
Role of Ion Channel Remodeling in Endothelial Dysfunction Induced by Pulmonary Arterial Hypertension

 , , ,
, , ,  and
and 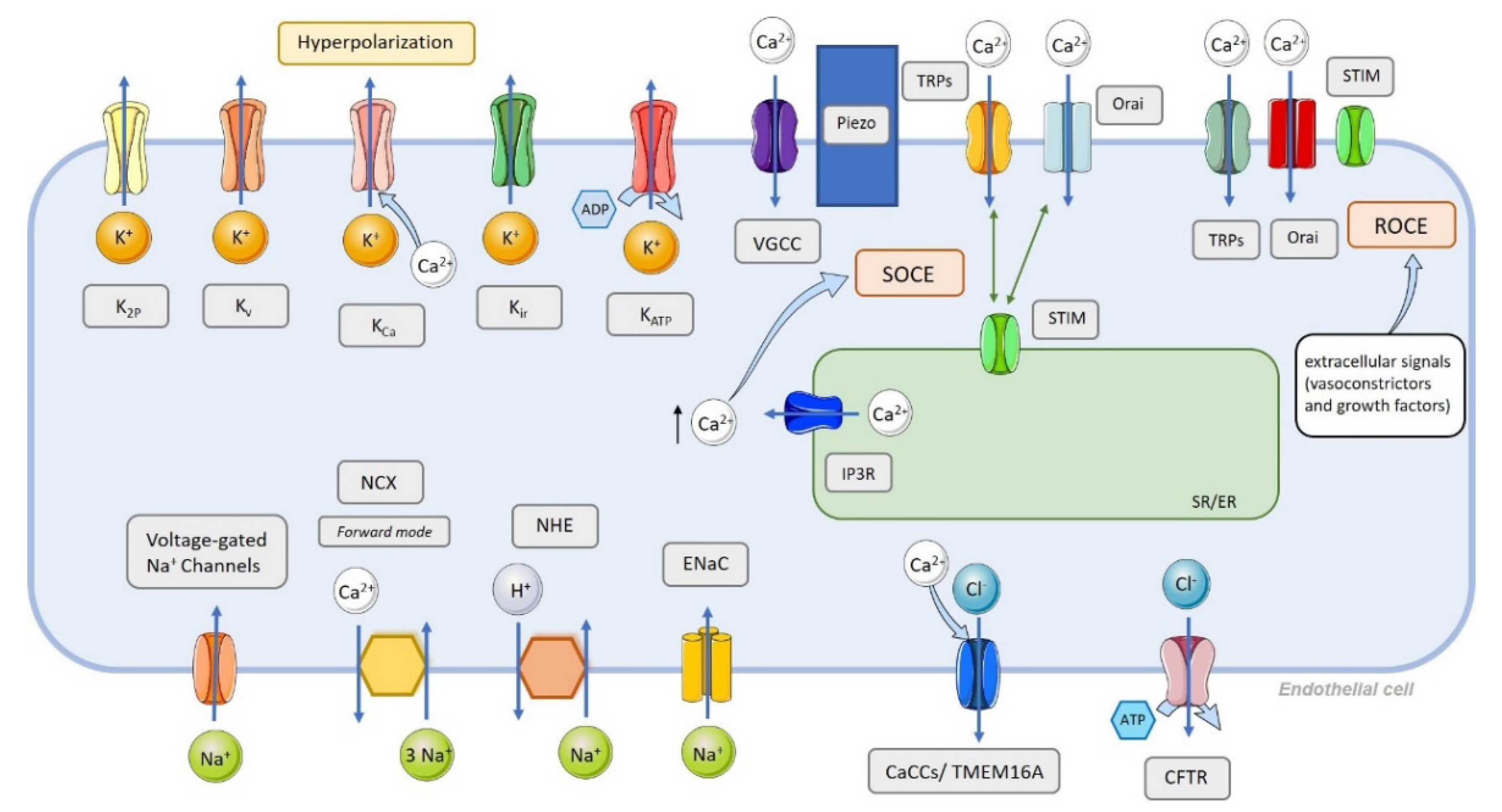{kind=link}
{kind=link}
{kind=link}
{kind=link}
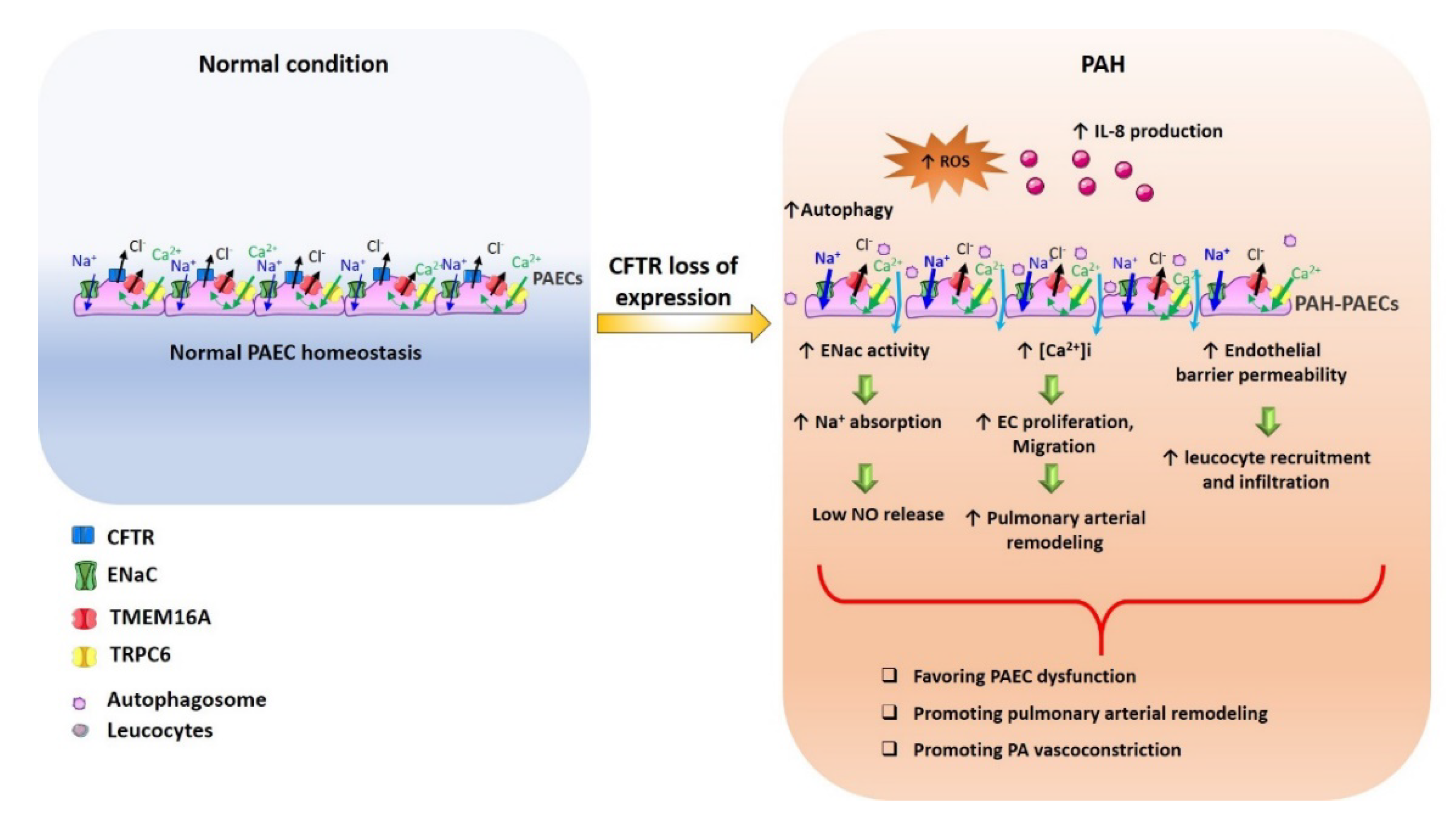{kind=link}
Abstract
:1. Introduction
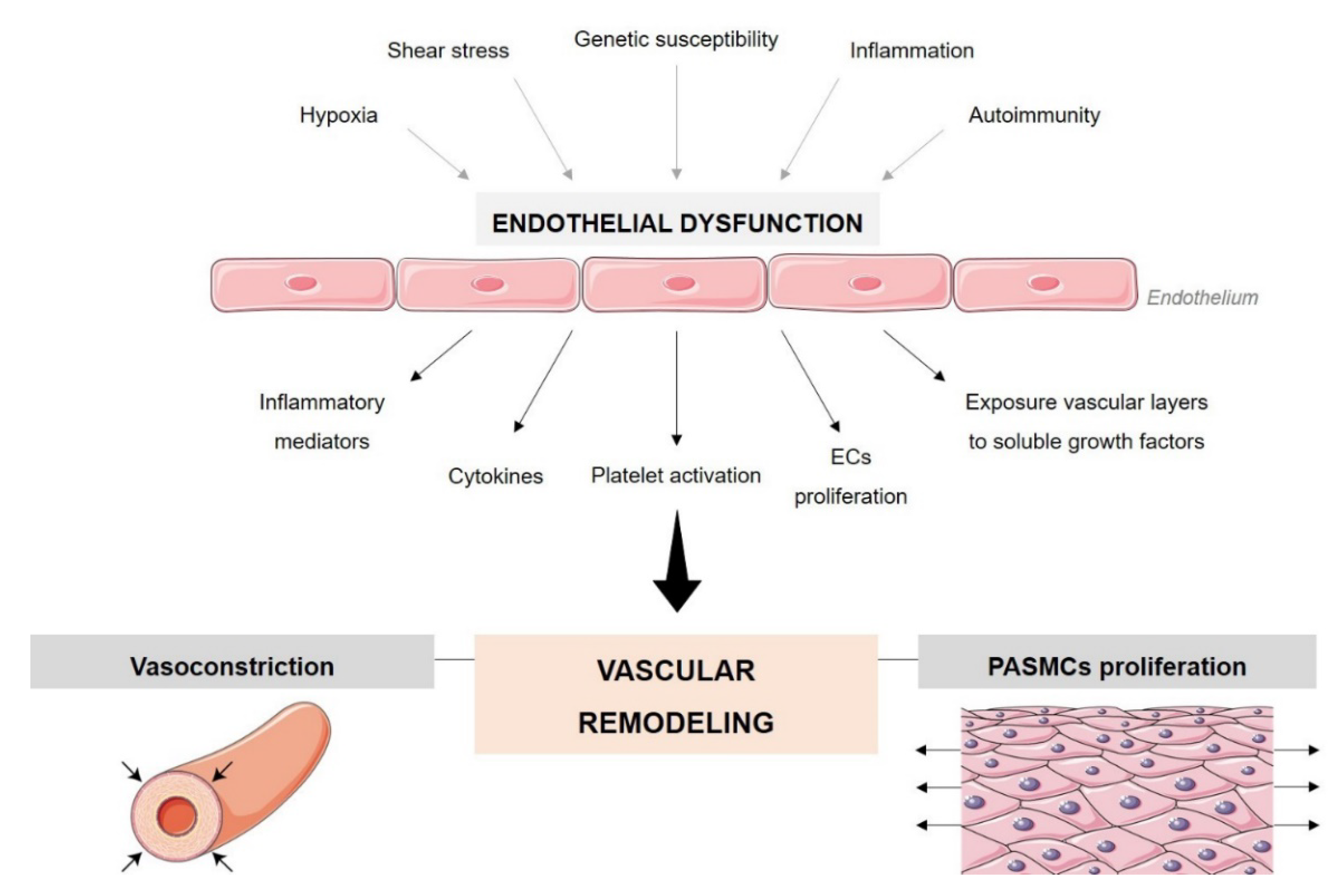
2. Endothelial Cell Dysfunction in PAH
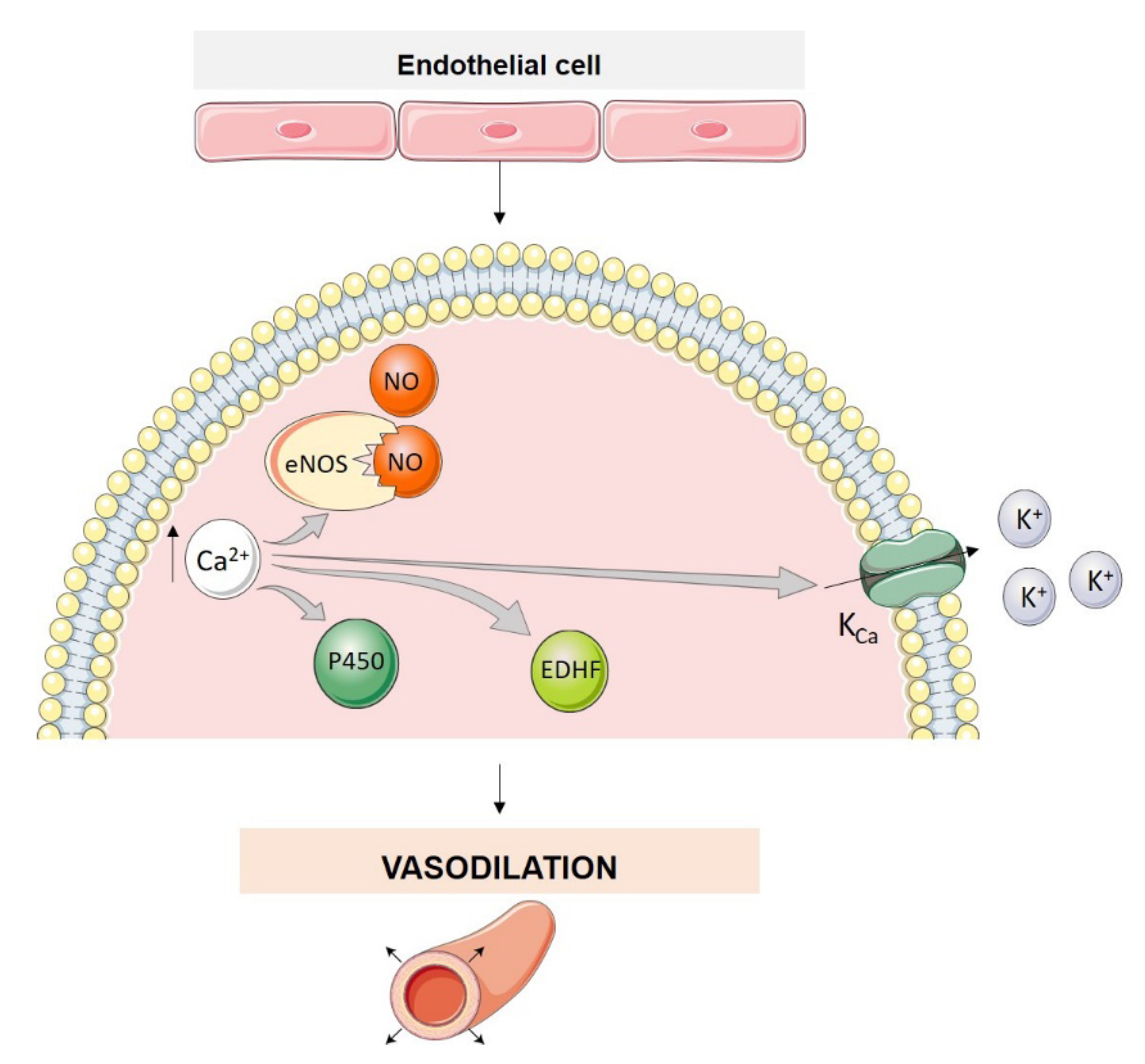
3. Endothelial Cell Ion Channel Function in Pulmonary Arterial Tone
