
Integrative Gene Expression and Metabolic Analysis Tool IgemRNA


Abstract
:
1. Introduction
2. Materials and Methods
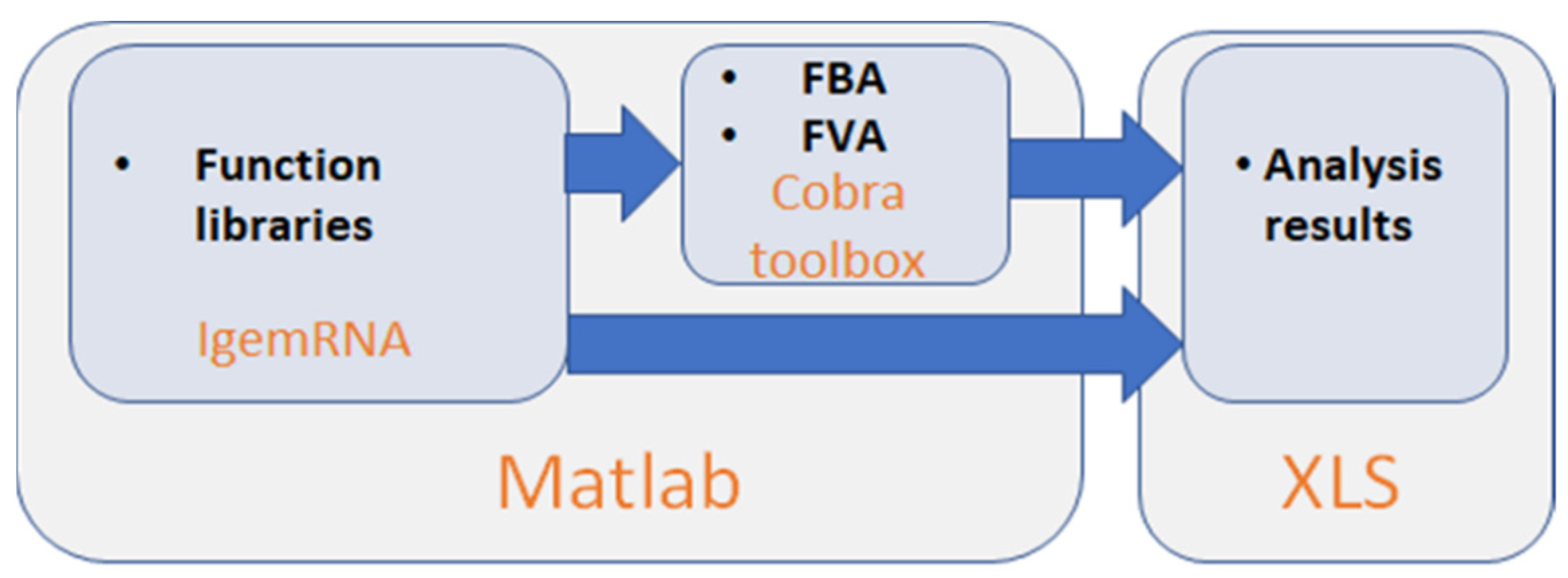
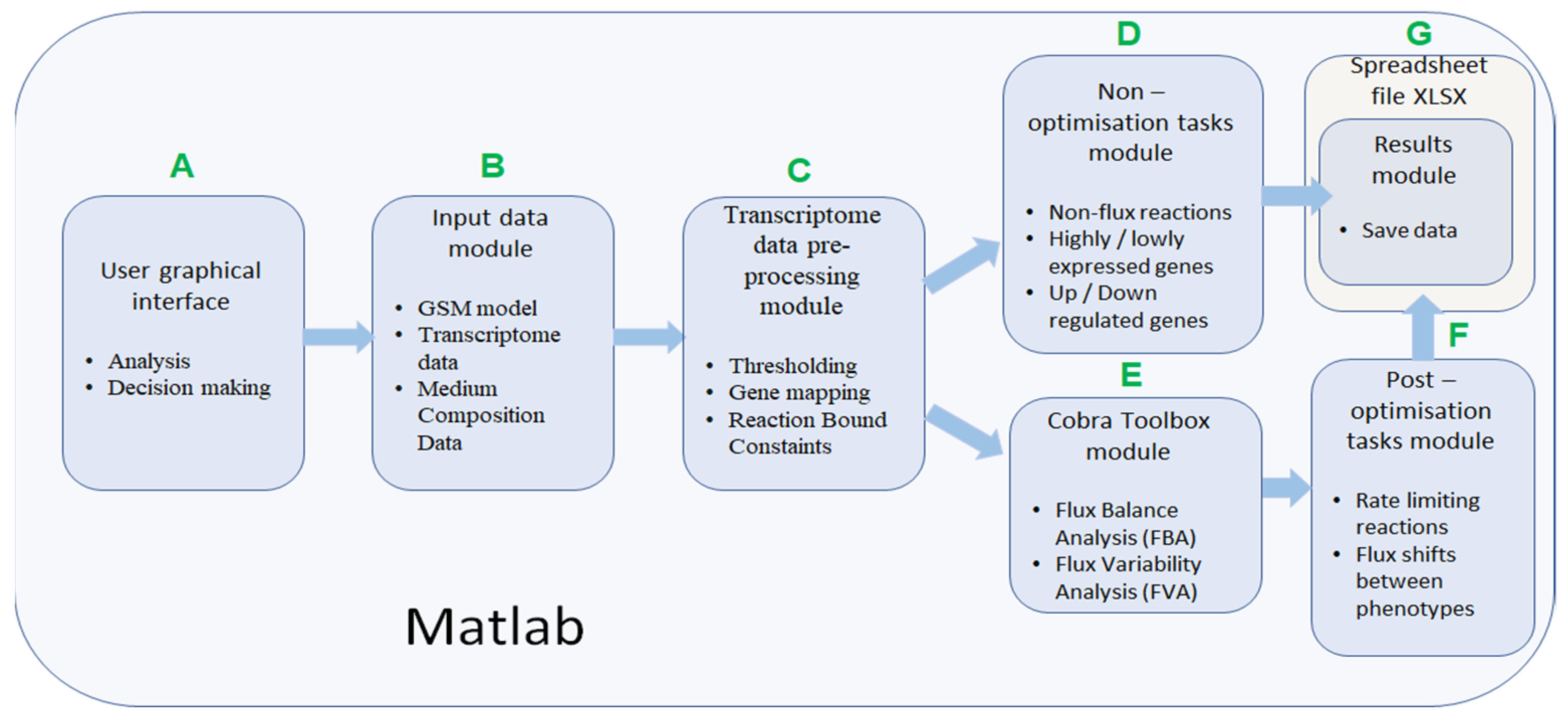
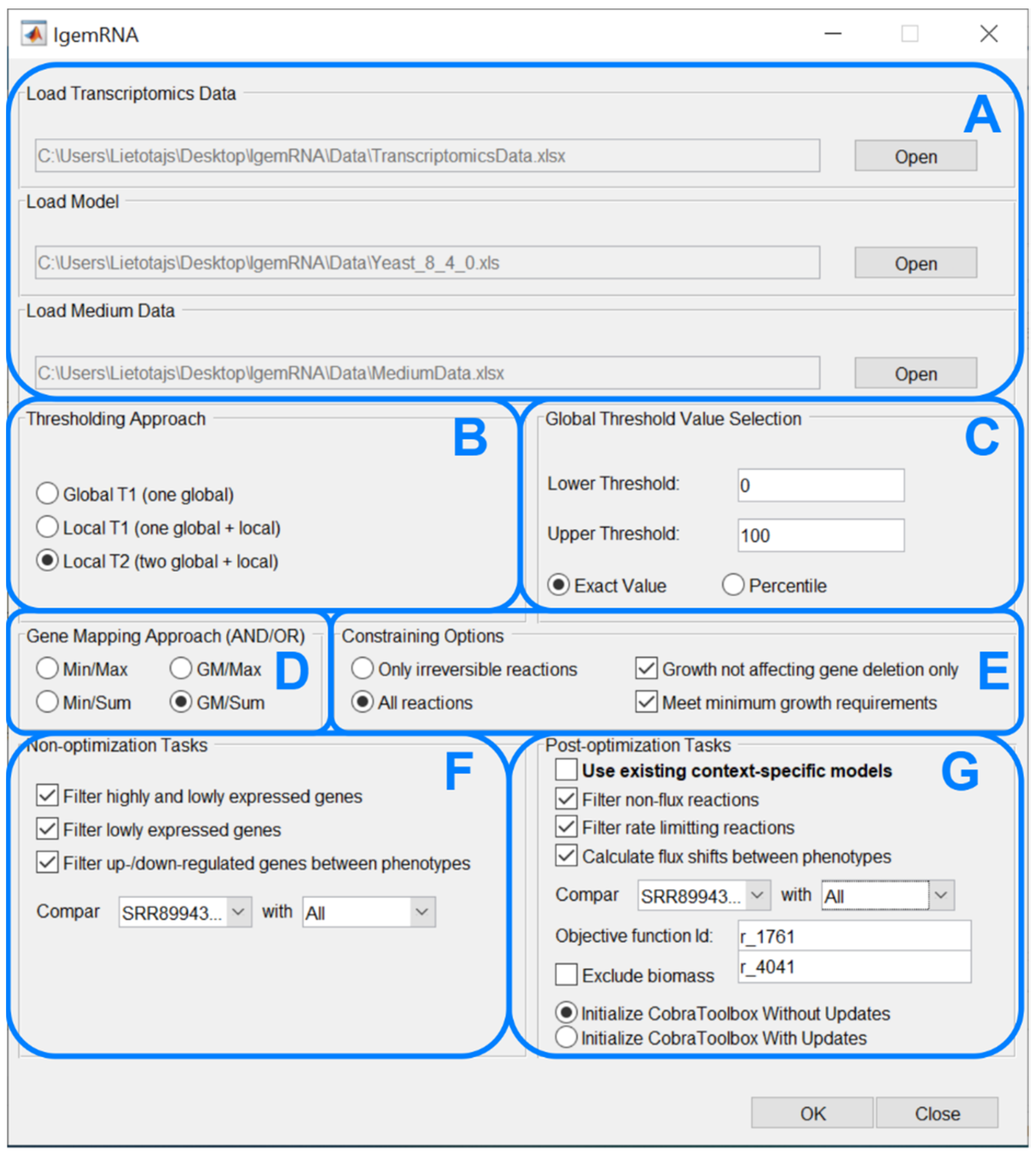
2.1. IgemRNA Architecture Description
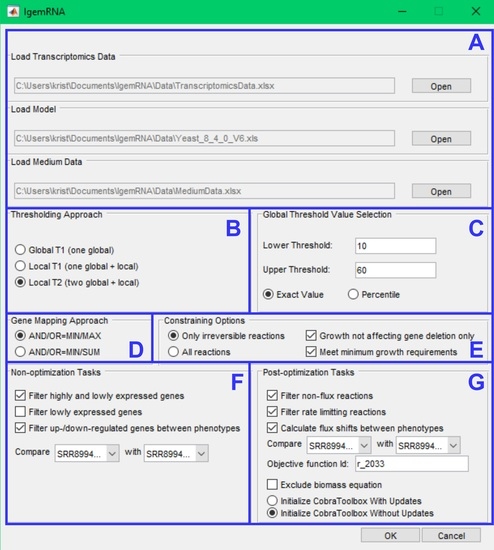
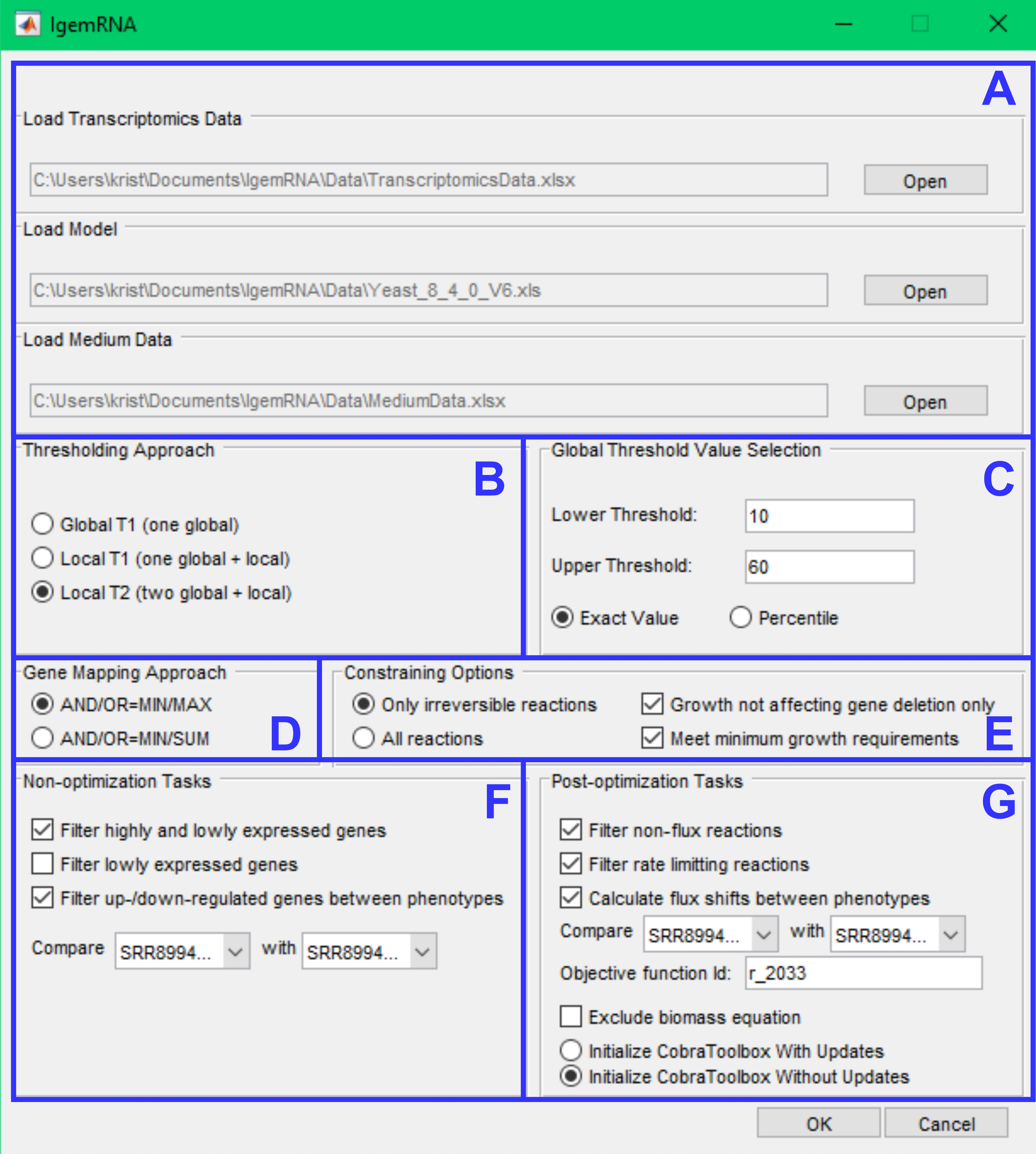
2.2. Tools Functionality Description
- Global T1 (GT1): is designed to analyze the transcriptome datasets using one global threshold. Example case shows that all transcriptome levels above 130 sequencing reads per gene (for a detailed description, see Section 2.3) are