylation
DNA Methyltransferases Inhibitor
- 1.
- Decitabine
- 2.
- 5-Azacytidine
- 3.
- Zebularine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNMT Inhibitor | Effect on Cancer Progression | References |
|---|---|---|
| Decitabine | Lung Cancer, Colorectal Cancer, Breast Cancer, Prostate Cancer, Liver Cancer, Acute Myeloid Leukemia | [147,148,149,150,151,152] |
| 5-Azacitidine | Gastric Cancer, Acute Myeloid Leukemia, Germ-Cell Tumor, Esophageal Cancer, Colon Cancer | [153,154,155,156,157,158,159] |
| Zebularine | Colon Cancer, Liver Cancer, Pancreatic Cancer, Prostate Cancer, Medulloblastoma | [140,141,142,143,144] |
| Guadecitabine (SGI-110) | Germ-Cell Tumor, Ovarian Cancer, Liver Cancer, Urothelial Cancer | [160,161,162,163] |
| 5-Fluro-2′ deoxycytidine | Urothelial Cancer, Colon Cancer | [164,165] |
| 5,6, dihydro 5 azacytidine | T-Cell Acute Lymphocytic Leukemia, Acute Myeloid Leukemia | [166] |
| CP-4200 | Acute Myeloid Leukemia, Breast Cancer, Colon Cancer | [167,168] |
| Gemcitabine | Cervical Cancer, Colorectal Cancer, Pancreatic Cancer, Bladder Cancer | [169,170,171,172,173,174] |
| Rx3117 | Pancreatic Cancer, Bladder Cancer, Lung Cancer, Leukemic Lymphoblasts | [175,176] |
| Hydralazine | Prostate Cancer, Solid Cancers, Osteosarcoma | [177,178,179] |
6. PARP1 Inhibitors in DNA Hypomethylation of Cancer Cells
6.1. PARP1 Inhibitors
6.2. PARP1 Inhibitors in Reversal of Tumor-Suppressor-Gene Expression
6.2.1. Increase in DNA Hypomethylation by an Increase in TET Activity
6.2.2. Maintenance of DNA Methylation by Poly(ADP-ribosyl)ation of CTCF and DNMT1
7. Combination Therapy of DNA Methyltransferase Inhibitor and PARP Inhibitor
8. Perspective and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boland, M.J.; Nazor, K.L.; Loring, J.F. Epigenetic regulation of pluripotency and differentiation. Circ. Res. 2014, 115, 311–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brookes, E.; Shi, Y. Diverse epigenetic mechanisms of human disease. Annu. Rev. Genet. 2014, 48, 237–268. [Google Scholar] [CrossRef]
- Oh, E.S.; Petronis, A. Origins of human disease: The chrono-epigenetic perspective. Nat. Rev. Genet. 2021, 22, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlberg, C.; Molnár, F. DNA Methylation. In Human Epigenomics; Carlberg, C., Molnár, F., Eds.; Springer Singapore: Singapore, 2018; pp. 57–73. [Google Scholar]
- Noroozi, R.; Ghafouri-Fard, S.; Pisarek, A.; Rudnicka, J.; Spolnicka, M.; Branicki, W.; Taheri, M.; Pospiech, E. DNA methylation-based age clocks: From age prediction to age reversion. Ageing Res. Rev. 2021, 68, 101314. [Google Scholar] [CrossRef]
- Angeloni, A.; Bogdanovic, O. Sequence determinants, function, and evolution of CpG islands. Biochem. Soc. Trans. 2021, 49, 1109–1119. [Google Scholar] [CrossRef]
- Kim, M.; Costello, J. DNA methylation: An epigenetic mark of cellular memory. Exp. Mol. Med. 2017, 49, e322. [Google Scholar] [CrossRef] [Green Version]
- Skvortsova, K.; Stirzaker, C.; Taberlay, P. The DNA methylation landscape in cancer. Essays Biochem. 2019, 63, 797–811. [Google Scholar] [CrossRef]
- Wilson, A.G. Epigenetic regulation of gene expression in the inflammatory response and relevance to common diseases. J. Periodontol 2008, 79, 1514–1519. [Google Scholar] [CrossRef] [Green Version]
- Ng, H.H.; Zhang, Y.; Hendrich, B.; Johnson, C.A.; Turner, B.M.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D.; Bird, A. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat. Genet. 1999, 23, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Magaña-Acosta, M.; Valadez-Graham, V. Chromatin Remodelers in the 3D Nuclear Compartment. Front. Genet. 2020, 11, 600615. [Google Scholar] [CrossRef] [PubMed]
- Fouse, S.D.; Nagarajan, R.O.; Costello, J.F. Genome-scale DNA methylation analysis. Epigenomics 2010, 2, 105–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomizawa, S.; Kobayashi, H.; Watanabe, T.; Andrews, S.; Hata, K.; Kelsey, G.; Sasaki, H. Dynamic stage-specific changes in imprinted differentially methylated regions during early mammalian development and prevalence of non-CpG methylation in oocytes. Development 2011, 138, 811–820. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014, 17, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Ramsahoye, B.H.; Biniszkiewicz, D.; Lyko, F.; Clark, V.; Bird, A.P.; Jaenisch, R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. USA 2000, 97, 5237–5242. [Google Scholar] [CrossRef] [Green Version]
- Stoccoro, A.; Coppede, F. Mitochondrial DNA Methylation and Human Diseases. Int. J. Mol. Sci. 2021, 22, 4594. [Google Scholar] [CrossRef]
- Srivastava, R.; Ahn, S.H. Modifications of RNA polymerase II CTD: Connections to the histone code and cellular function. Biotechnol. Adv. 2015, 33, 856–872. [Google Scholar] [CrossRef]
- Srivastava, R.; Duan, R.; Ahn, S.H. Multiple roles of CTDK-I throughout the cell. Cell Mol. Life Sci. 2019, 76, 2789–2797. [Google Scholar] [CrossRef]
- Srivastava, R.; Singh, U.M.; Dubey, N.K. Histone Modifications by different histone modifiers: Insights into histone writers and erasers during chromatin modification. J. Biol. Sci. Med. 2016, 2, 45–54. [Google Scholar]
- Srivastava, R.; Srivastava, R.; Ahn, S.H. The Epigenetic Pathways to Ribosomal DNA Silencing. Microbiol. Mol. Biol. Rev. 2016, 80, 545–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Beek, L.; McClay, E.; Patel, S.; Schimpl, M.; Spagnolo, L.; de Oliveira, T.M. PARP Power: A Structural Perspective on PARP1, PARP2, and PARP3 in DNA Damage Repair and Nucleosome Remodelling. Int. J. Mol. Sci. 2021, 22, 5112. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, J.O.; Van Houten, B.; Durrant, J.D. PARP1: Structural insights and pharmacological targets for inhibition. DNA Repair 2021, 103, 103125. [Google Scholar] [CrossRef] [PubMed]
- Ummarino, S.; Hausman, C.; Di Ruscio, A. The PARP Way to Epigenetic Changes. Genes 2021, 12, 446. [Google Scholar] [CrossRef]
- Tulin, A.; Spradling, A. Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puff loci. Science 2003, 299, 560–562. [Google Scholar] [CrossRef]
- Titcombe, P.; Murray, R.; Hewitt, M.; Antoun, E.; Cooper, C.; Inskip, H.M.; Holbrook, J.D.; Godfrey, K.M.; Lillycrop, K.; Hanson, M.; et al. Human non-CpG methylation patterns display both tissue-specific and inter-individual differences suggestive of underlying function. Epigenetics 2021, 1–12. [Google Scholar] [CrossRef]
- Rideout, W.M., 3rd; Coetzee, G.A.; Olumi, A.F.; Jones, P.A. 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science 1990, 249, 1288–1290. [Google Scholar] [CrossRef]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Schöler, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef]
- Aran, D.; Sabato, S.; Hellman, A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013, 14, R21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papin, C.; Le Gras, S.; Ibrahim, A.; Salem, H.; Karimi, M.M.; Stoll, I.; Ugrinova, I.; Schröder, M.; Fontaine-Pelletier, E.; Omran, Z.; et al. CpG Islands Shape the Epigenome Landscape. J. Mol. Biol. 2021, 433, 166659. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Bartolomei, M.S. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell 2013, 152, 1308–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Arand, J.; Spieler, D.; Karius, T.; Branco, M.R.; Meilinger, D.; Meissner, A.; Jenuwein, T.; Xu, G.; Leonhardt, H.; Wolf, V.; et al. In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012, 8, e1002750. [Google Scholar] [CrossRef] [Green Version]
- Day, J.J.; Kennedy, A.J.; Sweatt, J.D. DNA methylation and its implications and accessibility for neuropsychiatric therapeutics. Annu. Rev. Pharm. Toxicol. 2015, 55, 591–611. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.X.; Riggs, A.D. DNA methylation and demethylation in mammals. J. Biol. Chem. 2011, 286, 18347–18353. [Google Scholar] [CrossRef] [Green Version]