4. Classification of Ion Channels in the Pulmonary Circulation and Their Function in PAH Pathophysiology
4.1. Ca2+ Channels
- (i)
- Voltage-gated Ca2+ channels (VGCC);
- (ii)
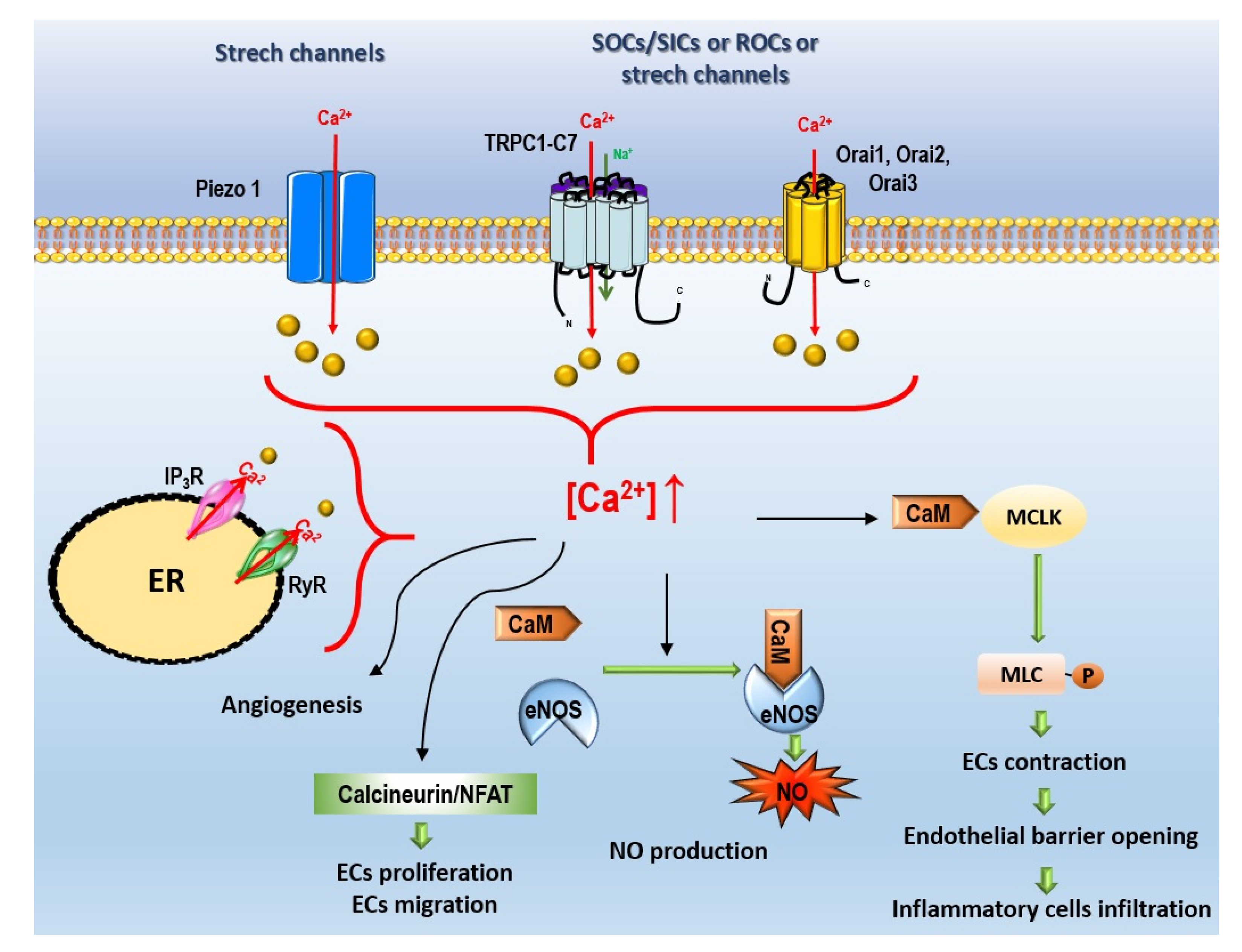
- Non-voltage-dependent Ca2+ channels (Figure 4):
- (a)
- Store-operated Ca2+ entry (SOCE);
- (b)
- Non-voltage-dependent store-independent Ca2+ entry (SICE), also called receptor-operated Ca2+ entry (ROCE);
- (c)
- Ca2+ stretch channels.