considered expressed, and others are considered suppressed. The global T1 threshold approach can be used for one or several phenotype transcriptome datasets.
- Local T1 (LT1): is designed to analyze transcriptome datasets having one global threshold value and a local rule. Local thresholds set a strict border for particular genes based on their varying gene expression levels across multiple samples to determine whether a gene is expressed or suppressed in a specific dataset. Local thresholds are only applied to those genes with expression levels above the global threshold since genes below the global threshold are automatically seen as suppressed. The example case shows that all transcriptome levels defined by the global threshold above 130 sequencing reads per gene are considered as possibly expressed, and below 130 sequencing reads per gene are considered suppressed. Local thresholds for specific genes determine expression or suppression for genes with expression levels above the global threshold.
- Local T2 (LT2) is designed to use two global threshold values: upper and lower thresholds. Transcriptome levels higher than the upper global threshold are considered expressed genes and are active, and transcriptome levels below the lower global threshold are considered inactive genes. All genes with expression levels between the upper and lower global thresholds are considered possibly active. Local rules for these genes are calculated across multiple gene expression datasets and applied to determine their activity levels. The Local T2 thresholding approach can be used if several transcriptome datasets are available. An example of Local T2 shows that all gene expression levels above the upper global threshold of 130 sequencing reads per gene are considered active. Gene expressions lower than the global threshold of 50 sequencing reads per gene are considered suppressed.