- Auclair, G.; Weber, M. Mechanisms of DNA methylation and demethylation in mammals. Biochimie 2012, 94, 2202–2211. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Ueda, Y.; Dodge, J.E.; Wang, Z.; Li, E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell Biol. 2003, 23, 5594–5605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, G.; Chan, M.F.; Tomigahara, Y.; Tsai, Y.C.; Gonzales, F.A.; Li, E.; Laird, P.W.; Jones, P.A. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol. Cell. Biol. 2002, 22, 480–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Y.; Chen, T. DNA Methylation Reprogramming during Mammalian Development. Genes 2019, 10, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kareta, M.S.; Botello, Z.M.; Ennis, J.J.; Chou, C.; Chedin, F. Reconstitution and mechanism of the stimulation of de novo methylation by human DNMT3L. J. Biol. Chem. 2006, 281, 25893–25902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, S.K.; Wolf, D.; Hartung, O.; Agarwal, S.; Daley, G.Q.; Goff, S.P.; Bestor, T.H. Dynamic instability of genomic methylation patterns in pluripotent stem cells. Epigenetics Chromatin. 2010, 3, 17. [Google Scholar] [CrossRef] [Green Version]
- Bourc’his, D.; Xu, G.L.; Lin, C.S.; Bollman, B.; Bestor, T.H. Dnmt3L and the establishment of maternal genomic imprints. Science 2001, 294, 2536–2539. [Google Scholar] [CrossRef] [Green Version]
- Chedin, F. The DNMT3 family of mammalian de novo DNA methyltransferases. Prog. Mol. Biol. Transl. Sci. 2011, 101, 255–285. [Google Scholar] [CrossRef]
- Hu, Y.G.; Hirasawa, R.; Hu, J.L.; Hata, K.; Li, C.L.; Jin, Y.; Chen, T.P.; Li, E.; Rigolet, M.; Viegas-Pequignot, E.; et al. Regulation of DNA methylation activity through Dnmt3L promoter methylation by Dnmt3 enzymes in embryonic development. Hum. Mol. Genet. 2008, 17, 2654–2664. [Google Scholar] [CrossRef] [Green Version]
- Gujar, H.; Weisenberger, D.J.; Liang, G. The Roles of Human DNA Methyltransferases and Their Isoforms in Shaping the Epigenome. Genes 2019, 10, 172. [Google Scholar] [CrossRef] [Green Version]
- Neri, F.; Krepelova, A.; Incarnato, D.; Maldotti, M.; Parlato, C.; Galvagni, F.; Matarese, F.; Stunnenberg, H.G.; Oliviero, S. Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs. Cell 2013, 155, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Meng, H.; Cao, Y.; Qin, J.; Song, X.; Zhang, Q.; Shi, Y.; Cao, L. DNA methylation, its mediators and genome integrity. Int. J. Biol. Sci. 2015, 11, 604–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goll, M.G.; Kirpekar, F.; Maggert, K.A.; Yoder, J.A.; Hsieh, C.L.; Zhang, X.; Golic, K.G.; Jacobsen, S.E.; Bestor, T.H. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 2006, 311, 395–398. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, X.; Shi, J.; Tuorto, F.; Li, X.; Liu, Y.; Liebers, R.; Zhang, L.; Qu, Y.; Qian, J.; et al. Dnmt2 mediates intergenerational transmission of paternally acquired metabolic disorders through sperm small non-coding RNAs. Nat. Cell Biol. 2018, 20, 535–540. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A.; Taylor, S.M. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980, 20, 85–93. [Google Scholar] [CrossRef]
- Mayer, W.; Niveleau, A.; Walter, J.; Fundele, R.; Haaf, T. Demethylation of the zygotic paternal genome. Nature 2000, 403, 501–502. [Google Scholar] [CrossRef]
- An, J.; Rao, A.; Ko, M. TET family dioxygenases and DNA demethylation in stem cells and cancers. Exp. Mol. Med. 2017, 49, e323. [Google Scholar] [CrossRef] [Green Version]
- Melamed, P.; Yosefzon, Y.; David, C.; Tsukerman, A.; Pnueli, L. Tet Enzymes, Variants, and Differential Effects on Function. Front. Cell Dev. Biol. 2018, 6, 22. [Google Scholar] [CrossRef] [Green Version]
- Dick, A.; Chen, A. The role of TET proteins in stress-induced neuroepigenetic and behavioural adaptations. Neurobiol. Stress 2021, 15, 100352. [Google Scholar] [CrossRef] [PubMed]
- Neri, F.; Incarnato, D.; Oliviero, S. DNA methylation and demethylation dynamics. Oncotarget 2015, 6, 34049–34050. [Google Scholar] [CrossRef] [PubMed]
- Nawy, T. Dynamics of DNA demethylation. Nat. Methods 2013, 10, 466. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, N.; Burns, D.M.; Blau, H.M. DNA demethylation dynamics. Cell 2011, 146, 866–872. [Google Scholar] [CrossRef] [Green Version]
- Pastor, W.A.; Aravind, L.; Rao, A. TETonic shift: Biological roles of TET proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.G.; Zhang, Z.M.; Dunwell, T.L.; Harter, M.R.; Wu, X.; Johnson, J.; Li, Z.; Liu, J.; Szabó, P.E.; Lu, Q.; et al. Tet3 Reads 5-Carboxylcytosine through Its CXXC Domain and Is a Potential Guardian against Neurodegeneration. Cell Rep. 2016, 14, 493–505. [Google Scholar] [CrossRef] [Green Version]
- Valencia, A.M.; Kadoch, C. Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nat. Cell Biol. 2019, 21, 152–161. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, C.; Wu, C.; Cui, W.; Wang, L. DNA Methyltransferases in Cancer: Biology, Paradox, Aberrations, and Targeted Therapy. Cancers 2020, 12, 2123. [Google Scholar] [CrossRef]
- Lakshminarasimhan, R.; Liang, G. The Role of DNA Methylation in Cancer. Adv. Exp. Med. Biol. 2016, 945, 151–172. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Xu, J.; Zheng, J.; Man, X.; Wu, H.; Jin, J.; Wang, K.; Xiao, H.; Li, S.; et al. Aberrant DNA methyltransferase expression in pancreatic ductal adenocarcinoma development and progression. J. Exp. Clin. Cancer Res. 2013, 32, 86. [Google Scholar] [CrossRef] [Green Version]
- Han, M.; Jia, L.; Lv, W.; Wang, L.; Cui, W. Epigenetic Enzyme Mutations: Role in Tumorigenesis and Molecular Inhibitors. Front. Oncol. 2019, 9, 194. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Kim, H.S.; Kim, J.Y.; Sun, J.M.; Ahn, J.S.; Ahn, M.J.; Park, K.; Esteller, M.; Lee, S.H.; Choi, J.K. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat. Commun. 2019, 10, 4278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheaffer, K.L.; Elliott, E.N.; Kaestner, K.H. DNA Hypomethylation Contributes to Genomic Instability and Intestinal Cancer Initiation. Cancer Prev. Res. 2016, 9, 534–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Yan, Q. Cancer Epigenetics, Tumor Immunity, and Immunotherapy. Trends Cancer 2020, 6, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Rao, A. TET family dioxygenases and the TET activator vitamin C in immune responses and cancer. Blood 2020, 136, 1394–1401. [Google Scholar] [CrossRef] [PubMed]
- Kamaletdinova, T.; Fanaei-Kahrani, Z.; Wang, Z.Q. The Enigmatic Function of PARP1: From PARylation Activity to PAR Readers. Cells 2019, 8, 1625. [Google Scholar] [CrossRef] [Green Version]
- Kotova, E.; Lodhi, N.; Jarnik, M.; Pinnola, A.D.; Ji, Y.; Tulin, A.V. Drosophila histone H2A variant (H2Av) controls poly(ADP-ribose) polymerase 1 (PARP1) activation in chromatin. Proc. Natl. Acad. Sci. USA 2011, 108, 6205–6210. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Thomas, C.; Tulin, N.; Lodhi, N.; Boamah, E.; Kolenko, V.; Tulin, A.V. Charon Mediates Immune Deficiency-Driven PARP-1-Dependent Immune Responses in Drosophila. J. Immunol. 2016, 197, 2382–2389. [Google Scholar] [CrossRef] [Green Version]
- Boamah, E.K.; Kotova, E.; Garabedian, M.; Jarnik, M.; Tulin, A.V. Poly(ADP-Ribose) polymerase 1 (PARP-1) regulates ribosomal biogenesis in Drosophila nucleoli. PLoS Genet. 2012, 8, e1002442. [Google Scholar] [CrossRef] [Green Version]
- Lodhi, N.; Ji, Y.; Tulin, A. Mitotic bookmarking: Maintaining post-mitotic reprogramming of transcription reactivation. Curr. Mol. Biol. Rep. 2016, 2, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Lodhi, N.; Kossenkov, A.V.; Tulin, A.V. Bookmarking promoters in mitotic chromatin: Poly(ADP-ribose)polymerase-1 as an epigenetic mark. Nucleic Acids Res. 2014, 42, 7028–7038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zardo, G.; DErme, M.; Reale, A.; Strom, R.; Perilli, M.; Caiafa, P. Does poly(ADP-ribosyl)ation regulate the DNA methylation pattern? Biochemistry 1997, 36, 7937–7943. [Google Scholar] [CrossRef] [PubMed]