4.1.1. Voltage-Gated Ca2+ Channels
4.1.2. Non-Voltage-Dependent Ca2+ Channels
Store-Operated Ca2+ Entry
Store-Independent Ca2+ Entry, Also Called Receptor-Operated Ca2+ Entry
Ca2+ Stretch Channels
4.2. Na+ Channels
4.2.1. Epithelial Na+ Channels
4.2.2. Voltage-Gated Na+ Channels
4.2.3. Sodium-Calcium Exchanger
4.2.4. Na+-H+ Exchanger
4.3. Cl− Channels
4.3.1. Ca2+-Activated Cl− Channels–TMEM16 Family
4.3.2. Cystic Fibrosis Transmembrane Conductance Regulator
4.4. K+ Channels
4.4.1. Voltage-Gated K+ Channels
4.4.2. Ca2+-Activated K+ Channels
4.4.3. Two Pore Potassium Channels
4.4.4. ATP-Sensitive K+ Channel
4.4.5. Inward Rectifier Channel Family
5. Emerging Ion Channel Targets for PAH Therapy
5.1. Ca2+ Channels
5.1.1. Direct Pharmacological Action
5.1.2. Indirect Pharmacological Action
5.2. Na+ Channels
Indirect Pharmacological Action
5.3. Cl− Channels
5.4. K+ Channels
5.4.1. Direct Pharmacological Action
5.4.2. Indirect Pharmacological Action
5.4.3. Untested Pharmacological Compounds
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jackson, W.F. Endothelial Cell Ion Channel Expression and Function in Arterioles and Resistance Arteries. In Vascular Ion Channels in Physiology and Disease; Levitan, P.I., Dopico, M.D.P.A.M., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 3–36. [Google Scholar]
- Santos-Ribeiro, D.; Mendes-Ferreira, P.; Maia-Rocha, C.; Adao, R.; Leite-Moreira, A.F.; Bras-Silva, C. Pulmonary arterial hypertension: Basic knowledge for clinicians. Arch. Cardiovasc. Dis. 2016, 109, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Wen, J.; Wang, N.; Wang, C.; Xu, Q.; Yang, Y. Ion Channels and Vascular Diseases. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e146–e156. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.; Capuano, V.; Olschewski, A.; Sabourin, J.; Nagaraj, C.; Girerd, B.; Weatherald, J.; Humbert, M.; Antigny, F. Ion Channels in Pulmonary Hypertension: A Therapeutic Interest? Int. J. Mol. Sci. 2018, 19, 3162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouen-Tachoire, T.R.H.; Tucker, S.J.; Tammaro, P. Ion channels as convergence points in the pathology of pulmonary arterial hypertension. Biochem. Soc. Trans. 2021, 49, 1855–1865. [Google Scholar] [CrossRef]
- Lambert, M.; Boet, A.; Rucker-Martin, C.; Mendes-Ferreira, P.; Capuano, V.; Hatem, S.; Adao, R.; Bras-Silva, C.; Hautefort, A.; Michel, J.B.; et al. Loss of KCNK3 is a hallmark of RV hypertrophy/dysfunction associated with pulmonary hypertension. Cardiovasc. Res. 2018, 114, 880–893. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Townsley, M.I.; Alexeyev, M.; Voelkel, N.F.; Stevens, T. Endothelial hyperpermeability in severe pulmonary arterial hypertension: Role of store-operated calcium entry. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L560–L569. [Google Scholar] [CrossRef] [Green Version]
- Pietra, G.G.; Capron, F.; Stewart, S.; Leone, O.; Humbert, M.; Robbins, I.M.; Reid, L.M.; Tuder, R.M. Pathologic assessment of vasculopathies in pulmonary hypertension. J. Am. Coll. Cardiol. 2004, 43, 25S–32S. [Google Scholar] [CrossRef] [Green Version]
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; Christman, B.W.; Weir, E.K.; Eickelberg, O.; Voelkel, N.F.; et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, 13S–24S. [Google Scholar] [CrossRef] [Green Version]
- Huertas, A.; Perros, F.; Tu, L.; Cohen-Kaminsky, S.; Montani, D.; Dorfmuller, P.; Guignabert, C.; Humbert, M. Immune dysregulation and endothelial dysfunction in pulmonary arterial hypertension: A complex interplay. Circulation 2014, 129, 1332–1340. [Google Scholar] [CrossRef]
- Morrell, N.W.; Adnot, S.; Archer, S.L.; Dupuis, J.; Lloyd Jones, P.; MacLean, M.R.; McMurtry, I.F.; Stenmark, K.R.; Thistlethwaite, P.A.; Weissmann, N.; et al. Cellular and molecular basis of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009, 54, S20–S31. [Google Scholar] [CrossRef] [Green Version]
- Makino, A.; Firth, A.L.; Yuan, J.X. Endothelial and smooth muscle cell ion channels in pulmonary vasoconstriction and vascular remodeling. Compr. Physiol. 2011, 1, 1555–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luksha, L.; Agewall, S.; Kublickiene, K. Endothelium-derived hyperpolarizing factor in vascular physiology and cardiovascular disease. Atherosclerosis 2009, 202, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Yamamura, A.; Zimnicka, A.M.; Voiriot, G.; Smith, K.A.; Tang, H.; Ayon, R.J.; Choudhury, M.S.; Ko, E.A.; Wang, J.; et al. Chronic hypoxia selectively enhances L- and T-type voltage-dependent Ca2+ channel activity in pulmonary artery by upregulating Cav1.2 and Cav3.2. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L154–L164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catterall, W.A. Voltage-Gated Calcium Channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef]
- Lang, B.; Pinto, A.; Giovannini, F.; Newsom-Davis, J.; Vincent, A. Pathogenic Autoantibodies in the Lambert-Eaton myasthenic syndrome. Ann. N. Y. Acad. Sci. 2003, 998, 187–195. [Google Scholar] [CrossRef]
- Gueguinou, M.; Chantome, A.; Fromont, G.; Bougnoux, P.; Vandier, C.; Potier-Cartereau, M. KCa and Ca2+ channels: The complex thought. Biochim. Biophys. Acta 2014, 1843, 2322–2333. [Google Scholar] [CrossRef] [Green Version]
- Firth, A.L.; Remillard, C.V.; Platoshyn, O.; Fantozzi, I.; Ko, E.A.; Yuan, J.X. Functional ion channels in human pulmonary artery smooth muscle cells: Voltage-dependent cation channels. Pulm. Circ. 2011, 1, 48–71. [Google Scholar] [CrossRef] [Green Version]
- Cribbs, L.L. T-type Ca2+ channels in vascular smooth muscle: Multiple functions. Cell Calcium 2006, 40, 221–230. [Google Scholar] [CrossRef]
- Zheng, Z.; Chen, H.; Xie, P.; Dickerson, C.A.; King, J.A.C.; Alexeyev, M.F.; Wu, S. α1G T-type calcium channel determines the angiogenic potential of pulmonary microvascular endothelial cells. Am. J. Physiol. Cell Physiol. 2019, 316, C353–C364. [Google Scholar] [CrossRef]
- Moccia, F.; Berra-Romani, R.; Tanzi, F. Update on vascular endothelial Ca2+ signalling: A tale of ion channels, pumps and transporters. World J. Biol. Chem. 2012, 3, 127–158. [Google Scholar] [CrossRef]
- Chevalier, M.; Gilbert, G.; Roux, E.; Lory, P.; Marthan, R.; Savineau, J.P.; Quignard, J.F. T-type calcium channels are involved in hypoxic pulmonary hypertension. Cardiovasc. Res. 2014, 103, 597–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodman, D.M.; Reese, K.; Harral, J.; Fouty, B.; Wu, S.; West, J.; Hoedt-Miller, M.; Tada, Y.; Li, K.X.; Cool, C.; et al. Low-voltage-activated (T-type) calcium channels control proliferation of human pulmonary artery myocytes. Circ. Res. 2005, 96, 864–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankhe, S.; Manousakidi, S.; Antigny, F.; Arthur Ataam, J.; Bentebbal, S.; Ruchon, Y.; Lecerf, F.; Sabourin, J.; Price, L.; Fadel, E.; et al. T-type Ca2+ channels elicit pro-proliferative and anti-apoptotic responses through impaired PP2A/Akt1 signaling in PASMCs from patients with pulmonary arterial hypertension. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Vinet, R.; Vargas, F.F. L- and T-type voltage-gated Ca2+ currents in adrenal medulla endothelial cells. Am. J. Physiol. 1999, 276, H1313–H1322. [Google Scholar] [CrossRef]
- Bossu, J.L.; Elhamdani, A.; Feltz, A.; Tanzi, F.; Aunis, D.; Thierse, D. Voltage-gated Ca entry in isolated bovine capillary endothelial cells: Evidence of a new type of BAY K 8644-sensitive channel. Pflug. Arch. 1992, 420, 200–207. [Google Scholar] [CrossRef]
- Kefaloyianni, E.; Coetzee, W.A. Transcriptional remodeling of ion channel subunits by flow adaptation in human coronary artery endothelial cells. J. Arch. Res. 2011, 48, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Haynes, J., Jr.; Taylor, J.T.; Obiako, B.O.; Stubbs, J.R.; Li, M.; Stevens, T. Cav3.1 (alpha1G) T-type Ca2+ channels mediate vaso-occlusion of sickled erythrocytes in lung microcirculation. Circ. Res. 2003, 93, 346–353. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, G.; Courtois, A.; Dubois, M.; Cussac, L.A.; Ducret, T.; Lory, P.; Marthan, R.; Savineau, J.P.; Quignard, J.F. T-type voltage gated calcium channels are involved in endothelium-dependent relaxation of mice pulmonary artery. BioChem. Pharmacol. 2017, 138, 61–72. [Google Scholar] [CrossRef]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [Green Version]