- Minimum (MIN) AND operands in the GPR association are calculated by taking the lowest gene expression value.
- Geometric mean (GM) [32] AND operands in the GPR association are calculated as the geometric mean of the gene expression values.
- Maximum (MAX) OR operands in the GPR association are calculated by taking the highest gene expression value.
- Sum (SUM) OR operands in the GPR association are calculated as the sum of all the gene expression values.
- Only irreversible reactions function. Enzymatic reactions have three different directions in metabolic models: irreversible, reversible, and backward irreversible. This approach constraints only irreversible and backward irreversible reactions in the respective direction.
- All reactions function constrains all reactions: irreversible and backward irreversible reactions in an oriented direction, but reversible reactions are constrained in both directions.
- Growth not affecting gene deletion only option allows for the deletion of only those genes with expression values below the given threshold and which deletion does not affect growth. Cobra Toolbox 3.0 singleGeneDeletion analysis with the FBA method is performed before executing gene deletion for those genes. Only if the returned output grRatio by singleGeneDeletion function is equal to 1 (meaning that the wild type growth is equal to the deletion strain growth) does the gene get deleted.
- Meet minimum growth requirements option allows constraining only those reactions where the gene mapping end value (which is set as a reaction bound) is not below the minimum growth requirements for that reaction. Minimum growth requirements are obtained by creating another context-specific model where only the gene deletion and medium exchange reaction constraining is applied to calculate the Cobra Toolbox 3.0 FBA (optimizeCbModel) minimization of growth.
- Filter high- and low-expression genes: this method uses chosen threshold data and sorts genes into high-expression and low-expression datasets.
- Filter low-expression genes: this method uses chosen threshold parameters, filters genes with expression levels below the supplied thresholds, and returns them as non-expressed datasets.
- Filter up-/down-regulated genes between phenotypes: This method uses chosen threshold data and filters up- and down-regulated genes from two or more transcriptome datasets. The gene names must match in all datasets.The resulting data are passed to the Spreadsheet module (Figure 2G).
- Cobra Toolbox module is called before post-optimization tasks to calculate FBA and FVA results using Cobra Toolbox 3.0 functions (Figure 2E). This module requires a metabolic model.
- Filter non-flux reactions: this functionality filters out enzymatic reactions that do not carry a flux because the coded gene transcription levels are below the chosen threshold value in the pre-processing module (Figure 2C).
- Filter rate-limiting reactions: This functionality finds maximum reaction rates equal to the calculated GPR value based on gene expression data. The function uses the FVA optimization method to calculate the minimal and maximal rate value for each reaction and then filters reactions with upper bounds of the same value as the FVA maximal results.
- Flux shifts between phenotypes: this function compares minimal and maximal fluxes (calculated by FVA) between different phenotypes or datasets, calculating ratios between them.