- Zardo, G.; Marenzi, S.; Perilli, M.; Caiafa, P. Inhibition of poly(ADP-ribosyl)ation introduces an anomalous methylation pattern in transfected foreign DNA. FASEB J. 1999, 13, 1518–1522. [Google Scholar] [CrossRef] [PubMed]
- Kraus, W.L.; Hottiger, M.O. PARP-1 and gene regulation: Progress and puzzles. Mol. Asp. Med. 2013, 34, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, A.; Krüger, A.; Hampp, S.; Assmann, G.; Rank, L.; Hufnagel, M.; Stöckl, M.T.; Fischer, J.M.F.; Veith, S.; Rossatti, P.; et al. The C-terminal domain of p53 orchestrates the interplay between non-covalent and covalent poly(ADP-ribosyl)ation of p53 by PARP1. Nucleic Acids Res. 2018, 46, 804–822. [Google Scholar] [CrossRef] [Green Version]
- Reale, A.; Matteis, G.D.; Galleazzi, G.; Zampieri, M.; Caiafa, P. Modulation of DNMT1 activity by ADP-ribose polymers. Oncogene 2005, 24, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Ciccarone, F.; Klinger, F.G.; Catizone, A.; Calabrese, R.; Zampieri, M.; Bacalini, M.G.; De Felici, M.; Caiafa, P. Poly(ADP-ribosyl)ation Acts in the DNA Demethylation of Mouse Primordial Germ Cells Also with DNA Damage-Independent Roles. PLoS ONE 2012, 7, e46927. [Google Scholar] [CrossRef] [Green Version]
- Zampieri, M.; Passananti, C.; Calabrese, R.; Perilli, M.; Corbi, N.; De Cave, F.; Guastafierro, T.; Bacalini, M.G.; Reale, A.; Amicosante, G.; et al. Parp1 localizes within the Dnmt1 promoter and protects its unmethylated state by its enzymatic activity. PLoS ONE 2009, 4, e4717. [Google Scholar] [CrossRef]
- Ciccarone, F.; Zampieri, M.; Caiafa, P. PARP1 orchestrates epigenetic events setting up chromatin domains. Semin. Cell Dev. Biol. 2017, 63, 123–134. [Google Scholar] [CrossRef]
- Nocchi, L.; Tomasetti, M.; Amati, M.; Neuzil, J.; Santarelli, L.; Saccucci, F. Thrombomodulin Is Silenced in Malignant Mesothelioma by a Poly(ADP-ribose) Polymerase-1-mediated Epigenetic Mechanism. J. Biol. Chem. 2011, 286, 19478–19488. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Wu, Z.; Hergert, P.; Henke, C.A.; Bitterman, P.B.; Phan, S.H. Regulation of Myofibroblast Differentiation by Poly(ADP-Ribose) Polymerase 1. Am. J. Pathol. 2013, 182, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccarone, F.; Valentini, E.; Bacalini, M.G.; Zampieri, M.; Calabrese, R.; Guastafierro, T.; Mariano, G.; Reale, A.; Franceschi, C.; Caiafa, P. Poly(ADP-ribosyl)ation is involved in the epigenetic control of TET1 gene transcription. Oncotarget 2014, 5, 10356–10367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampieri, M.; Guastafierro, T.; Calabrese, R.; Ciccarone, F.; Bacalini, M.G.; Reale, A.; Perilli, M.; Passananti, C.; Caiafa, P. ADP-ribose polymers localized on Ctcf-Parp1-Dnmt1 complex prevent methylation of Ctcf target sites. Biochem. J. 2012, 441, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Doege, C.A.; Inoue, K.; Yamashita, T.; Rhee, D.B.; Travis, S.; Fujita, R.; Guarnieri, P.; Bhagat, G.; Vanti, W.B.; Shih, A.; et al. Early-stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature 2012, 488, 652–655. [Google Scholar] [CrossRef] [Green Version]
- Roper, S.J.; Chrysanthou, S.; Senner, C.E.; Sienerth, A.; Gnan, S.; Murray, A.; Masutani, M.; Latos, P.; Hemberger, M. ADP-ribosyltransferases Parp1 and Parp7 safeguard pluripotency of ES cells. Nucleic Acids Res. 2014, 42, 8914–8927. [Google Scholar] [CrossRef] [Green Version]
- Witcher, M.; Emerson, B.M. Epigenetic Silencing of the p16INK4a Tumor Suppressor Is Associated with Loss of CTCF Binding and a Chromatin Boundary. Mol. Cell 2009, 34, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [CrossRef]
- Gottipati, P.; Vischioni, B.; Schultz, N.; Solomons, J.; Bryant, H.E.; Djureinovic, T.; Issaeva, N.; Sleeth, K.; Sharma, R.A.; Helleday, T. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 2010, 70, 5389–5398. [Google Scholar] [CrossRef] [Green Version]
- Karpova, Y.; Wu, C.; Divan, A.; McDonnell, M.E.; Hewlett, E.; Makhov, P.; Gordon, J.; Ye, M.; Reitz, A.B.; Childers, W.E.; et al. Non-NAD-like PARP-1 inhibitors in prostate cancer treatment. Biochem. Pharm. 2019, 167, 149–162. [Google Scholar] [CrossRef]
- Ciccarone, F.; Valentini, E.; Zampieri, M.; Caiafa, P. 5mC-hydroxylase activity is influenced by the PARylation of TET1 enzyme. Oncotarget 2015, 6, 24333–24347. [Google Scholar] [CrossRef] [Green Version]
- Berger, N.A. Poly(ADP-ribose) in the cellular response to DNA damage. Radiat. Res. 1985, 101, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillai, J.B.; Gupta, M.; Rajamohan, S.B.; Lang, R.; Raman, J.; Gupta, M.P. Poly(ADP-ribose) polymerase-1-deficient mice are protected from angiotensin II-induced cardiac hypertrophy. Am. J. Physiol Heart Circ. Physiol. 2006, 291, H1545–H1553. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.D.; Wei, S.J.; Wang, X.P.; Wang, J.; Wang, W.K.; Liu, F.; Gong, L.; Yan, F.; Zhang, Y.; Zhang, M. Poly(ADP-ribose) polymerase 1 inhibition protects against low shear stress induced inflammation. Biochim. Biophys. Acta 2013, 1833, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Rajamohan, S.B.; Pillai, V.B.; Gupta, M.; Sundaresan, N.R.; Birukov, K.G.; Samant, S.; Hottiger, M.O.; Gupta, M.P. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol. Cell Biol. 2009, 29, 4116–4129. [Google Scholar] [CrossRef] [Green Version]
- Aerts, I.; Lumbroso-Le Rouic, L.; Gauthier-Villars, M.; Brisse, H.; Doz, F.; Desjardins, L. Retinoblastoma. Orphanet J. Rare Dis. 2006, 1, 31. [Google Scholar] [CrossRef]
- Wesierska-Gadek, J.; Wojciechowski, J.; Schmid, G. Central and carboxy-terminal regions of human p53 protein are essential for interaction and complex formation with PARP-1. J. Cell Biochem. 2003, 89, 220–232. [Google Scholar] [CrossRef]
- Simbulan-Rosenthal, C.M.; Rosenthal, D.S.; Luo, R.; Smulson, M.E. Poly(ADP-ribosyl)ation of p53 during apoptosis in human osteosarcoma cells. Cancer Res. 1999, 59, 2190–2194. [Google Scholar]
- Ayyappan, V.; Wat, R.; Barber, C.; Vivelo, C.A.; Gauch, K.; Visanpattanasin, P.; Cook, G.; Sazeides, C.; Leung, A.K.L. ADPriboDB 2.0: An updated database of ADP-ribosylated proteins. Nucleic Acids Res. 2021, 49, D261–D265. [Google Scholar] [CrossRef]
- Malanga, M.; Pleschke, J.M.; Kleczkowska, H.E.; Althaus, F.R. Poly(ADP-ribose) binds to specific domains of p53 and alters its DNA binding functions. J. Biol Chem. 1998, 273, 11839–11843. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Shen, S.; Verma, I.M. NF-kappaB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, A.S., Jr. The NF-kappa B and I kappa B proteins: New discoveries and insights. Annu. Rev. Immunol. 1996, 14, 649–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M.; Delhase, M. The I kappa B kinase (IKK) and NF-kappa B: Key elements of proinflammatory signalling. Semin. Immunol. 2000, 12, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef]
- Weaver, A.N.; Yang, E.S. Beyond DNA Repair: Additional Functions of PARP-1 in Cancer. Front. Oncol. 2013, 3, 290. [Google Scholar] [CrossRef] [Green Version]
- Hassa, P.O.; Covic, M.; Hasan, S.; Imhof, R.; Hottiger, M.O. The enzymatic and DNA binding activity of PARP-1 are not required for NF-kappa B coactivator function. J. Biol. Chem. 2001, 276, 45588–45597. [Google Scholar] [CrossRef] [Green Version]
- Le Page, C.; Sanceau, J.; Drapier, J.C.; Wietzerbin, J. Inhibitors of ADP-ribosylation impair inducible nitric oxide synthase gene transcription through inhibition of NF kappa B activation. Biochem. Biophys. Res. Commun. 1998, 243, 451–457. [Google Scholar] [CrossRef]
- Kagoya, Y.; Yoshimi, A.; Kataoka, K.; Nakagawa, M.; Kumano, K.; Arai, S.; Kobayashi, H.; Saito, T.; Iwakura, Y.; Kurokawa, M. Positive feedback between NF-κB and TNF-α promotes leukemia-initiating cell capacity. J. Clin. Investig. 2014, 124, 528–542. [Google Scholar] [CrossRef]