- Putney, J.W., Jr. Formation and actions of calcium-mobilizing messenger, inositol 1,4,5-trisphosphate. Am. J. Physiol. 1987, 252, G149–G157. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masson, B.; Montani, D.; Humbert, M.; Capuano, V.; Antigny, F. Role of Store-Operated Ca2+ Entry in the Pulmonary Vascular Remodeling Occurring in Pulmonary Arterial Hypertension. Biomolecules 2021, 11, 1781. [Google Scholar] [CrossRef]
- Eder, P.; Molkentin, J.D. TRPC channels as effectors of cardiac hypertrophy. Circ. Res. 2011, 108, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Garland, C.J. Recent developments in vascular endothelial cell transient receptor potential channels. Circ. Res. 2005, 97, 853–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilius, B.; Droogmans, G. Ion channels and their functional role in vascular endothelium. Physiol. Rev. 2001, 81, 1415–1459. [Google Scholar] [CrossRef]
- Sundivakkam, P.C.; Freichel, M.; Singh, V.; Yuan, J.P.; Vogel, S.M.; Flockerzi, V.; Malik, A.B.; Tiruppathi, C. The Ca2+ sensor stromal interaction molecule 1 (STIM1) is necessary and sufficient for the store-operated Ca2+ entry function of transient receptor potential canonical (TRPC) 1 and 4 channels in endothelial cells. Mol. Pharmacol. 2012, 81, 510–526. [Google Scholar] [CrossRef] [Green Version]
- Garcia, R.L.; Schilling, W.P. Differential expression of mammalian TRP homologues across tissues and cell lines. BioChem. Biophys. Res. Commun. 1997, 239, 279–283. [Google Scholar] [CrossRef]
- Kamouchi, M.; Philipp, S.; Flockerzi, V.; Wissenbach, U.; Mamin, A.; Raeymaekers, L.; Eggermont, J.; Droogmans, G.; Nilius, B. Properties of heterologously expressed hTRP3 channels in bovine pulmonary artery endothelial cells. J. Physiol. 1999, 518 Pt 2, 345–358. [Google Scholar] [CrossRef]
- Kohler, R.; Brakemeier, S.; Kuhn, M.; Degenhardt, C.; Buhr, H.; Pries, A.; Hoyer, J. Expression of ryanodine receptor type 3 and TRP channels in endothelial cells: Comparison of in situ and cultured human endothelial cells. Cardiovasc. Res. 2001, 51, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Nilius, B.; Voets, T. Diversity of TRP channel activation. In Novartis Foundation Symposium; Wiley: New York, NY, USA, 2004; Volume 258, pp. 140–149, discussion 149–159, 263–146. [Google Scholar]
- Fernandez, R.A.; Sundivakkam, P.; Smith, K.A.; Zeifman, A.S.; Drennan, A.R.; Yuan, J.X. Pathogenic role of store-operated and receptor-operated Ca2+ channels in pulmonary arterial hypertension. J. Signal. Transduct. 2012, 2012, 951497. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, A.; Yamamura, H.; Zeifman, A.; Yuan, J.X. Activity of Ca2+-activated Cl− channels contributes to regulating receptor- and store-operated Ca2+ entry in human pulmonary artery smooth muscle cells. Pulm. Circ. 2011, 1, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Rinne, A.; Banach, K.; Blatter, L.A. Regulation of nuclear factor of activated T cells (NFAT) in vascular endothelial cells. J. Mol. Cell. Cardiol. 2009, 47, 400–410. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Jousset, H.; Konig, S.; Frieden, M. Thapsigargin activates Ca2+ entry both by store-dependent, STIM1/Orai1-mediated, and store-independent, TRPC3/PLC/PKC-mediated pathways in human endothelial cells. Cell Calcium 2011, 49, 115–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beech, D.J. Characteristics of transient receptor potential canonical calcium-permeable channels and their relevance to vascular physiology and disease. Circ. J. 2013, 77, 570–579. [Google Scholar] [CrossRef] [Green Version]
- Hecquet, C.M.; Ahmmed, G.U.; Vogel, S.M.; Malik, A.B. Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ. Res. 2008, 102, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Tauseef, M.; Knezevic, N.; Chava, K.R.; Smith, M.; Sukriti, S.; Gianaris, N.; Obukhov, A.G.; Vogel, S.M.; Schraufnagel, D.E.; Dietrich, A.; et al. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J. Exp. Med. 2012, 209, 1953–1968. [Google Scholar] [CrossRef]
- Ferrera, L.; Barbieri, R.; Picco, C.; Zuccolini, P.; Remigante, A.; Bertelli, S.; Fumagalli, M.R.; Zifarelli, G.; La Porta, C.A.M.; Gavazzo, P.; et al. TRPM2 Oxidation Activates Two Distinct Potassium Channels in Melanoma Cells through Intracellular Calcium Increase. Int. J. Mol. Sci. 2021, 22, 8359. [Google Scholar] [CrossRef]
- Willette, R.N.; Bao, W.; Nerurkar, S.; Yue, T.L.; Doe, C.P.; Stankus, G.; Turner, G.H.; Ju, H.; Thomas, H.; Fishman, C.E.; et al. Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: Part 2. J. Pharmacol. Exp. Ther. 2008, 326, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Negri, S.; Faris, P.; Berra-Romani, R.; Guerra, G.; Moccia, F. Endothelial Transient Receptor Potential Channels and Vascular Remodeling: Extracellular Ca2+ Entry for Angiogenesis, Arteriogenesis and Vasculogenesis. Front. Physiol. 2019, 10, 1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, D.; Ahmmed, G.U.; Paria, B.C.; Holinstat, M.; Voyno-Yasenetskaya, T.; Tiruppathi, C.; Minshall, R.D.; Malik, A.B. RhoA interaction with inositol 1,4,5-trisphosphate receptor and transient receptor potential channel-1 regulates Ca2+ entry. Role in signaling increased endothelial permeability. J. Biol. Chem. 2003, 278, 33492–33500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paria, B.C.; Vogel, S.M.; Ahmmed, G.U.; Alamgir, S.; Shroff, J.; Malik, A.B.; Tiruppathi, C. Tumor necrosis factor-alpha-induced TRPC1 expression amplifies store-operated Ca2+ influx and endothelial permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L1303–L1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzoubi, A.; Almalouf, P.; Toba, M.; O’Neill, K.; Qian, X.; Francis, M.; Taylor, M.S.; Alexeyev, M.; McMurtry, I.F.; Oka, M.; et al. TRPC4 inactivation confers a survival benefit in severe pulmonary arterial hypertension. Am. J. Pathol. 2013, 183, 1779–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, M.; Xu, N.; Zhou, C.; Stevens, T. Transient Receptor Potential Channel 4 Encodes a Vascular Permeability Defect and High-Frequency Ca2+ Transients in Severe Pulmonary Arterial Hypertension. Am. J. Pathol. 2016, 186, 1701–1709. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Fantozzi, I.; Remillard, C.V.; Landsberg, J.W.; Kunichika, N.; Platoshyn, O.; Tigno, D.D.; Thistlethwaite, P.A.; Rubin, L.J.; Yuan, J.X. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc. Natl. Acad. Sci. USA 2004, 101, 13861–13866. [Google Scholar] [CrossRef] [Green Version]
- Fantozzi, I.; Zhang, S.; Platoshyn, O.; Remillard, C.V.; Cowling, R.T.; Yuan, J.X. Hypoxia increases AP-1 binding activity by enhancing capacitative Ca2+ entry in human pulmonary artery endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L1233–L1245. [Google Scholar] [CrossRef] [Green Version]
- Firth, A.L.; Remillard, C.V.; Yuan, J.X. TRP channels in hypertension. Biochim. Biophys. Acta 2007, 1772, 895–906. [Google Scholar] [CrossRef] [Green Version]
- Freichel, M.; Suh, S.H.; Pfeifer, A.; Schweig, U.; Trost, C.; Weissgerber, P.; Biel, M.; Philipp, S.; Freise, D.; Droogmans, G.; et al. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4−/− mice. Nat. Cell Biol. 2001, 3, 121–127. [Google Scholar] [CrossRef]
- Tiruppathi, C.; Freichel, M.; Vogel, S.M.; Paria, B.C.; Mehta, D.; Flockerzi, V.; Malik, A.B. Impairment of store-operated Ca2+ entry in TRPC4−/− mice interferes with increase in lung microvascular permeability. Circ. Res. 2002, 91, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, A.; Mederos, Y.S.M.; Gollasch, M.; Gross, V.; Storch, U.; Dubrovska, G.; Obst, M.; Yildirim, E.; Salanova, B.; Kalwa, H.; et al. Increased vascular smooth muscle contractility in TRPC6−/− mice. Mol. Cell. Biol. 2005, 25, 6980–6989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.Y.; Makino, A.; Yuan, J.X. STIM2 Contributes to Enhanced Store-operated Ca Entry in Pulmonary Artery Smooth Muscle Cells from Patients with Idiopathic Pulmonary Arterial Hypertension. Pulm. Circ. 2011, 1, 84–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, R.A.; Wan, J.; Song, S.; Smith, K.A.; Gu, Y.; Tauseef, M.; Tang, H.; Makino, A.; Mehta, D.; Yuan, J.X. Upregulated expression of STIM2, TRPC6, and Orai2 contributes to the transition of pulmonary arterial smooth muscle cells from a contractile to proliferative phenotype. Am. J. Physiol. Cell Physiol. 2015, 308, C581–C593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saygin, D.; Tabib, T.; Bittar, H.E.T.; Valenzi, E.; Sembrat, J.; Chan, S.Y.; Rojas, M.; Lafyatis, R. Transcriptional profiling of lung cell populations in idiopathic pulmonary arterial hypertension. Pulm. Circ. 2020, 10. [Google Scholar] [CrossRef] [Green Version]