2.3. RNA Sequencing Data Analysis
3. Results
3.1. The Comparison of Available Transcriptome Data Integration Tools
3.2. IgemRNA Demonstration
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- López de Maturana, E.; Alonso, L.; Alarcón, P.; Martín-Antoniano, I.A.; Pineda, S.; Piorno, L.; Calle, M.L.; Malats, N. Challenges in the Integration of Omics and Non-Omics Data. Genes 2019, 10, 238. [Google Scholar] [CrossRef] [Green Version]
- Krassowski, M.; Das, V.; Sahu, S.K.; Misra, B.B. State of the Field in Multi-Omics Research: From Computational Needs to Data Mining and Sharing. Front. Genet. 2020, 11, 610798. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Boren, J.; Smith, U.; Uhlen, M.; Nielsen, J. Systems Biology in Hepatology: Approaches and Applications. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 365–377. [Google Scholar] [CrossRef]
- Benfeitas, R.; Uhlen, M.; Nielsen, J.; Mardinoglu, A. New Challenges to Study Heterogeneity in Cancer Redox Metabolism. Front. Cell Dev. Biol. 2017, 5, 65. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Nielsen, J. New Paradigms for Metabolic Modeling of Human Cells. Curr. Opin. Biotechnol. 2015, 34, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.; Song, C.W.; Shin, J.H.; Lee, S.Y. Biorefineries for the Production of Top Building Block Chemicals and Their Derivatives. Metab. Eng. 2015, 28, 223–239. [Google Scholar] [CrossRef]
- Kalnenieks, U.; Pentjuss, A.; Rutkis, R.; Stalidzans, E.; Fell, D.A. Modeling of Zymomonas Mobilis Central Metabolism for Novel Metabolic Engineering Strategies. Front. Microbiol. 2014, 5, 42. [Google Scholar] [CrossRef] [Green Version]
- Kalnenieks, U.; Balodite, E.; Strähler, S.; Strazdina, I.; Rex, J.; Pentjuss, A.; Fuchino, K.; Bruheim, P.; Rutkis, R.; Pappas, K.M.; et al. Improvement of Acetaldehyde Production in Zymomonas Mobilis by Engineering of Its Aerobic Metabolism. Front. Microbiol. 2019, 10, 2533. [Google Scholar] [CrossRef] [Green Version]
- McNally, C.P.; Borenstein, E. Metabolic Model-Based Analysis of the Emergence of Bacterial Cross-Feeding via Extensive Gene Loss. BMC Syst. Biol. 2018, 12, 69. [Google Scholar] [CrossRef]
- Borer, B.; Ataman, M.; Hatzimanikatis, V.; Or, D. Modeling Metabolic Networks of Individual Bacterial Agents in Heterogeneous and Dynamic Soil Habitats (IndiMeSH). PLoS Comput. Biol. 2019, 15, e1007127. [Google Scholar] [CrossRef]
- Marinos, G.; Kaleta, C.; Waschina, S. Defining the Nutritional Input for Genome-Scale Metabolic Models: A Roadmap. PLoS ONE 2020, 15, e0236890. [Google Scholar] [CrossRef] [PubMed]
- Stalidzans, E.; Seiman, A.; Peebo, K.; Komasilovs, V.; Pentjuss, A. Model-Based Metabolism Design: Constraints for Kinetic and Stoichiometric Models. Biochem. Soc. Trans. 2018, 46, 261–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pentjuss, A.; Odzina, I.; Kostromins, A.; Fell, D.A.; Stalidzans, E.; Kalnenieks, U. Biotechnological Potential of Respiring Zymomonas Mobilis: A Stoichiometric Analysis of Its Central Metabolism. J. Biotechnol. 2013, 165, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pentjuss, A.; Stalidzans, E.; Liepins, J.; Kokina, A.; Martynova, J.; Zikmanis, P.; Mozga, I.; Scherbaka, R.; Hartman, H.; Poolman, M.G.; et al. Model-Based Biotechnological Potential Analysis of Kluyveromyces marxianus Central Metabolism. J. Ind. Microbiol. Biotechnol. 2017, 44, 1177–1190. [Google Scholar] [CrossRef] [Green Version]
- Ramata-Stunda, A.; Valkovska, V.; Borodušķis, M.; Livkiša, D.; Kaktiņa, E.; Silamikele, B.; Borodušķe, A.; Pentjušs, A.; Rostoks, N. Development of Metabolic Engineering Approaches to Regulate the Content of Total Phenolics, Antiradical Activity and Organic Acids in Callus Cultures of the Highbush Blueberry (Vaccinium corymbosum L.). Agron. Res. 2020, 18, 1860–1872. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-Omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 117793221989905. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Lee, S.; Bidkhori, G.; Benfeitas, R.; Lovric, A.; Chen, S.; Uhlen, M.; Nielsen, J.; Mardinoglu, A. RMetD2: A Tool for Integration of Relative Transcriptomics Data into Genome-Scale Metabolic Models. bioRxiv 2019, 663096. [Google Scholar] [CrossRef]