- Li, D.; Luo, Y.; Chen, X.; Zhang, L.; Wang, T.; Zhuang, Y.; Fan, Y.; Xu, J.; Chen, Y.; Wu, L. NF-κB and Poly (ADP-ribose) Polymerase 1 Form a Positive Feedback Loop that Regulates DNA Repair in Acute Myeloid Leukemia Cells. Mol. Cancer Res. 2019, 17, 761–772. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Yu, J.; Qin, B.; Moyer, A.M.; Nowsheen, S.; Liu, T.; Qin, S.; Zhuang, Y.; Liu, D.; Lu, S.W.; Kalari, K.R.; et al. DNA methyltransferase expression in triple-negative breast cancer predicts sensitivity to decitabine. J. Clin. Investig. 2018, 128, 2376–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christman, J.K. 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jüttermann, R.; Li, E.; Jaenisch, R. Toxicity of 5-aza-2’-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc. Natl. Acad. Sci. USA 1994, 91, 11797–11801. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M. Relevance of DNA methylation in the management of cancer. Lancet Oncol. 2003, 4, 351–358. [Google Scholar] [CrossRef]
- Sakai, T.; Toguchida, J.; Ohtani, N.; Yandell, D.W.; Rapaport, J.M.; Dryja, T.P. Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Am. J. Hum. Genet. 1991, 48, 880–888. [Google Scholar] [PubMed]
- Herman, J.G.; Latif, F.; Weng, Y.; Lerman, M.I.; Zbar, B.; Liu, S.; Samid, D.; Duan, D.S.; Gnarra, J.R.; Linehan, W.M.; et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc. Natl. Acad. Sci. USA 1994, 91, 9700–9704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, S.; Heil, O.; Lyko, F.; Brueckner, B. Azacytidine and Decitabine Induce Gene-Specific and Non-Random DNA Demethylation in Human Cancer Cell Lines. PLoS ONE 2011, 6, e17388. [Google Scholar] [CrossRef]
- Schneider-Stock, R.; Diab-Assef, M.; Rohrbeck, A.; Foltzer-Jourdainne, C.; Boltze, C.; Hartig, R.; Schönfeld, P.; Roessner, A.; Gali-Muhtasib, H. 5-aza-Cytidine Is a Potent Inhibitor of DNA Methyltransferase 3a and Induces Apoptosis in HCT-116 Colon Cancer Cells via Gadd45- and p53-Dependent Mechanisms. J. Pharmacol. Exp. Ther. 2005, 312, 525–536. [Google Scholar] [CrossRef] [Green Version]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef] [Green Version]
- Walter, M.J.; Ding, L.; Shen, D.; Shao, J.; Grillot, M.; McLellan, M.; Fulton, R.; Schmidt, H.; Kalicki-Veizer, J.; O’Laughlin, M.; et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 2011, 25, 1153–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, M.; Meacham, A.M.; Hamazaki, T.; Rodic, N.; Chang, L.J.; Terada, N. De novo DNA methyltransferases Dnmt3a and Dnmt3b primarily mediate the cytotoxic effect of 5-aza-2’-deoxycytidine. Oncogene 2005, 24, 3091–3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, G.; Brown, A.; Mudd, G.; Huxley, P.; Van Rietschoten, K.; Pavan, S.; Chen, L.; Watcham, S.; Lahdenranta, J.; Keen, N. MMAE Delivery Using the Bicycle Toxin Conjugate BT5528. Mol. Cancer Ther. 2020, 19, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Tohme, R.; Tomlinson, B.; Sakre, N.; Hasipek, M.; Durkin, L.; Schuerger, C.; Grabowski, D.; Zidan, A.M.; Radivoyevitch, T.; et al. Decitabine- and 5-azacytidine resistance emerges from adaptive responses of the pyrimidine metabolism network. Leukemia 2021, 35, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Silverman, L.R.; Demakos, E.P.; Peterson, B.L.; Kornblith, A.B.; Holland, J.C.; Odchimar-Reissig, R.; Stone, R.M.; Nelson, D.; Powell, B.L.; DeCastro, C.M.; et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: A study of the cancer and leukemia group B. J. Clin. Oncol. 2002, 20, 2429–2440. [Google Scholar] [CrossRef]
- Fenaux, P.; Mufti, G.J.; Hellström-Lindberg, E.; Santini, V.; Gattermann, N.; Germing, U.; Sanz, G.; List, A.F.; Gore, S.; Seymour, J.F.; et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 562–569. [Google Scholar] [CrossRef]
- Adès, L.; Sekeres, M.A.; Wolfromm, A.; Teichman, M.L.; Tiu, R.V.; Itzykson, R.; Maciejewski, J.P.; Dreyfus, F.; List, A.F.; Fenaux, P.; et al. Predictive factors of response and survival among chronic myelomonocytic leukemia patients treated with azacitidine. Leuk. Res. 2013, 37, 609–613. [Google Scholar] [CrossRef]
- Campbell, K.J.; Dhayade, S.; Ferrari, N.; Sims, A.H.; Johnson, E.; Mason, S.M.; Dickson, A.; Ryan, K.M.; Kalna, G.; Edwards, J.; et al. MCL-1 is a prognostic indicator and drug target in breast cancer. Cell Death Dis. 2018, 9, 19. [Google Scholar] [CrossRef]
- Sabatino, M.A.; Geroni, C.; Ganzinelli, M.; Ceruti, R.; Broggini, M. Zebularine partially reverses GST methylation in prostate cancer cells and restores sensitivity to the DNA minor groove binder brostallicin. Epigenetics 2013, 8, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Sanaei, M.; Kavoosi, F.; Behjoo, H. Effect of valproic acid and zebularine on SOCS-1 and SOCS-3 gene expression in colon carcinoma SW48 cell line. Exp. Oncol. 2020, 42, 183–187. [Google Scholar] [CrossRef]
- Sanaei, M.; Kavoosi, F.; Hosseini, F. Effect of Zebularine on p16INK4a, p14ARF, p15INK4b, and DNA Methyltransferase 1 Gene Expression, Cell Growth Inhibition, and Apoptosis Induction in Human Hepatocellular Carcinoma PLC/PRF5 and Pancreatic Cancer PA-TU-8902 Cell Lines. Iran. J. Pharm. Res. IJPR 2020, 19, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Andrade, A.F.; Borges, K.S.; Suazo, V.K.; Geron, L.; Corrêa, C.A.; Castro-Gamero, A.M.; de Vasconcelos, E.J.; de Oliveira, R.S.; Neder, L.; Yunes, J.A.; et al. The DNA methyltransferase inhibitor zebularine exerts antitumor effects and reveals BATF2 as a poor prognostic marker for childhood medulloblastoma. Investig. New Drugs 2017, 35, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liang, N.; Yang, T.; Li, Y.; Li, J.; Huang, Q.; Wu, C.; Sun, L.; Zhou, X.; Cheng, X.; et al. DNMT1-mediated methylation of BEX1 regulates stemness and tumorigenicity in liver cancer. J. Hepatol. 2021, 75, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-S. Constructing host/pathogen genetic-and-epigenetic networks for investigating molecular mechanisms to identify drug targets in the infection of Epstein–Barr virus via big data mining and genome-wide NGS data identification. In Systems Immunology and Infection Microbiology, 1st ed.; Academic Press: Salt Lake City, UT, USA, 2021; pp. 489–557. [Google Scholar]
- Cheng, J.C.; Matsen, C.B.; Gonzales, F.A.; Ye, W.; Greer, S.; Marquez, V.E.; Jones, P.A.; Selker, E.U. Inhibition of DNA Methylation and Reactivation of Silenced Genes by Zebularine. JNCI J. Natl. Cancer Inst. 2003, 95, 399–409. [Google Scholar] [CrossRef]
- Momparler, R.L.; Ayoub, J. Potential of 5-aza-2’-deoxycytidine (Decitabine) a potent inhibitor of DNA methylation for therapy of advanced non-small cell lung cancer. Lung Cancer 2001, 34 (Suppl. 4), S111–S115. [Google Scholar] [CrossRef]
- Bu, F.; Zhu, X.; Liu, S.; Lin, K.; Zhu, J.; Huang, J. Comprehensive analysis of Syk gene methylation in colorectal cancer. Immun. Inflamm. Dis. 2021, 9, 923–931. [Google Scholar] [CrossRef]
- Bévant, K.; Desoteux, M.; Abdel Wahab, A.H.A.; Abdel Wahab, S.A.; Metwally, A.M.; Coulouarn, C. DNA Methylation of TGFβ Target Genes: Epigenetic Control of TGFβ Functional Duality in Liver Cancer. Cells 2021, 10, 2207. [Google Scholar] [CrossRef]
- Buocikova, V.; Longhin, E.M.; Pilalis, E.; Mastrokalou, C.; Miklikova, S.; Cihova, M.; Poturnayova, A.; Mackova, K.; Babelova, A.; Trnkova, L.; et al. Decitabine potentiates efficacy of doxorubicin in a preclinical trastuzumab-resistant HER2-positive breast cancer models. Biomed. Pharm. 2022, 147, 112662. [Google Scholar] [CrossRef]
- Su, Y.; Huang, Q.; Lu, L.; Qu, H.; Wang, D.; Qiu, J.; Li, W.; Lin, M.; Liu, H.; Wang, Z.; et al. Promoter Methylation-Mediated NPTX2 Silencing Promotes Tumor Growth in Human Prostate Cancer. J. Cancer 2022, 13, 706–714. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, L.W.; Lu, C.Q.; Wang, T.Z.; Shan, M.; Xiao, J.Y.; Tian, H.; Ma, X.; Xu, Y.; Wu, D.P. Efficacy and safety of venetoclax combined with azacitidine versus CAG regimen combined with decitabine in elderly patients with relapsed acute myeloid leukemia. Zhonghua Nei Ke Za Zhi 2022, 61, 157–163. [Google Scholar] [CrossRef]