- Antigny, F.; Girardin, N.; Frieden, M. Transient receptor potential canonical channels are required for in vitro endothelial tube formation. J. Biol. Chem. 2012, 287, 5917–5927. [Google Scholar] [CrossRef] [Green Version]
- Zergane, M.; Kuebler, W.M.; Michalick, L. Heteromeric TRP Channels in Lung Inflammation. Cells 2021, 10, 1654. [Google Scholar] [CrossRef]
- Tiruppathi, C.; Minshall, R.D.; Paria, B.C.; Vogel, S.M.; Malik, A.B. Role of Ca2+ signaling in the regulation of endothelial permeability. Vasc. Pharmacol. 2002, 39, 173–185. [Google Scholar] [CrossRef]
- Coste, B.; Murthy, S.E.; Mathur, J.; Schmidt, M.; Mechioukhi, Y.; Delmas, P.; Patapoutian, A. Piezo1 ion channel pore properties are dictated by C-terminal region. Nat. Commun. 2015, 6, 7223. [Google Scholar] [CrossRef]
- Barbeau, S.; Gilbert, G.; Cardouat, G.; Baudrimont, I.; Freund-Michel, V.; Guibert, C.; Marthan, R.; Vacher, P.; Quignard, J.F.; Ducret, T. Mechanosensitivity in Pulmonary Circulation: Pathophysiological Relevance of Stretch-Activated Channels in Pulmonary Hypertension. Biomolecules 2021, 11, 1389. [Google Scholar] [CrossRef]
- Li, J.; Hou, B.; Tumova, S.; Muraki, K.; Bruns, A.; Ludlow, M.J.; Sedo, A.; Hyman, A.J.; McKeown, L.; Young, R.S.; et al. Piezo1 integration of vascular architecture with physiological force. Nature 2014, 515, 279–282. [Google Scholar] [CrossRef]
- Wang, S.; Chennupati, R.; Kaur, H.; Iring, A.; Wettschureck, N.; Offermanns, S. Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J. Clin. Investig. 2016, 126, 4527–4536. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.J. Hypertension: Mechanosensation by PIEZO1 in blood pressure control. Nat. Rev. Nephrol. 2017, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Retailleau, K.; Duprat, F. Polycystins and partners: Proposed role in mechanosensitivity. J. Physiol. 2014, 592, 2453–2471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Chen, J.; Babicheva, A.; Jain, P.P.; Rodriguez, M.; Ayon, R.J.; Ravellette, K.S.; Wu, L.; Balistrieri, F.; Tang, H.; et al. Endothelial upregulation of mechanosensitive channel Piezo1 in pulmonary hypertension. Am. J. Physiol. Cell Physiol. 2021, 321, C1010–C1027. [Google Scholar] [CrossRef]
- Lhomme, A.; Gilbert, G.; Pele, T.; Deweirdt, J.; Henrion, D.; Baudrimont, I.; Campagnac, M.; Marthan, R.; Guibert, C.; Ducret, T.; et al. Stretch-activated Piezo1 Channel in Endothelial Cells Relaxes Mouse Intrapulmonary Arteries. Am. J. Respir. Cell Mol. Biol. 2019, 60, 650–658. [Google Scholar] [CrossRef]
- Liao, J.; Lu, W.; Chen, Y.; Duan, X.; Zhang, C.; Luo, X.; Lin, Z.; Chen, J.; Liu, S.; Yan, H.; et al. Upregulation of Piezo1 (Piezo Type Mechanosensitive Ion Channel Component 1) Enhances the Intracellular Free Calcium in Pulmonary Arterial Smooth Muscle Cells from Idiopathic Pulmonary Arterial Hypertension Patients. Hypertension 2021, 77, 1974–1989. [Google Scholar] [CrossRef]
- Bhalla, V.; Hallows, K.R. Mechanisms of ENaC regulation and clinical implications. J. Am. Soc. Nephrol. 2008, 19, 1845–1854. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Meng, F.; Mohan, S.; Champaneri, B.; Gu, Y. Functional ENaC channels expressed in endothelial cells: A new candidate for mediating shear force. Microcirculation 2009, 16, 276–287. [Google Scholar] [CrossRef]
- Staruschenko, A.; Adams, E.; Booth, R.E.; Stockand, J.D. Epithelial Na+ channel subunit stoichiometry. Biophys. J. 2005, 88, 3966–3975. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhang, J.N.; Zhao, D.; Wang, Q.S.; Gu, Y.C.; Ma, H.P.; Zhang, Z.R. Role of the epithelial sodium channel in salt-sensitive hypertension. Acta Pharmacol. Sin. 2011, 32, 789–797. [Google Scholar] [CrossRef] [Green Version]
- Jernigan, N.L.; Drummond, H.A. Vascular ENaC proteins are required for renal myogenic constriction. Am. J. Physiol. Ren. Physiol. 2005, 289, F891–F901. [Google Scholar] [CrossRef] [Green Version]
- Drummond, H.A.; Gebremedhin, D.; Harder, D.R. Degenerin/epithelial Na+ channel proteins: Components of a vascular mechanosensor. Hypertension 2004, 44, 643–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockand, J.D. The role of the epithelial Na+ channel (ENaC) in high AVP but low aldosterone states. Front. Physiol. 2012, 3, 304. [Google Scholar] [CrossRef] [Green Version]
- Kusche-Vihrog, K.; Jeggle, P.; Oberleithner, H. The role of ENaC in vascular endothelium. Pflug. Arch. 2014, 466, 851–859. [Google Scholar] [CrossRef] [PubMed]
- De Man, F.S.; Tu, L.; Handoko, M.L.; Rain, S.; Ruiter, G.; Francois, C.; Schalij, I.; Dorfmuller, P.; Simonneau, G.; Fadel, E.; et al. Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 780–789. [Google Scholar] [CrossRef] [Green Version]
- Zaika, O.; Mamenko, M.; Staruschenko, A.; Pochynyuk, O. Direct activation of ENaC by angiotensin II: Recent advances and new insights. Curr. Hypertens. Rep. 2013, 15, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becq, F.; Mall, M.A.; Sheppard, D.N.; Conese, M.; Zegarra-Moran, O. Pharmacological therapy for cystic fibrosis: From bench to bedside. J. Cyst. Fibros. 2011, 10 (Suppl. 2), S129–S145. [Google Scholar] [CrossRef] [Green Version]
- Horisberger, J.D. ENaC-CFTR interactions: The role of electrical coupling of ion fluxes explored in an epithelial cell model. Pflug. Arch. 2003, 445, 522–528. [Google Scholar] [CrossRef]
- Le Ribeuz, H.; To, L.; Ghigna, M.R.; Martin, C.; Nagaraj, C.; Dreano, E.; Rucker-Martin, C.; Girerd, B.; Bouligand, J.; Pechoux, C.; et al. Involvement of CFTR in the pathogenesis of pulmonary arterial hypertension. Eur. Respir. J. 2021, 58, 2000653. [Google Scholar] [CrossRef]
- Declercq, M.; de Zeeuw, P.; Conchinha, N.V.; Geldhof, V.; Ramalho, A.S.; Garcia-Caballero, M.; Brepoels, K.; Ensinck, M.; Carlon, M.S.; Bird, M.J.; et al. Transcriptomic analysis of CFTR-impaired endothelial cells reveals a pro-inflammatory phenotype. Eur. Respir. J. 2021, 57, 2000261. [Google Scholar] [CrossRef]
- Mall, M.A. ENaC inhibition in cystic fibrosis: Potential role in the new era of CFTR modulator therapies. Eur. Respir. J. 2020, 56, 2000946. [Google Scholar] [CrossRef] [PubMed]
- Platoshyn, O.; Remillard, C.V.; Fantozzi, I.; Sison, T.; Yuan, J.X. Identification of functional voltage-gated Na+ channels in cultured human pulmonary artery smooth muscle cells. Pflug. Arch. 2005, 451, 380–387. [Google Scholar] [CrossRef] [Green Version]
- De Lera Ruiz, M.; Kraus, R.L. Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J. Med. Chem. 2015, 58, 7093–7118. [Google Scholar] [CrossRef] [PubMed]
- Kuriyama, H.; Kitamura, K.; Nabata, H. Pharmacological and physiological significance of ion channels and factors that modulate them in vascular tissues. Pharmacol. Rev. 1995, 47, 387–573. [Google Scholar] [PubMed]
- Geraci, M.W.; Moore, M.; Gesell, T.; Yeager, M.E.; Alger, L.; Golpon, H.; Gao, B.; Loyd, J.E.; Tuder, R.M.; Voelkel, N.F. Gene expression patterns in the lungs of patients with primary pulmonary hypertension: A gene microarray analysis. Circ. Res. 2001, 88, 555–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.M.; Wang, Y.X. Sodium-calcium exchanger in pulmonary artery smooth muscle cells. Ann. N. Y. Acad. Sci. 2007, 1099, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T. Sodium-calcium exchange inhibitors: Therapeutic potential in cardiovascular diseases. Future Cardiol. 2005, 1, 519–529. [Google Scholar] [CrossRef]
- Zhang, J. New insights into the contribution of arterial NCX to the regulation of myogenic tone and blood pressure. Adv. Exp. Med. Biol. 2013, 961, 329–343. [Google Scholar] [CrossRef]
- Lillo, M.A.; Gaete, P.S.; Puebla, M.; Ardiles, N.M.; Poblete, I.; Becerra, A.; Simon, F.; Figueroa, X.F. Critical contribution of Na+-Ca2+ exchanger to the Ca2+-mediated vasodilation activated in endothelial cells of resistance arteries. FASEB J. 2018, 32, 2137–2147. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, S.; Demura, Y.; Ameshima, S.; Okamura, S.; Miyamori, I.; Ishizaki, T. Alkalosis stimulates endothelial nitric oxide synthase in cultured human pulmonary arterial endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L113–L119. [Google Scholar] [CrossRef]