- Åkesson, M.; Förster, J.; Nielsen, J. Integration of Gene Expression Data into Genome-Scale Metabolic Models. Metab. Eng. 2004, 6, 285–293. [Google Scholar] [CrossRef]
- Colijn, C.; Brandes, A.; Zucker, J.; Lun, D.S.; Weiner, B.; Farhat, M.R.; Cheng, T.-Y.; Moody, D.B.; Murray, M.; Galagan, J.E. Interpreting Expression Data with Metabolic Flux Models: Predicting Mycobacterium Tuberculosis Mycolic Acid Production. PLoS Comput. Biol. 2009, 5, e1000489. [Google Scholar] [CrossRef]
- Becker, S.A.; Palsson, B.O. Context-Specific Metabolic Networks Are Consistent with Experiments. PLoS Comput. Biol. 2008, 4, e1000082. [Google Scholar] [CrossRef] [PubMed]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and Analysis of Biochemical Constraint-Based Models Using the COBRA Toolbox v.3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Tarca, A.L.; Romero, R.; Draghici, S. Analysis of Microarray Experiments of Gene Expression Profiling. Am. J. Obstet. Gynecol. 2006, 195, 373–388. [Google Scholar] [CrossRef] [Green Version]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What Is Flux Balance Analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef]
- Fell, D.A.; Small, J.R. Fat Synthesis in Adipose Tissue. An Examination of Stoichiometric Constraints. Biochem. J. 1986, 238, 781–786. [Google Scholar] [CrossRef] [Green Version]
- Gudmundsson, S.; Thiele, I. Computationally Efficient Flux Variability Analysis. BMC Bioinform. 2010, 11, 489. [Google Scholar] [CrossRef] [Green Version]
- Nogales, J.; Agudo, L. A Practical Protocol for Integration of Transcriptomics Data into Genome-Scale Metabolic Reconstructions. In Hydrocarbon and Lipid Microbiology Protocols; Springer: Berlin/Heidelberg, Germany, 2015; pp. 135–152. [Google Scholar] [CrossRef]
- Blazier, A.S.; Papin, J.A. Integration of Expression Data in Genome-Scale Metabolic Network Reconstructions. Front. Physiol. 2012, 3, 299. [Google Scholar] [CrossRef] [Green Version]
- Lewis, N.E.; Nagarajan, H.; Palsson, B.O. Constraining the Metabolic Genotype–Phenotype Relationship Using a Phylogeny of in Silico Methods. Nat. Rev. Microbiol. 2012, 10, 291–305. [Google Scholar] [CrossRef] [Green Version]
- Richelle, A.; Joshi, C.; Lewis, N.E. Assessing Key Decisions for Transcriptomic Data Integration in Biochemical Networks. PLoS Comput. Biol. 2019, 15, e1007185. [Google Scholar] [CrossRef] [Green Version]
- Salvy, P.; Hatzimanikatis, V. The ETFL Formulation Allows Multi-Omics Integration in Thermodynamics-Compliant Metabolism and Expression Models. Nat. Commun. 2020, 11, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viéitez, C.; Martínez-Cebrián, G.; Solé, C.; Böttcher, R.; Potel, C.M.; Savitski, M.M.; Onnebo, S.; Fabregat, M.; Shilatifard, A.; Posas, F.; et al. A Genetic Analysis Reveals Novel Histone Residues Required for Transcriptional Reprogramming upon Stress. Nucleic Acids Res. 2020, 48, 3455–3475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2016 Update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential Expression Analysis of Multifactor RNA-Seq Experiments with Respect to Biological Variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Jerby, L.; Shlomi, T.; Ruppin, E. Computational Reconstruction of Tissue-specific Metabolic Models: Application to Human Liver Metabolism. Mol. Syst. Biol. 2010, 6, 401. [Google Scholar] [CrossRef]
- Jensen, P.A.; Papin, J.A. Functional Integration of a Metabolic Network Model and Expression Data without Arbitrary Thresholding. Bioinformatics 2011, 27, 541–547. [Google Scholar] [CrossRef]