- El Khawanky, N.; Hughes, A.; Yu, W.; Myburgh, R.; Matschulla, T.; Taromi, S.; Aumann, K.; Clarson, J.; Vinnakota, J.M.; Shoumariyeh, K.; et al. Demethylating therapy increases anti-CD123 CAR T cell cytotoxicity against acute myeloid leukemia. Nat. Commun. 2021, 12, 6436. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Cojocari, D.; Purkal, J.J.; Popovic, R.; Talaty, N.N.; Xiao, Y.; Solomon, L.R.; Boghaert, E.R.; Leverson, J.D.; Phillips, D.C. 5-Azacitidine Induces NOXA to Pri.ime AML Cells for Venetoclax-Mediated Apoptosis. Clin. Cancer Res. 2020, 26, 3371–3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezaei, S.; Hosseinpourfeizi, M.A.; Moaddab, Y.; Safaralizadeh, R. Contribution of DNA methylation and EZH2 in SRBC down-regulation in gastric cancer. Mol. Biol. Rep. 2020, 47, 5721–5727. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.T.; Tao, Q.; Robertson, K.D.; Flinn, I.W.; Mann, R.B.; Klencke, B.; Kwan, W.H.; Leung, T.W.; Johnson, P.J.; Ambinder, R.F. Azacitidine induces demethylation of the Epstein-Barr virus genome in tumors. J. Clin. Oncol. 2004, 22, 1373–1381. [Google Scholar] [CrossRef]
- Oing, C.; Verem, I.; Mansour, W.Y.; Bokemeyer, C.; Dyshlovoy, S.; Honecker, F. 5-Azacitidine Exerts Prolonged Pro-Apoptotic Effects and Overcomes Cisplatin-Resistance in Non-Seminomatous Germ Cell Tumor Cells. Int. J. Mol. Sci. 2018, 20, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, B.J.; Shah, M.A.; Klute, K.; Ocean, A.; Popa, E.; Altorki, N.; Lieberman, M.; Schreiner, A.; Yantiss, R.; Christos, P.J.; et al. Phase I Study of Epigenetic Priming with Azacitidine Prior to Standard Neoadjuvant Chemotherapy for Patients with Resectable Gastric and Esophageal Adenocarcinoma: Evidence of Tumor Hypomethylation as an Indicator of Major Histopathologic Response. Clin. Cancer Res. 2017, 23, 2673–2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, N.; Sajed, D.; Arora, K.S.; Solovyov, A.; Rajurkar, M.; Bledsoe, J.R.; Sil, S.; Amri, R.; Tai, E.; MacKenzie, O.C.; et al. Diverse repetitive element RNA expression defines epigenetic and immunologic features of colon cancer. JCI Insight 2017, 2, e91078. [Google Scholar] [CrossRef] [Green Version]
- Fang, F.; Munck, J.; Tang, J.; Taverna, P.; Wang, Y.; Miller, D.F.; Pilrose, J.; Choy, G.; Azab, M.; Pawelczak, K.S.; et al. The novel, small-molecule DNA methylation inhibitor SGI-110 as an ovarian cancer chemosensitizer. Clin. Cancer Res. 2014, 20, 6504–6516. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Oliveras, A.; Izquierdo-Torres, E.; Hernández-Martínez, G.; Zarain-Herzberg, Á.; Santiago-García, J. Transcriptional and epigenetic landscape of Ca(2+)-signaling genes in hepatocellular carcinoma. J. Cell Commun. Signal. 2021, 15, 433–445. [Google Scholar] [CrossRef]
- Crabb, S.J.; Danson, S.; Catto, J.W.F.; Hussain, S.; Chan, D.; Dunkley, D.; Downs, N.; Marwood, E.; Day, L.; Saunders, G.; et al. Phase I Trial of DNA Methyltransferase Inhibitor Guadecitabine Combined with Cisplatin and Gemcitabine for Solid Malignancies Including Urothelial Carcinoma (SPIRE). Clin. Cancer Res. 2021, 27, 1882–1892. [Google Scholar] [CrossRef]
- Albany, C.; Fazal, Z.; Singh, R.; Bikorimana, E.; Adra, N.; Hanna, N.H.; Einhorn, L.H.; Perkins, S.M.; Sandusky, G.E.; Christensen, B.C.; et al. A phase 1 study of combined guadecitabine and cisplatin in platinum refractory germ cell cancer. Cancer Med. 2021, 10, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Fan, J.; Hong, W.; Li, L.; Wu, M. Inhibition of cancer cell proliferation by 5-fluoro-2′-deoxycytidine, a DNA methylation inhibitor, through activation of DNA damage response pathway. SpringerPlus 2012, 1, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coyne, G.O.S.; Wang, L.; Zlott, J.; Juwara, L.; Covey, J.M.; Beumer, J.H.; Cristea, M.C.; Newman, E.M.; Koehler, S.; Nieva, J.J.; et al. Intravenous 5-fluoro-2′-deoxycytidine administered with tetrahydrouridine increases the proportion of p16-expressing circulating tumor cells in patients with advanced solid tumors. Cancer Chemother. Pharm. 2020, 85, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Matoušová, M.; Votruba, I.; Otmar, M.; Tloušťová, E.; Günterová, J.; Mertlíková-Kaiserová, H. 2′-deoxy-5,6-dihydro-5-azacytidine-a less toxic alternative of 2′-deoxy-5-azacytidine. Epigenetics 2011, 6, 769–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hummel-Eisenbeiss, J.; Hascher, A.; Hals, P.A.; Sandvold, M.L.; Müller-Tidow, C.; Lyko, F.; Rius, M. The role of human equilibrative nucleoside transporter 1 on the cellular transport of the DNA methyltransferase inhibitors 5-azacytidine and CP-4200 in human leukemia cells. Mol. Pharmacol. 2013, 84, 438–450. [Google Scholar] [CrossRef] [Green Version]
- Brueckner, B.; Rius, M.; Markelova, M.R.; Fichtner, I.; Hals, P.-A.; Sandvold, M.L.; Lyko, F. Delivery of 5-Azacytidine to Human Cancer Cells by Elaidic Acid Esterification Increases Therapeutic Drug Efficacy. Mol. Cancer Ther. 2010, 9, 1256–1264. [Google Scholar] [CrossRef] [Green Version]
- Candelaria, M.; de la Cruz-Hernandez, E.; Taja-Chayeb, L.; Perez-Cardenas, E.; Trejo-Becerril, C.; Gonzalez-Fierro, A.; Chavez-Blanco, A.; Soto-Reyes, E.; Dominguez, G.; Trujillo, J.E.; et al. DNA methylation-independent reversion of gemcitabine resistance by hydralazine in cervical cancer cells. PLoS ONE 2012, 7, e29181. [Google Scholar] [CrossRef]
- Lu, C.; Yang, D.; Sabbatini, M.E.; Colby, A.H.; Grinstaff, M.W.; Oberlies, N.H.; Pearce, C.; Liu, K. Contrasting roles of H3K4me3 and H3K9me3 in regulation of apoptosis and gemcitabine resistance in human pancreatic cancer cells. BMC Cancer 2018, 18, 149. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Zhang, B.; Gu, G.; Yang, X.; Qian, Z. Metformin increases the chemosensitivity of pancreatic cancer cells to gemcitabine by reversing EMT through regulation DNA methylation of miR-663. OncoTargets Ther. 2020, 13, 10417. [Google Scholar] [CrossRef]
- Stubbe, B.E.; Henriksen, S.D.; Madsen, P.H.; Larsen, A.C.; Krarup, H.B.; Pedersen, I.S.; Johansen, M.N.; Thorlacius-Ussing, O. Validation of SFRP1 Promoter Hypermethylation in Plasma as a Prognostic Marker for Survival and Gemcitabine Effectiveness in Patients with Stage IV Pancreatic Adenocarcinoma. Cancers 2021, 13, 5717. [Google Scholar] [CrossRef]
- Baretti, M.; Karunasena, E.; Zahurak, M.; Walker, R.; Zhao, Y.; Pisanic, T.R., 2nd; Wang, T.-H.; Greten, T.F.; Duffy, A.G.; Gootjes, E.; et al. A phase 2 trial of gemcitabine and docetaxel in patients with metastatic colorectal adenocarcinoma with methylated checkpoint with forkhead and ring finger domain promoter and/or microsatellite instability phenotype. Clin. Transl. Sci. 2021, 14, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Enokida, H.; Osako, Y.; Nohata, N.; Yonemori, M.; Sugita, S.; Kuroshima, K.; Tsuruda, M.; Tatarano, S.; Nakagawa, M. Characterization of PHGDH expression in bladder cancer: Potential targeting therapy with gemcitabine/cisplatin and the contribution of promoter DNA hypomethylation. Mol. Oncol. 2020, 14, 2190–2202. [Google Scholar] [CrossRef] [PubMed]
- Honeywell, R.J.; Sarkisjan, D.; Kristensen, M.H.; de Klerk, D.J.; Peters, G.J. DNA methyltransferases expression in normal tissues and various human cancer cell lines, xenografts and tumors. Nucleosides Nucleotides Nucleic Acids 2018, 37, 696–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkisjan, D.; Julsing, J.R.; El Hassouni, B.; Honeywell, R.J.; Kathmann, I.; Matherly, L.H.; Lee, Y.B.; Kim, D.J.; Peters, G.J. RX-3117 (Fluorocyclopentenyl-Cytosine)-Mediated Down-Regulation of DNA Methyltransferase 1 Leads to Protein Expression of Tumor-Suppressor Genes and Increased Functionality of the Proton-Coupled Folate Carrier. Int. J. Mol. Sci. 2020, 21, 2717. [Google Scholar] [CrossRef] [PubMed]
- Lopes, N.; Pacheco, M.B.; Soares-Fernandes, D.; Correia, M.P.; Camilo, V.; Henrique, R.; Jerónimo, C. Hydralazine and Enzalutamide: Synergistic Partners against Prostate Cancer. Biomedicines 2021, 9, 976. [Google Scholar] [CrossRef]