- Wang, Y.X.; Dhulipala, P.K.; Kotlikoff, M.I. Hypoxia inhibits the Na+/Ca2+ exchanger in pulmonary artery smooth muscle cells. FASEB J. 2000, 14, 1731–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Yuan, J.X.; Barrett, K.E.; Dong, H. Role of Na+/Ca2+ exchange in regulating cytosolic Ca2+ in cultured human pulmonary artery smooth muscle cells. Am. J. Physiol. Cell Physiol. 2005, 288, C245–C252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Dong, H.; Rubin, L.J.; Yuan, J.X. Upregulation of Na+/Ca2+ exchanger contributes to the enhanced Ca2+ entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Am. J. Physiol. Cell Physiol. 2007, 292, C2297–C2305. [Google Scholar] [CrossRef]
- Valles, P.G.; Bocanegra, V.; Gil Lorenzo, A.; Costantino, V.V. Physiological Functions and Regulation of the Na+/H+ Exchanger [NHE1] in Renal Tubule Epithelial Cells. Kidney Blood Press. Res. 2015, 40, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Huetsch, J.; Shimoda, L.A. Na+/H+ exchange and hypoxic pulmonary hypertension. Pulm. Circ. 2015, 5, 228–243. [Google Scholar] [CrossRef] [Green Version]
- Karmazyn, M.; Gan, X.T.; Humphreys, R.A.; Yoshida, H.; Kusumoto, K. The myocardial Na+-H+ exchange: Structure, regulation, and its role in heart disease. Circ. Res. 1999, 85, 777–786. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, I.; Poteser, M.; Groschner, K. Intracellular pH as a determinant of vascular smooth muscle function. J. Vasc. Res. 2006, 43, 238–250. [Google Scholar] [CrossRef]
- Rios, E.J.; Fallon, M.; Wang, J.; Shimoda, L.A. Chronic hypoxia elevates intracellular pH and activates Na+/H+ exchange in pulmonary arterial smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L867–L874. [Google Scholar] [CrossRef] [Green Version]
- Quinn, D.A.; Du, H.K.; Thompson, B.T.; Hales, C.A. Amiloride analogs inhibit chronic hypoxic pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1998, 157, 1263–1268. [Google Scholar] [CrossRef]
- Yu, L.; Quinn, D.A.; Garg, H.G.; Hales, C.A. Deficiency of the NHE1 gene prevents hypoxia-induced pulmonary hypertension and vascular remodeling. Am. J. Respir. Crit. Care Med. 2008, 177, 1276–1284. [Google Scholar] [CrossRef]
- Yu, L.; Hales, C.A. Silencing of sodium-hydrogen exchanger 1 attenuates the proliferation, hypertrophy, and migration of pulmonary artery smooth muscle cells via E2F1. Am. J. Respir. Cell Mol. Biol. 2011, 45, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutaia, M.V.; Parks, N.; Centracchio, J.; Rounds, S.; Yip, K.P.; Sun, A.M. Effect of hypoxic exposure on Na+/H+ antiport activity, isoform expression, and localization in endothelial cells. Am. J. Physiol. 1998, 275, L442–L451. [Google Scholar] [CrossRef] [PubMed]
- Caputo, A.; Caci, E.; Ferrera, L.; Pedemonte, N.; Barsanti, C.; Sondo, E.; Pfeffer, U.; Ravazzolo, R.; Zegarra-Moran, O.; Galietta, L.J. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 2008, 322, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.D.; Cho, H.; Koo, J.Y.; Tak, M.H.; Cho, Y.; Shim, W.S.; Park, S.P.; Lee, J.; Lee, B.; Kim, B.M.; et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 2008, 455, 1210–1215. [Google Scholar] [CrossRef]
- Schroeder, B.C.; Cheng, T.; Jan, Y.N.; Jan, L.Y. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 2008, 134, 1019–1029. [Google Scholar] [CrossRef] [Green Version]
- Gyobu, S.; Ishihara, K.; Suzuki, J.; Segawa, K.; Nagata, S. Characterization of the scrambling domain of the TMEM16 family. Proc. Natl. Acad. Sci. USA 2017, 114, 6274–6279. [Google Scholar] [CrossRef] [Green Version]
- Manoury, B.; Tamuleviciute, A.; Tammaro, P. TMEM16A/anoctamin 1 protein mediates calcium-activated chloride currents in pulmonary arterial smooth muscle cells. J. Physiol. 2010, 588, 2305–2314. [Google Scholar] [CrossRef]
- Ma, M.M.; Gao, M.; Guo, K.M.; Wang, M.; Li, X.Y.; Zeng, X.L.; Sun, L.; Lv, X.F.; Du, Y.H.; Wang, G.L.; et al. TMEM16A Contributes to Endothelial Dysfunction by Facilitating Nox2 NADPH Oxidase-Derived Reactive Oxygen Species Generation in Hypertension. Hypertension 2017, 69, 892–901. [Google Scholar] [CrossRef]
- Xie, J.Y.; Liu, W.Y.; Lv, W.J.; Han, X.H.; Kong, Q.N.; Wu, Y.H.; Liu, X.; Han, Y.; Shi, C.Y.; Jia, X.J. Transmembrane protein 16A/anoctamin 1 inhibitor T16A(inh)-A01 reversed monocrotaline-induced rat pulmonary arterial hypertension. Pulm. Circ. 2020, 10, 2045894020946670. [Google Scholar] [CrossRef]
- Liu, D.; Wang, K.; Su, D.; Huang, Y.; Shang, L.; Zhao, Y.; Huang, J.; Pang, Y. TMEM16A Regulates Pulmonary Arterial Smooth Muscle Cells Proliferation via p38MAPK/ERK Pathway in High Pulmonary Blood Flow-Induced Pulmonary Arterial Hypertension. J. Vasc. Res. 2020, 58, 27–37. [Google Scholar] [CrossRef]
- Papp, R.; Nagaraj, C.; Zabini, D.; Nagy, B.M.; Lengyel, M.; Skofic Maurer, D.; Sharma, N.; Egemnazarov, B.; Kovacs, G.; Kwapiszewska, G.; et al. Targeting TMEM16A to reverse vasoconstriction and remodelling in idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1800965. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Wang, K.; Liu, D.; Qin, S.; Huang, J.; Zhao, Y.; Pang, Y. TMEM16A regulates the cell cycle of pulmonary artery smooth muscle cells in high-flow-induced pulmonary arterial hypertension rat model. Exp. Ther. Med. 2020, 19, 3275–3281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skofic Maurer, D.; Zabini, D.; Nagaraj, C.; Sharma, N.; Lengyel, M.; Nagy, B.M.; Frank, S.; Klepetko, W.; Gschwandtner, E.; Enyedi, P.; et al. Endothelial Dysfunction Following Enhanced TMEM16A Activity in Human Pulmonary Arteries. Cells 2020, 9, 1984. [Google Scholar] [CrossRef] [PubMed]
- Allawzi, A.M.; Vang, A.; Clements, R.T.; Jhun, B.S.; Kue, N.R.; Mancini, T.J.; Landi, A.K.; Terentyev, D.; O-Uchi, J.; Comhair, S.A.; et al. Activation of Anoctamin-1 Limits Pulmonary Endothelial Cell Proliferation via p38-Mitogen-activated Protein Kinase-Dependent Apoptosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 658–667. [Google Scholar] [CrossRef]
- Tousson, A.; Van Tine, B.A.; Naren, A.P.; Shaw, G.M.; Schwiebert, L.M. Characterization of CFTR expression and chloride channel activity in human endothelia. Am. J. Physiol. 1998, 275, C1555–C1564. [Google Scholar] [CrossRef] [Green Version]
- Robert, R.; Savineau, J.P.; Norez, C.; Becq, F.; Guibert, C. Expression and function of cystic fibrosis transmembrane conductance regulator in rat intrapulmonary arteries. Eur. Respir. J. 2007, 30, 857–864. [Google Scholar] [CrossRef] [Green Version]
- Tabeling, C.; Yu, H.; Wang, L.; Ranke, H.; Goldenberg, N.M.; Zabini, D.; Noe, E.; Krauszman, A.; Gutbier, B.; Yin, J.; et al. CFTR and sphingolipids mediate hypoxic pulmonary vasoconstriction. Proc. Natl. Acad. Sci. USA 2015, 112, E1614–E1623. [Google Scholar] [CrossRef] [Green Version]
- Totani, L.; Plebani, R.; Piccoli, A.; Di Silvestre, S.; Lanuti, P.; Recchiuti, A.; Cianci, E.; Dell’Elba, G.; Sacchetti, S.; Patruno, S.; et al. Mechanisms of endothelial cell dysfunction in cystic fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3243–3253. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, Z.; Liao, J.; Zhang, J.; He, W.; Zhang, C.; Yang, K.; Lu, W.; Hong, C.; Liu, X.; et al. The causality between CFTR and pulmonary hypertension: Insights from Mendelian randomization studies. Hypertens. Res. 2021, 44, 1230–1232. [Google Scholar] [CrossRef]
- Ousingsawat, J.; Kongsuphol, P.; Schreiber, R.; Kunzelmann, K. CFTR and TMEM16A are separate but functionally related Cl− channels. Cell. Physiol. BioChem. 2011, 28, 715–724. [Google Scholar] [CrossRef]
- Antigny, F.; Norez, C.; Dannhoffer, L.; Bertrand, J.; Raveau, D.; Corbi, P.; Jayle, C.; Becq, F.; Vandebrouck, C. Transient receptor potential canonical channel 6 links Ca2+ mishandling to cystic fibrosis transmembrane conductance regulator channel dysfunction in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 44, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, J.; Dannhoffer, L.; Antigny, F.; Vachel, L.; Jayle, C.; Vandebrouck, C.; Becq, F.; Norez, C. A functional tandem between transient receptor potential canonical channels 6 and calcium-dependent chloride channels in human epithelial cells. Eur. J. Pharmacol. 2015, 765, 337–345. [Google Scholar] [CrossRef]
- Jackson, W.F. KV channels and the regulation of vascular smooth muscle tone. Microcirculation 2018, 25, e12421. [Google Scholar] [CrossRef]