- Bauman, J.; Shaheen, M.; Verschraegen, C.F.; Belinsky, S.A.; Houman Fekrazad, M.; Lee, F.-C.; Rabinowitz, I.; Ravindranathan, M.; Jones, D.V. A Phase I Protocol of Hydralazine and Valproic Acid in Advanced, Previously Treated Solid Cancers. Transl. Oncol. 2014, 7, 349–354. [Google Scholar] [CrossRef]
- Kumanishi, S.; Yamanegi, K.; Nishiura, H.; Fujihara, Y.; Kobayashi, K.; Nakasho, K.; Futani, H.; Yoshiya, S. Epigenetic modulators hydralazine and sodium valproate act synergistically in VEGI-mediated anti-angiogenesis and VEGF interference in human osteosarcoma and vascular endothelial cells. Int. J. Oncol 2019, 55, 167–178. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Choi, W.; Lee, E.S. Therapeutic Targ.geting of DNA Damage Response in Cancer. Int. J. Mol. Sci. 2022, 23, 1701. [Google Scholar] [CrossRef]
- Hanna, D.; Chopra, N.; Hochhauser, D.; Khan, K. The role of PARP inhibitors in gastrointestinal cancers. Crit. Rev. Oncol. 2022, 171, 103621. [Google Scholar] [CrossRef]
- Li, F.; Wu, X.; Fu, X.; Liu, J.; Song, W.; Xiao, G.G.; Lu, A.; Zhang, G. Poly (ADP-ribose) polymerase 1 (PARP1) inhibition promotes pulmonary metastasis of osteosarcoma by boosting ezrin phosphorylation. Int. J. Biol. Sci. 2022, 18, 1238–1253. [Google Scholar] [CrossRef] [PubMed]
- Sha, H.; Gan, Y.; Zou, R.; Wu, J.; Feng, J. Research Advances in the Role of the Poly ADP Ribose Polymerase Family in Cancer. Front. Oncol. 2021, 11, 790967. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, K.; Xiong, H.; Huang, Y.; Chen, X.; Zhou, Y.; Qin, W.; Su, J.; Chen, R.; Qiu, H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Provides a New Combination Therapy in Pancreatic Cancer. Front. Immunol. 2021, 12, 762989. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Wang, M.; Wang, Q.; Han, Z. Dual targeting, a new strategy for novel PARP inhibitor discovery. Drug Discov. Ther. 2021, 15, 300–309. [Google Scholar] [CrossRef]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Chen, C.; Yu, Y. Avoid the trap: Targeting PARP1 beyond human malignancy. Cell Chem. Biol. 2021, 28, 456–462. [Google Scholar] [CrossRef]
- Martínez-Morcillo, F.J.; Cantón-Sandoval, J.; Martínez-Menchón, T.; Corbalán-Vélez, R.; Mesa-Del-Castillo, P.; Pérez-Oliva, A.B.; García-Moreno, D.; Mulero, V. Non-canonical roles of NAMPT and PARP in inflammation. Dev. Comp. Immunol. 2021, 115, 103881. [Google Scholar] [CrossRef]
- Biegała, Ł.; Gajek, A.; Marczak, A.; Rogalska, A. PARP inhibitor resistance in ovarian cancer: Underlying mechanisms and therapeutic approaches targeting the ATR/CHK1 pathway. Biochim. Biophys. Acta. 2021, 1876, 188633. [Google Scholar] [CrossRef]
- Kim, D.S.; Camacho, C.V.; Kraus, W.L. Alternate therapeutic pathways for PARP inhibitors and potential mechanisms of resistance. Exp. Mol. Med. 2021, 53, 42–51. [Google Scholar] [CrossRef]
- Ngoi, N.Y.L.; Leo, E.; O’Connor, M.J.; Yap, T.A. Development of Next-Generation Poly(ADP-Ribose) Polymerase 1-Selective Inhibitors. Cancer J. 2021, 27, 521–528. [Google Scholar] [CrossRef]
- Bird, A.W.; Yu, D.Y.; Pray-Grant, M.G.; Qiu, Q.; Harmon, K.E.; Megee, P.C.; Grant, P.A.; Smith, M.M.; Christman, M.F. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature 2002, 419, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.J.; Kotova, E.; Andrake, M.; Adolf-Bryfogle, J.; Glaser, R.; Regnard, C.; Tulin, A.V. Kinase-mediated changes in nucleosome conformation trigger chromatin decondensation via poly(ADP-ribosyl)ation. Mol. Cell 2014, 53, 831–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caracciolo, D.; Riillo, C.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P. Alternative Non-Homologous End-Joining: Error-Prone DNA Repair as Cancer’s Achilles’ Heel. Cancers 2021, 13, 1392. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, J. Targeted Cancer Therapy Based on Acetylation and Deacetylation of Key Proteins Involved in Double-Strand Break Repair. Cancer Manag. Res. 2022, 14, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Makhov, P.; Uzzo, R.G.; Tulin, A.V.; Kolenko, V.M. Histone-dependent PARP-1 inhibitors: A novel therapeutic modality for the treatment of prostate and renal cancers. Urol. Oncol. 2021, 39, 312–315. [Google Scholar] [CrossRef]
- Thomas, C.; Ji, Y.; Lodhi, N.; Kotova, E.; Pinnola, A.D.; Golovine, K.; Makhov, P.; Pechenkina, K.; Kolenko, V.; Tulin, A.V. Non-NAD-Like poly(ADP-Ribose) Polymerase-1 Inhibitors effectively Eliminate Cancer in vivo. EBioMedicine 2016, 13, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Karpova, Y.; Guo, D.; Makhov, P.; Haines, A.M.; Markov, D.A.; Kolenko, V.; Tulin, A.V. Poly(ADP)-Ribosylation Inhibition: A Promising Approach for Clear Cell Renal Cell Carcinoma Therapy. Cancers 2021, 13, 4973. [Google Scholar] [CrossRef]
- Nieborowska-Skorska, M.; Maifrede, S.; Ye, M.; Toma, M.; Hewlett, E.; Gordon, J.; Le, B.V.; Sliwinski, T.; Zhao, H.; Piwocka, K.; et al. Non-NAD-like PARP1 inhibitor enhanced synthetic lethal effect of NAD-like PARP inhibitors against BRCA1-deficient leukemia. Leuk. Lymphoma. 2019, 60, 1098–1101. [Google Scholar] [CrossRef]
- Vidal, E.; Sayols, S.; Moran, S.; Guillaumet-Adkins, A.; Schroeder, M.P.; Royo, R.; Orozco, M.; Gut, M.; Gut, I.; Lopez-Bigas, N.; et al. A DNA methylation map of human cancer at single base-pair resolution. Oncogene 2017, 36, 5648–5657. [Google Scholar] [CrossRef]
- Burton, G.W.; Foster, D.O.; Perly, B.; Slater, T.F.; Smith, I.C.; Ingold, K.U. Biological antioxidants. Philos. Trans. R. Soc. B. Biol. Sci. 1985, 311, 565–578. [Google Scholar] [CrossRef]
- Chiappa, M.; Guffanti, F.; Bertoni, F.; Colombo, I.; Damia, G. Overcoming PARPi resistance: Preclinical and clinical evidence in ovarian cancer. Drug Resist. Updat. 2021, 55, 100744. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, E.A.; Litton, J.K.; Yap, T.A. Development of the PARP inhibitor talazoparib for the treatment of advanced BRCA1 and BRCA2 mutated breast cancer. Expert Opin. Pharmacother. 2021, 22, 1825–1837. [Google Scholar] [CrossRef]
- Zhou, P.; Wang, J.; Mishail, D.; Wang, C.Y. Recent advancements in PARP inhibitors-based targeted cancer therapy. Precis. Clin. Med. 2020, 3, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Bamezai, S.; Demir, D.; Pulikkottil, A.J.; Ciccarone, F.; Fischbein, E.; Sinha, A.; Borga, C.; Te Kronnie, G.; Meyer, L.H.; Mohr, F.; et al. TET1 promotes growth of T-cell acute lymphoblastic leukemia and can be antagonized via PARP inhibition. Leukemia 2021, 35, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Farrar, D.; Rai, S.; Chernukhin, I.; Jagodic, M.; Ito, Y.; Yammine, S.; Ohlsson, R.; Murrell, A.; Klenova, E. Mutational analysis of the poly(ADP-ribosyl)ation sites of the transcription factor CTCF provides an insight into the mechanism of its regulation by poly(ADP-ribosyl)ation. Mol. Cell Biol. 2010, 30, 1199–1216. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Ginjala, V.; Pant, V.; Chernukhin, I.; Whitehead, J.; Docquier, F.; Farrar, D.; Tavoosidana, G.; Mukhopadhyay, R.; Kanduri, C.; et al. Poly(ADP-ribosyl)ation regulates CTCF-dependent chromatin insulation. Nat. Genet. 2004, 36, 1105–1110. [Google Scholar] [CrossRef] [Green Version]
- Zardo, G.; Caiafa, P. The unmethylated state of CpG islands in mouse fibroblasts depends on the poly(ADP-ribosyl)ation process. J. Biol. Chem. 1998, 273, 16517–16520. [Google Scholar] [CrossRef] [Green Version]
- de Capoa, A.; Febbo, F.R.; Giovannelli, F.; Niveleau, A.; Zardo, G.; Marenzi, S.; Caiafa, P. Reduced levels of poly(ADP-ribosyl)ation result in chromatin compaction and hypermethylation as shown by cell-by-cell computer-assisted quantitative analysis. FASEB J. 1999, 13, 89–93. [Google Scholar] [CrossRef]
- Camps, M.; Eichman, B.F. Unraveling a connection between DNA demethylation repair and cancer. Mol. Cell 2011, 44, 343–344. [Google Scholar] [CrossRef] [Green Version]
- Christmann, M.; Kaina, B. Epigenetic regulation of DNA repair genes and implications for tumor therapy. Mutat. Res. Rev. Mutat. Res. 2019, 780, 15–28. [Google Scholar] [CrossRef]
- Srivastava, R.; Mishra, N.; Singh, U.M.; Srivastava, R. Genotoxicity: Mechanisms and its impact on human diseases. Octa J. Biosci. 2016, 4, 67–70. [Google Scholar]
- Scardocci, A.; Guidi, F.; D’Alo, F.; Gumiero, D.; Fabiani, E.; Diruscio, A.; Martini, M.; Larocca, L.M.; Zollino, M.; Hohaus, S.; et al. Reduced BRCA1 expression due to promoter hypermethylation in therapy-related acute myeloid leukaemia. Br. J. Cancer 2006, 95, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents-A Potential Therapy for Cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontandreopoulou, C.N.; Diamantopoulos, P.T.; Tiblalexi, D.; Giannakopoulou, N.; Viniou, N.A. PARP1 as a therapeutic target in acute myeloid leukemia and myelodysplastic syndrome. Blood Adv. 2021, 5, 4794–4805. [Google Scholar] [CrossRef] [PubMed]
- Baer, M.R.; Kogan, A.A.; Bentzen, S.M.; Mi, T.; Lapidus, R.G.; Duong, V.H.; Emadi, A.; Niyongere, S.; O’Connell, C.L.; Youngblood, B.A.; et al. Phase I clinical trial of DNA methyltransferase inhibitor decitabine and PARP inhibitor talazoparib combination therapy in relapsed/refractory acute myeloid leukemia. Clin. Cancer Res. 2022. [Google Scholar] [CrossRef]
- Li, X.; Li, C.; Jin, J.; Wang, J.; Huang, J.; Ma, Z.; Huang, X.; He, X.; Zhou, Y.; Xu, Y.; et al. High PARP-1 expression predicts poor survival in acute myeloid leukemia and PARP-1 inhibitor and SAHA-bendamustine hybrid inhibitor combination treatment synergistically enhances anti-tumor effects. EBioMedicine 2018, 38, 47–56. [Google Scholar] [CrossRef]
- Iacobucci, I.; Qu, C.; Varotto, E.; Janke, L.J.; Yang, X.; Seth, A.; Shelat, A.; Friske, J.D.; Fukano, R.; Yu, J.; et al. Modeling and targeting of erythroleukemia by hematopoietic genome editing. Blood 2021, 137, 1628–1640. [Google Scholar] [CrossRef]
- Barber, L.J.; Sandhu, S.; Chen, L.; Campbell, J.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Rodrigues, D.N.; Reis Filho, J.S.; Moreno, V.; et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J. Pathol. 2013, 229, 422–429. [Google Scholar] [CrossRef]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat. Med. 2013, 19, 1381–1388. [Google Scholar] [CrossRef]
- Pulliam, N.; Fang, F.; Ozes, A.R.; Tang, J.; Adewuyi, A.; Keer, H.; Lyons, J.; Baylin, S.B.; Matei, D.; Nakshatri, H.; et al. An Effective Epigenetic-PARP Inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA Mutations. Clin. Cancer Res. 2018, 24, 3163–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Li, X.; Cassady, K.; Zou, Z.; Zhang, X. TET2 Function in Hematopoietic Malignancies, Immune Regulation, and DNA Repair. Front. Oncol 2019, 9, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattopadhyaya, S.; Ghosal, S. DNA methylation: A saga of genome maintenance in hematological perspective. Hum. Cell 2022, 35, 448–461. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Seong, H.; Liu, Y.; Yu, X.; Xu, S.; Li, Y. Ten-eleven translocation proteins (TETs): Tumor suppressors or tumor enhancers? Front. Biosci. Landmark Ed. 2021, 26, 895–915. [Google Scholar] [CrossRef]
- Harrision, D.; Gravells, P.; Thompson, R.; Bryant, H.E. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP)-Function in Genome Maintenance and Relevance of Inhibitors for Anti-cancer Therapy. Front. Mol. Biosci. 2020, 7, 191. [Google Scholar] [CrossRef]
- Slade, D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef] [Green Version]
- Tanuma, S.I.; Shibui, Y.; Oyama, T.; Uchiumi, F.; Abe, H. Targeting poly(ADP-ribose) glycohydrolase to draw apoptosis codes in cancer. Biochem. Pharm. 2019, 167, 163–172. [Google Scholar] [CrossRef]
- Agarwal, N.; Azad, A.; Shore, N.D.; Carles, J.; Fay, A.P.; Dunshee, C.; Karsh, L.I.; Paccagnella, M.L.; Santo, N.D.; Elmeliegy, M.; et al. Talazoparib plus enzalutamide in metastatic castration-resistant prostate cancer: TALAPRO-2 phase III study design. Future Oncol. 2022, 18, 425–436. [Google Scholar] [CrossRef]
- Fenton, S.E.; Chalmers, Z.R.; Hussain, M. PARP Inhibition in Advanced Prostate Cancer. Cancer J. 2021, 27, 457–464. [Google Scholar] [CrossRef]
- Rao, A.; Moka, N.; Hamstra, D.A.; Ryan, C.J. Co-Inhibition of Androgen Receptor and PARP as a Novel Treatment Paradigm in Prostate Cancer-Where Are We Now? Cancers 2022, 14, 801. [Google Scholar] [CrossRef]
- Mandumpala, J.J.; Baby, S.; Tom, A.A.; Godugu, C.; Shankaraiah, N. Role of histone demethylases and histone methyltransferases in triple-negative breast cancer: Epigenetic mnemonics. Life Sci. 2022, 292, 120321. [Google Scholar] [CrossRef] [PubMed]
- Good, C.R.; Panjarian, S.; Kelly, A.D.; Madzo, J.; Patel, B.; Jelinek, J.; Issa, J.J. TET1-Mediated Hypomethylation Activates Oncogenic Signaling in Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 4126–4137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ossovskaya, V.; Koo, I.C.; Kaldjian, E.P.; Alvares, C.; Sherman, B.M. Upregulation of Poly (ADP-Ribose) Polymerase-1 (PARP1) in Triple-Negative Breast Cancer and Other Primary Human Tumor Types. Genes Cancer 2010, 1, 812–821. [Google Scholar] [CrossRef]
- Sultan, M.; Schulz, M.H.; Richard, H.; Magen, A.; Klingenhoff, A.; Scherf, M.; Seifert, M.; Borodina, T.; Soldatov, A.; Parkhomchuk, D.; et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 2008, 321, 956–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.Q.; Auer, B.; Stingl, L.; Berghammer, H.; Haidacher, D.; Schweiger, M.; Wagner, E.F. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995, 9, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ménissier de Murcia, J.; Ricoul, M.; Tartier, L.; Niedergang, C.; Huber, A.; Dantzer, F.; Schreiber, V.; Amé, J.C.; Dierich, A.; LeMeur, M.; et al. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003, 22, 2255–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342, 249–268. [Google Scholar] [CrossRef]
- Csizmar, C.M.; Saliba, A.N.; Swisher, E.M.; Kaufmann, S.H. PARP Inhibitors and Myeloid Neoplasms: A Double-Edged Sword. Cancers 2021, 13, 6385. [Google Scholar] [CrossRef]
- Faraoni, I.; Giansanti, M.; Voso, M.T.; Lo-Coco, F.; Graziani, G. Targeting ADP-ribosylation by PARP inhibitors in acute myeloid leukaemia and related disorders. Biochem. Pharm. 2019, 167, 133–148. [Google Scholar] [CrossRef]
- Padella, A.; Ghelli Luserna Di Rorà, A.; Marconi, G.; Ghetti, M.; Martinelli, G.; Simonetti, G. Targeting PARP proteins in acute leukemia: DNA damage response inhibition and therapeutic strategies. J. Hematol. Oncol. 2022, 15, 10. [Google Scholar] [CrossRef]
- Park, S.; Kim, Y.J.; Huh, H.J.; Chung, H.S.; Lee, M.; Park, Y.M.; Mun, Y.C.; Seong, C.M.; Huh, J. Comprehensive DNA repair gene expression analysis and its prognostic significance in acute myeloid leukemia. Hematology 2021, 26, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Collet, L.; Péron, J.; Penault-Llorca, F.; Pujol, P.; Lopez, J.; Freyer, G.; You, B. PARP Inhibitors: A Major Therapeutic Option in Endocrine-Receptor Positive Breast Cancers. Cancers 2022, 14, 599. [Google Scholar] [CrossRef] [PubMed]
- Mekhaeil, M.; Dev, K.K.; Conroy, M.J. Existing Evidence for the Repurposing of PARP-1 Inhibitors in Rare Demyelinating Diseases. Cancers 2022, 14, 687. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Peng, Y.; Wu, L.C.; Xie, Z.; Deng, Y.; Hughes, T.; He, S.; Mo, X.; Chiu, M.; Wang, Q.E.; et al. Curcumin down-regulates DNA methyltransferase 1 and plays an anti-leukemic role in acute myeloid leukemia. PLoS ONE 2013, 8, e55934. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Meeran, S.M.; Patel, S.N.; Chen, H.; Hardy, T.M.; Tollefsbol, T.O. Epigenetic reactivation of estrogen receptor-α (ERα) by genistein enhances hormonal therapy sensitivity in ERα-negative breast cancer. Mol. Cancer 2013, 12, 9. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Wang, Z.; Wang, Y.; Xie, X.; Shen, J.; Peng, C.; You, J.; Peng, F.; Tang, H.; Guan, X.; et al. Dietary compound isoliquiritigenin prevents mammary carcinogenesis by inhibiting breast cancer stem cells through WIF1 demethylation. Oncotarget 2015, 6, 9854–9876. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.; Zhang, K.; Clarke, K.; Weiland, T.; Sauter, E.R. Methylation and miRNA effects of resveratrol on mammary tumors vs. normal tissue. Nutr. Cancer 2014, 66, 270–277. [Google Scholar] [CrossRef]