- Ko, E.A.; Han, J.; Jung, I.D.; Park, W.S. Physiological roles of K+ channels in vascular smooth muscle cells. J. Smooth Muscle Res. 2008, 44, 65–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheong, A.; Dedman, A.M.; Beech, D.J. Expression and function of native potassium channel [K(V)alpha1] subunits in terminal arterioles of rabbit. J. Physiol. 2001, 534, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Walsh, K.B. Mechanical stimulation regulates voltage-gated potassium currents in cardiac microvascular endothelial cells. Circ. Res. 1999, 84, 451–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheong, A.; Dedman, A.M.; Xu, S.Z.; Beech, D.J. K(V)alpha1 channels in murine arterioles: Differential cellular expression and regulation of diameter. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1057–H1065. [Google Scholar] [CrossRef]
- Baldwin, S.N.; Sandow, S.L.; Mondejar-Parreno, G.; Stott, J.B.; Greenwood, I.A. Kv7 Channel Expression and Function Within Rat Mesenteric Endothelial Cells. Front. Physiol. 2020, 11, 598779. [Google Scholar] [CrossRef] [PubMed]
- Hogg, D.S.; McMurray, G.; Kozlowski, R.Z. Endothelial cells freshly isolated from small pulmonary arteries of the rat possess multiple distinct k+ current profiles. Lung 2002, 180, 203–214. [Google Scholar] [CrossRef]
- Babicheva, A.; Ayon, R.J.; Zhao, T.; Ek Vitorin, J.F.; Pohl, N.M.; Yamamura, A.; Yamamura, H.; Quinton, B.A.; Ba, M.; Wu, L.; et al. MicroRNA-mediated downregulation of K+ channels in pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L10–L26. [Google Scholar] [CrossRef]
- Sheng, J.Z.; Braun, A.P. Small- and intermediate-conductance Ca2+-activated K+ channels directly control agonist-evoked nitric oxide synthesis in human vascular endothelial cells. Am. J. Physiol. Cell Physiol. 2007, 293, C458–C467. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yan, J. Regulation of BK channels by auxiliary gamma subunits. Front. Physiol. 2014, 5, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, D.L.; Bai, Y.L.; Cai, B.Z. Calcium-Activated Potassium Channels: Potential Target for Cardiovascular Diseases. Adv. Protein Chem. Struct. Biol. 2016, 104, 233–261. [Google Scholar] [CrossRef] [PubMed]
- Kohler, R.; Degenhardt, C.; Kuhn, M.; Runkel, N.; Paul, M.; Hoyer, J. Expression and function of endothelial Ca2+-activated K+ channels in human mesenteric artery: A single-cell reverse transcriptase-polymerase chain reaction and electrophysiological study in situ. Circ. Res. 2000, 87, 496–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bychkov, R.; Burnham, M.P.; Richards, G.R.; Edwards, G.; Weston, A.H.; Feletou, M.; Vanhoutte, P.M. Characterization of a charybdotoxin-sensitive intermediate conductance Ca2+-activated K+ channel in porcine coronary endothelium: Relevance to EDHF. Br. J. Pharmacol. 2002, 137, 1346–1354. [Google Scholar] [CrossRef] [Green Version]
- Burnham, M.P.; Bychkov, R.; Feletou, M.; Richards, G.R.; Vanhoutte, P.M.; Weston, A.H.; Edwards, G. Characterization of an apamin-sensitive small-conductance Ca2+-activated K+ channel in porcine coronary artery endothelium: Relevance to EDHF. Br. J. Pharmacol. 2002, 135, 1133–1143. [Google Scholar] [CrossRef] [Green Version]
- Gluais, P.; Edwards, G.; Weston, A.H.; Falck, J.R.; Vanhoutte, P.M.; Feletou, M. Role of SK(Ca) and IK(Ca) in endothelium-dependent hyperpolarizations of the guinea-pig isolated carotid artery. Br. J. Pharmacol. 2005, 144, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Eichler, I.; Wibawa, J.; Grgic, I.; Knorr, A.; Brakemeier, S.; Pries, A.R.; Hoyer, J.; Kohler, R. Selective blockade of endothelial Ca2+-activated small- and intermediate-conductance K+-channels suppresses EDHF-mediated vasodilation. Br. J. Pharmacol. 2003, 138, 594–601. [Google Scholar] [CrossRef] [Green Version]
- Vang, A.; Mazer, J.; Casserly, B.; Choudhary, G. Activation of endothelial BKCa channels causes pulmonary vasodilation. Vasc. Pharmacol. 2010, 53, 122–129. [Google Scholar] [CrossRef]
- Guntur, D.; Olschewski, H.; Enyedi, P.; Csaki, R.; Olschewski, A.; Nagaraj, C. Revisiting the Large-Conductance Calcium-Activated Potassium (BKCa) Channels in the Pulmonary Circulation. Biomolecules 2021, 11, 1629. [Google Scholar] [CrossRef]
- Kroigaard, C.; Dalsgaard, T.; Nielsen, G.; Laursen, B.E.; Pilegaard, H.; Kohler, R.; Simonsen, U. Activation of endothelial and epithelial K(Ca) 2.3 calcium-activated potassium channels by NS309 relaxes human small pulmonary arteries and bronchioles. Br. J. Pharmacol. 2012, 167, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroigaard, C.; Kudryavtseva, O.; Dalsgaard, T.; Wandall-Frostholm, C.; Olesen, S.P.; Simonsen, U. K(Ca)3.1 channel downregulation and impaired endothelium-derived hyperpolarization-type relaxation in pulmonary arteries from chronically hypoxic rats. Exp. Physiol. 2013, 98, 957–969. [Google Scholar] [CrossRef]
- Zyrianova, T.; Lopez, B.; Liao, A.; Gu, C.; Wong, L.; Ottolia, M.; Olcese, R.; Schwingshackl, A. BK Channels Regulate LPS-induced CCL-2 Release from Human Pulmonary Endothelial Cells. Am. J. Respir. Cell Mol. Biol. 2021, 64, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, A.P.; Seara, F.A.C.; Baptista, E.F.; Barenco, T.S.; Sottani, T.B.B.; Souza, N.S.C.; Domingos, A.E.; Barbosa, R.A.Q.; Takiya, C.M.; Couto, M.T.; et al. BKCa Channel Activation Attenuates the Pathophysiological Progression of Monocrotaline-Induced Pulmonary Arterial Hypertension in Wistar Rats. Cardiovasc. Drugs Ther. 2021, 35, 719–732. [Google Scholar] [CrossRef]
- Renigunta, V.; Schlichthorl, G.; Daut, J. Much more than a leak: Structure and function of K2p-channels. Pflug. Arch. 2015, 467, 867–894. [Google Scholar] [CrossRef] [PubMed]
- Gardener, M.J.; Johnson, I.T.; Burnham, M.P.; Edwards, G.; Heagerty, A.M.; Weston, A.H. Functional evidence of a role for two-pore domain potassium channels in rat mesenteric and pulmonary arteries. Br. J. Pharmacol. 2004, 142, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Roman-Campos, D.; Austin, E.D.; Eyries, M.; Sampson, K.S.; Soubrier, F.; Germain, M.; Tregouet, D.A.; Borczuk, A.; Rosenzweig, E.B.; et al. A novel channelopathy in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Le Ribeuz, H.; Capuano, V.; Girerd, B.; Humbert, M.; Montani, D.; Antigny, F. Implication of Potassium Channels in the Pathophysiology of Pulmonary Arterial Hypertension. Biomolecules 2020, 10, 1261. [Google Scholar] [CrossRef]
- Antigny, F.; Hautefort, A.; Meloche, J.; Belacel-Ouari, M.; Manoury, B.; Rucker-Martin, C.; Pechoux, C.; Potus, F.; Nadeau, V.; Tremblay, E.; et al. Potassium Channel Subfamily K Member 3 (KCNK3) Contributes to the Development of Pulmonary Arterial Hypertension. Circulation 2016, 133, 1371–1385. [Google Scholar] [CrossRef]
- Lambert, M.; Capuano, V.; Boet, A.; Tesson, L.; Bertero, T.; Nakhleh, M.K.; Remy, S.; Anegon, I.; Pechoux, C.; Hautefort, A.; et al. Characterization of Kcnk3-Mutated Rat, a Novel Model of Pulmonary Hypertension. Circ. Res. 2019, 125, 678–695. [Google Scholar] [CrossRef]
- Lambert, M.; Mendes-Ferreira, P.; Ghigna, M.R.; LeRibeuz, H.; Adao, R.; Boet, A.; Capuano, V.; Rucker-Martin, C.; Bras-Silva, C.; Quarck, R.; et al. Kcnk3 Dysfunction Exaggerates the Development of Pulmonary Hypertension Induced by Left Ventricular Pressure Overload. Cardiovasc. Res. 2021, 117, 2474–2488. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Feig, J.E.; Morrissey, A.; Ghiu, I.A.; Artman, M.; Coetzee, W.A. KATP channels of primary human coronary artery endothelial cells consist of a heteromultimeric complex of Kir6.1, Kir6.2, and SUR2B subunits. J. Mol. Cell. Cardiol. 2004, 37, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Mederos y Schnitzler, M.; Derst, C.; Daut, J.; Preisig-Muller, R. ATP-sensitive potassium channels in capillaries isolated from guinea-pig heart. J. Physiol. 2000, 525 Pt 2, 307–317. [Google Scholar] [CrossRef]
- Janigro, D.; West, G.A.; Gordon, E.L.; Winn, H.R. ATP-sensitive K+ channels in rat aorta and brain microvascular endothelial cells. Am. J. Physiol. 1993, 265, C812–C821. [Google Scholar] [CrossRef] [PubMed]
- Katnik, C.; Adams, D.J. Characterization of ATP-sensitive potassium channels in freshly dissociated rabbit aortic endothelial cells. Am. J. Physiol. 1997, 272, H2507–H2511. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.; Chancellor, J.D. Adenosine potentiates flow-induced dilation of coronary arterioles by activating KATP channels in endothelium. Am. J. Physiol. 1995, 269, H541–H549. [Google Scholar] [CrossRef]
- Aziz, Q.; Li, Y.; Anderson, N.; Ojake, L.; Tsisanova, E.; Tinker, A. Molecular and functional characterization of the endothelial ATP-sensitive potassium channel. J. Biol. Chem. 2017, 292, 17587–17597. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Aziz, Q.; Anderson, N.; Ojake, L.; Tinker, A. Endothelial ATP-Sensitive Potassium Channel Protects Against the Development of Hypertension and Atherosclerosis. Hypertension 2020, 76, 776–784. [Google Scholar] [CrossRef]
- Chatterjee, S.; Al-Mehdi, A.B.; Levitan, I.; Stevens, T.; Fisher, A.B. Shear stress increases expression of a KATP channel in rat and bovine pulmonary vascular endothelial cells. Am. J. Physiol. Cell Physiol. 2003, 285, C959–C967. [Google Scholar] [CrossRef] [Green Version]
- Bohnen, M.S.; Ma, L.; Zhu, N.; Qi, H.; McClenaghan, C.; Gonzaga-Jauregui, C.; Dewey, F.E.; Overton, J.D.; Reid, J.G.; Shuldiner, A.R.; et al. Loss-of-Function ABCC8 Mutations in Pulmonary Arterial Hypertension. Circ. Genom. Precis. Med. 2018, 11, e002087. [Google Scholar] [CrossRef] [Green Version]
- Le Ribeuz, H.; Boet, A.; Lambert, M.; Chung, W.K.; Montani, D.; Humbert, M.; Antigny, F. Sur1/kir6.2 Potassium Channel a New Actor Involved in Pulmonary Arterial Hypertension. Circulation 2019, 140, A10804. [Google Scholar]
- Li, J.; Long, C.; Cui, W.; Wang, H. Iptakalim ameliorates monocrotaline-induced pulmonary arterial hypertension in rats. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Sahara, M.; Sata, M.; Morita, T.; Hirata, Y.; Nagai, R. Nicorandil attenuates monocrotaline-induced vascular endothelial damage and pulmonary arterial hypertension. PLoS ONE 2012, 7, e33367. [Google Scholar] [CrossRef]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, L.; Yu, L.; Wang, Y.; Jin, X.; Zhang, Q.; Lu, P.; Yu, X.; Zhong, W.; Zheng, X.; Cui, N.; et al. Inward Rectifier K+ Currents are Regulated by CaMKII in Endothelial Cells of Primarily Cultured Bovine Pulmonary Arteries. PLoS ONE 2015, 10, e0145508. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.J.; Fancher, I.S.; Bian, J.T.; Zhang, C.X.; Schwab, S.; Gaffin, R.; Phillips, S.A.; Levitan, I. Inwardly rectifying K+ channels are major contributors to flow-induced vasodilatation in resistance arteries. J. Physiol. 2017, 595, 2339–2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitbon, O.; Humbert, M.; Jagot, J.L.; Taravella, O.; Fartoukh, M.; Parent, F.; Herve, P.; Simonneau, G. Inhaled nitric oxide as a screening agent for safely identifying responders to oral calcium-channel blockers in primary pulmonary hypertension. Eur. Respir. J. 1998, 12, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Sitbon, O.; Humbert, M.; Jais, X.; Ioos, V.; Hamid, A.M.; Provencher, S.; Garcia, G.; Parent, F.; Herve, P.; Simonneau, G. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005, 111, 3105–3111. [Google Scholar] [CrossRef] [Green Version]
- McMurtry, I.F.; Davidson, A.B.; Reeves, J.T.; Grover, R.F. Inhibition of hypoxic pulmonary vasoconstriction by calcium antagonists in isolated rat lungs. Circ. Res. 1976, 38, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Sastry, B.K.; Narasimhan, C.; Reddy, N.K.; Raju, B.S. Clinical efficacy of sildenafil in primary pulmonary hypertension: A randomized, placebo-controlled, double-blind, crossover study. J. Am. Coll. Cardiol. 2004, 43, 1149–1153. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Tymchak, W.; Noga, M.; Webster, L.; Wu, X.C.; Lien, D.; Wang, S.H.; Modry, D.; Archer, S.L. Long-term treatment with oral sildenafil is safe and improves functional capacity and hemodynamics in patients with pulmonary arterial hypertension. Circulation 2003, 108, 2066–2069. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Frantz, R.P.; Severson, C.J.; Durst, L.A.; McGoon, M.D. Immediate and long-term hemodynamic and clinical effects of sildenafil in patients with pulmonary arterial hypertension receiving vasodilator therapy. Mayo Clin. Proc. 2003, 78, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, J.; Zhao, L.; Wang, Y.; Liu, J.; Shi, L.; Xu, M.; Wang, C. Sildenafil inhibits human pulmonary artery smooth muscle cell proliferation by decreasing capacitative Ca2+ entry. J. Pharmacol. Sci. 2008, 108, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, H.; Kuwahara, K.; Kiyonaka, S.; Mori, Y.; Kuwabara, Y.; Usami, S.; Nakagawa, Y.; Nishikimi, T.; Ueshima, K.; Nakao, K. TRPC3/6 as Potentially Novel Therapeutic Targets for The Treatment of Pulmonary Arterial Hypertension. J. Card. Fail. 2011, 17, S153. [Google Scholar] [CrossRef]
- Zhang, S.; Patel, H.H.; Murray, F.; Remillard, C.V.; Schach, C.; Thistlethwaite, P.A.; Insel, P.A.; Yuan, J.X.J. Pulmonary artery smooth muscle cells from normal subjects and IPAH patients show divergent cAMP-mediated effects on TRPC expression and capacitative Ca2+ entry. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1202–L1210. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Doods, H.; Stassen, J.M. Inhibition of human pulmonary artery smooth muscle cell proliferation and migration by sabiporide, a new specific NHE-1 inhibitor. J. Cardiovasc. Pharmacol. 2006, 48, 34–40. [Google Scholar] [CrossRef]
- Chen, L.; Gan, X.T.; Haist, J.V.; Feng, Q.; Lu, X.; Chakrabarti, S.; Karmazyn, M. Attenuation of compensatory right ventricular hypertrophy and heart failure following monocrotaline-induced pulmonary vascular injury by the Na+-H+ exchange inhibitor cariporide. J. Pharmacol. Exp. Ther. 2001, 298, 469–476. [Google Scholar]
- Abud, E.M.; Maylor, J.; Undem, C.; Punjabi, A.; Zaiman, A.L.; Myers, A.C.; Sylvester, J.T.; Semenza, G.L.; Shimoda, L.A. Digoxin inhibits development of hypoxic pulmonary hypertension in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 1239–1244. [Google Scholar] [CrossRef] [Green Version]
- Pozeg, Z.I.; Michelakis, E.D.; McMurtry, M.S.; Thebaud, B.; Wu, X.C.; Dyck, J.R.; Hashimoto, K.; Wang, S.; Moudgil, R.; Harry, G.; et al. In vivo gene transfer of the O2-sensitive potassium channel Kv1.5 reduces pulmonary hypertension and restores hypoxic pulmonary vasoconstriction in chronically hypoxic rats. Circulation 2003, 107, 2037–2044. [Google Scholar] [CrossRef] [Green Version]
- Brevnova, E.E.; Platoshyn, O.; Zhang, S.; Yuan, J.X. Overexpression of human KCNA5 increases IK V and enhances apoptosis. Am. J. Physiol. Cell Physiol. 2004, 287, C715–C722. [Google Scholar] [CrossRef] [Green Version]
- Zuo, X.; Zong, F.; Wang, H.; Wang, Q.; Xie, W.; Wang, H. Iptakalim, a novel ATP-sensitive potassium channel opener, inhibits pulmonary arterial smooth muscle cell proliferation by downregulation of PKC-alpha. J. Biomed. Res. 2011, 25, 392–401. [Google Scholar] [CrossRef] [Green Version]
- Zhu, R.; Bi, L.Q.; Wu, S.L.; Li, L.; Kong, H.; Xie, W.P.; Wang, H.; Meng, Z.L. Iptakalim attenuates hypoxia-induced pulmonary arterial hypertension in rats by endothelial function protection. Mol. Med. Rep. 2015, 12, 2945–2952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revermann, M.; Schloss, M.; Mieth, A.; Babelova, A.; Schroder, K.; Neofitidou, S.; Buerkl, J.; Kirschning, T.; Schermuly, R.T.; Hofstetter, C.; et al. Levosimendan attenuates pulmonary vascular remodeling. Intensive Care Med. 2011, 37, 1368–1377. [Google Scholar] [CrossRef]
- Morecroft, I.; Murray, A.; Nilsen, M.; Gurney, A.M.; MacLean, M.R. Treatment with the Kv7 potassium channel activator flupirtine is beneficial in two independent mouse models of pulmonary hypertension. Br. J. Pharmacol. 2009, 157, 1241–1249. [Google Scholar] [CrossRef] [Green Version]
- Sedivy, V.; Joshi, S.; Ghaly, Y.; Mizera, R.; Zaloudikova, M.; Brennan, S.; Novotna, J.; Herget, J.; Gurney, A.M. Role of Kv7 channels in responses of the pulmonary circulation to hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L48–L57. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos-Gomes, J.; Le Ribeuz, H.; Brás-Silva, C.; Antigny, F.; Adão, R. Role of Ion Channel Remodeling in Endothelial Dysfunction Induced by Pulmonary Arterial Hypertension. Biomolecules 2022, 12, 484. https://doi.org/10.3390/biom12040484
Santos-Gomes J, Le Ribeuz H, Brás-Silva C, Antigny F, Adão R. Role of Ion Channel Remodeling in Endothelial Dysfunction Induced by Pulmonary Arterial Hypertension. Biomolecules. 2022; 12(4):484. https://doi.org/10.3390/biom12040484
Chicago/Turabian StyleSantos-Gomes, Joana, Hélène Le Ribeuz, Carmen Brás-Silva, Fabrice Antigny, and Rui Adão. 2022. "Role of Ion Channel Remodeling in Endothelial Dysfunction Induced by Pulmonary Arterial Hypertension" Biomolecules 12, no. 4: 484. https://doi.org/10.3390/biom12040484
APA StyleSantos-Gomes, J., Le Ribeuz, H., Brás-Silva, C., Antigny, F., & Adão, R. (2022). Role of Ion Channel Remodeling in Endothelial Dysfunction Induced by Pulmonary Arterial Hypertension. Biomolecules, 12(4), 484. https://doi.org/10.3390/biom12040484