- Kanai, Y.; Ushijima, S.; Nakanishi, Y.; Sakamoto, M.; Hirohashi, S. Mutation of the DNA methyltransferase (DNMT) 1 gene in human colorectal cancers. Cancer Lett. 2003, 192, 75–82. [Google Scholar] [CrossRef]
- Kanai, Y.; Ushijima, S.; Kondo, Y.; Nakanishi, Y.; Hirohashi, S. DNA methyltransferase expression and DNA methylation of CPG islands and peri-centromeric satellite regions in human colorectal and stomach cancers. Int. J. Cancer 2001, 91, 205–212. [Google Scholar] [CrossRef]
- Etoh, T.; Kanai, Y.; Ushijima, S.; Nakagawa, T.; Nakanishi, Y.; Sasako, M.; Kitano, S.; Hirohashi, S. Increased DNA methyltransferase 1 (DNMT1) protein expression correlates significantly with poorer tumor differentiation and frequent DNA hypermethylation of multiple CpG islands in gastric cancers. Am. J. Pathol. 2004, 164, 689–699. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.-K.; Wu, C.-Y.; Chang, J.-W.; Juan, L.-J.; Hsu, H.-S.; Chen, C.-Y.; Lu, Y.-Y.; Tang, Y.-A.; Yang, Y.-C.; Yang, P.-C. Dysregulation of p53/Sp1 control leads to DNA methyltransferase-1 overexpression in lung cancer. Cancer Res. 2010, 70, 5807–5817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Tang, Q.; Xiao, Q.; Yang, L.; Hann, S.S. Targeting EP4 downstream c-Jun through ERK1/2-mediated reduction of DNMT1 reveals novel mechanism of solamargine-inhibited growth of lung cancer cells. J. Cell Mol. Med. 2017, 21, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Hu, B.; Ni, J.; Wu, J.; Jiang, W.; Chen, C.; Yang, L.; Zeng, Y.; Wan, R.; Hu, G.; et al. Transcriptional repression of SOCS3 mediated by IL-6/STAT3 signaling via DNMT1 promotes pancreatic cancer growth and metastasis. J. Exp. Clin. Cancer Res. 2016, 35, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.S.; Wang, E.L.; Xu, W.F.; Yamada, S.; Yoshimoto, K.; Qian, Z.R.; Shi, L.; Liu, L.L.; Li, X.H. Overexpression of DNA (Cytosine-5)-Methyltransferase 1 (DNMT1) And DNA (Cytosine-5)-Methyltransferase 3A (DNMT3A) Is Associated with Aggressive Behavior and Hypermethylation of Tumor Suppressor Genes in Human Pituitary Adenomas. Med. Sci. Monit. 2018, 24, 4841–4850. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, B.; Huang, Z.; Zhao, D.-W.; Zeng, Q. Shikonin inhibites migration and invasion of thyroid cancer cells by downregulating DNMT1. Med. Sci. Monit. 2018, 24, 661. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.-J.; Xu, J.; Gu, Z.-H.; Pan, C.-M.; Lu, G.; Shen, Y.; Shi, J.-Y.; Zhu, Y.-M.; Tang, L.; Zhang, X.-W. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet. 2011, 43, 309–315. [Google Scholar] [CrossRef]
- Zhou, L.; Fu, L.; Lv, N.; Liu, J.; Li, Y.; Chen, X.; Xu, Q.; Chen, G.; Pang, B.; Wang, L. Methylation-associated silencing of BASP1 contributes to leukemogenesis in t (8; 21) acute myeloid leukemia. Exp. Mol. Med. 2018, 50, 1–8. [Google Scholar]
- Leonard, S.; Pereira, M.; Fox, R.; Gordon, N.; Yap, J.; Kehoe, S.; Luesley, D.; Woodman, C.; Ganesan, R. Over-expression of DNMT3A predicts the risk of recurrent vulvar squamous cell carcinomas. Gynecol. Oncol. 2016, 143, 414–420. [Google Scholar] [CrossRef] [Green Version]
- Qiu, W.; Lin, J.; Zhu, Y.; Zhang, J.; Zeng, L.; Su, M.; Tian, Y. Kaempferol modulates DNA methylation and downregulates DNMT3B in bladder cancer. Cell. Physiol. Biochem. 2017, 41, 1325–1335. [Google Scholar] [CrossRef]
- Roll, J.D.; Rivenbark, A.G.; Jones, W.D.; Coleman, W.B. DNMT3b overexpression contributes to a hypermethylator phenotype in human breast cancer cell lines. Mol. Cancer 2008, 7, 15. [Google Scholar] [CrossRef] [Green Version]
- Lai, S.-C.; Su, Y.-T.; Chi, C.-C.; Kuo, Y.-C.; Lee, K.-F.; Wu, Y.-C.; Lan, P.-C.; Yang, M.-H.; Chang, T.-S.; Huang, Y.-H. DNMT3b/OCT4 expression confers sorafenib resistance and poor prognosis of hepatocellular carcinoma through IL-6/STAT3 regulation. J. Exp. Clin. Cancer Res. 2019, 38, 474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuck, D.; Caulfield, T.; Lyko, F.; Medina-Franco, J.L. Nanaomycin A selectively inhibits DNMT3B and reactivates silenced tumor suppressor genes in human cancer cells. Mol. Cancer Ther. 2010, 9, 3015–3023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Shou, H.; Chen, L.; Gao, W.; Fang, C.; Zhang, P. Effects of ginsenoside Rg3 on epigenetic modification in ovarian cancer cells. Oncol. Rep. 2019, 41, 3209–3218. [Google Scholar] [CrossRef] [PubMed]
- Dolnik, A.; Engelmann, J.C.; Scharfenberger-Schmeer, M.; Mauch, J.; Kelkenberg-Schade, S.; Haldemann, B.; Fries, T.; Kronke, J.; Kuhn, M.W.; Paschka, P.; et al. Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood 2012, 120, e83–e92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gui, Y.; Guo, G.; Huang, Y.; Hu, X.; Tang, A.; Gao, S.; Wu, R.; Chen, C.; Li, X.; Zhou, L.; et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 2011, 43, 875–878. [Google Scholar] [CrossRef]
- Quesada, V.; Conde, L.; Villamor, N.; Ordonez, G.R.; Jares, P.; Bassaganyas, L.; Ramsay, A.J.; Bea, S.; Pinyol, M.; Martinez-Trillos, A.; et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Genet. 2011, 44, 47–52. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef]
- Scourzic, L.; Mouly, E.; Bernard, O.A. TET proteins and the control of cytosine demethylation in cancer. Genome Med. 2015, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Kan, Z.; Jaiswal, B.S.; Stinson, J.; Janakiraman, V.; Bhatt, D.; Stern, H.M.; Yue, P.; Haverty, P.M.; Bourgon, R.; Zheng, J.; et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010, 466, 869–873. [Google Scholar] [CrossRef] [Green Version]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, J.S.; Ju, Y.S.; Lee, W.C.; Shin, J.Y.; Lee, J.K.; Bleazard, T.; Lee, J.; Jung, Y.J.; Kim, J.O.; Shin, J.Y.; et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012, 22, 2109–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]






| PARP1 Inhibitor | Cancer Type | NCT Number * |
|---|---|---|
| Lynparza/Olaparib | Ovarian Cancer Breast Cancer | NCT04041128 NCT04826198 NCT04774406 NCT03462342 NCT04065269 NCT03150576 NCT04582552 NCT04774406 |
| Cyh33 | Ovarian Cancer Breast Cancer Solid Tumor Prostate Cancer Endometrial Cancer | NCT04586335 |
| Talazoparib | Neuroendocrine Tumors | NCT05053854 |
| Rp12146 | Solid Tumor Lung Cancer Breast Cancer Ovarian Cancer | NCT05002868 |
| Niraparib | Advanced Solid Tumors (Excluding Prostate Cancer) Ovarian Cancer Head And Neck Squamous Cell Carcinoma | NCT04267939 NCT04826198 NCT04774406 NCT04734665 NCT04681469 NCT04837209 NCT04774406 |
| Idx-1197 | Solid Tumors | NCT04174716 |
| Talazoparib | Breast Cancer | NCT03990896 NCT04774406 |
| Rucaparib | Solid Tumor | NCT04276376 NCT04774406 |
| Veliparib | Solid Tumors Liver Tumors Lymphomas Prostate Cancer | NCT01434316 NCT01618357 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, R.; Lodhi, N. DNA Methylation Malleability and Dysregulation in Cancer Progression: Understanding the Role of PARP1. Biomolecules 2022, 12, 417. https://doi.org/10.3390/biom12030417
Srivastava R, Lodhi N. DNA Methylation Malleability and Dysregulation in Cancer Progression: Understanding the Role of PARP1. Biomolecules. 2022; 12(3):417. https://doi.org/10.3390/biom12030417
Chicago/Turabian StyleSrivastava, Rakesh, and Niraj Lodhi. 2022. "DNA Methylation Malleability and Dysregulation in Cancer Progression: Understanding the Role of PARP1" Biomolecules 12, no. 3: 417. https://doi.org/10.3390/biom12030417
APA StyleSrivastava, R., & Lodhi, N. (2022). DNA Methylation Malleability and Dysregulation in Cancer Progression: Understanding the Role of PARP1. Biomolecules, 12(3), 417. https://doi.org/10.3390/biom12030417