- Jensen, P.A.; Lutz, K.A.; Papin, J.A. TIGER: Toolbox for Integrating Genome-Scale Metabolic Models, Expression Data, and Transcriptional Regulatory Networks. BMC Syst. Biol. 2011, 5, 147. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekaran, S.; Price, N.D. Probabilistic Integrative Modeling of Genome-Scale Metabolic and Regulatory Networks in Escherichia coli and Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2010, 107, 17845–17850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motamedian, E.; Mohammadi, M.; Shojaosadati, S.A.; Heydari, M. TRFBA: An Algorithm to Integrate Genome-Scale Metabolic and Transcriptional Regulatory Networks with Incorporation of Expression Data. Bioinformatics 2017, 29, 1399–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agren, R.; Bordel, S.; Mardinoglu, A.; Pornputtapong, N.; Nookaew, I.; Nielsen, J. Reconstruction of Genome-Scale Active Metabolic Networks for 69 Human Cell Types and 16 Cancer Types Using INIT. PLoS Comput. Biol. 2012, 8, e1002518. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Smallbone, K.; Dunn, W.B.; Murabito, E.; Winder, C.L.; Kell, D.B.; Mendes, P.; Swainston, N. Improving Metabolic Flux Predictions Using Absolute Gene Expression Data. BMC Syst. Biol. 2012, 6, 73. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Eddy, J.A.; Price, N.D. Reconstruction of Genome-Scale Metabolic Models for 126 Human Tissues Using MCADRE. BMC Syst. Biol. 2012, 6, 153. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Wallqvist, A.; Reifman, J. Modeling Phenotypic Metabolic Adaptations of Mycobacterium Tuberculosis H37Rv under Hypoxia. PLoS Comput. Biol. 2012, 8, e1002688. [Google Scholar] [CrossRef]
- Ravi, S.; Gunawan, R. ΔFBA—Predicting Metabolic Flux Alterations Using Genome-Scale Metabolic Models and Differential Transcriptomic Data. PLoS Comput. Biol. 2021, 17, e1009589. [Google Scholar] [CrossRef]
- Kim, J.; Reed, J.L. RELATCH: Relative Optimality in Metabolic Networks Explains Robust Metabolic and Regulatory Responses to Perturbations. Genome Biol. 2012, 13, R78. [Google Scholar] [CrossRef] [Green Version]
- Collins, S.B.; Reznik, E.; Segrè, D. Temporal Expression-Based Analysis of Metabolism. PLoS Comput. Biol. 2012, 8, e1002781. [Google Scholar] [CrossRef] [Green Version]
- Navid, A.; Almaas, E. Genome-Level Transcription Data of Yersinia Pestis Analyzed with a New Metabolic Constraint-Based Approach. BMC Syst. Biol. 2012, 6, 150. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Li, F.; Sánchez, B.J.; Zhu, Z.; Li, G.; Domenzain, I.; Marcišauskas, S.; Anton, P.M.; Lappa, D.; Lieven, C.; et al. A Consensus S. cerevisiae Metabolic Model Yeast8 and Its Ecosystem for Comprehensively Probing Cellular Metabolism. Nat. Commun. 2019, 10, 3586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, B.J.; Quan, C.W.; Mendoza, S.; Li, F.; Lu, H.; Beber, M.; Christian, L.; Kerkhoven, E.J. SysBioChalmers/Yeast-GEM: Yeast 8.4.0 (Version v8.4.0). 2020. Available online: https://zenodo.org/record/3894510#.Yld8NOhByUk (accessed on 11 April 2022). [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approach | Threshold Input | Visual Representation | Examples | |
|---|---|---|---|---|
| Global T1 | One global threshold:
|  | Lower threshold: 130 | |
| YDL227C | 871 | |||
| YDL226C | 126 | |||
| YDL225W | 319 | |||
| YDL224C | 56 | |||
| YDL223C | 13 | |||
| YDL222C | 3 | |||
| YDL221W | 135 | |||
| Local T1 | One global threshold:
|  | Lower threshold: 130 | |
| Local for YDL227C: 600 | ||||
| Local for YDL225W: 350 | ||||
| YDL227C | 871 | |||
| YDL226C | 126 | |||
| YDL225W | 319 | |||
| YDL223C | 56 | |||
| YDL223C | 13 | |||
| YDL222C | 3 | |||
| YDL221W | 135 | |||
| Local T2 | Two global thresholds:
|  | Lower Threshold: 50 | |
| Upper Threshold: 130 | ||||
| Local for YDL224C: 60 | ||||
| Local for YDL226C: 70 | ||||
| YDL227C | 871 | |||
| YDL226C | 126 | |||
| YDL225W | 319 | |||
| YDL224C | 56 | |||
| YDL223C | 13 | |||
| YDL222C | 3 | |||
| YDL221W | 135 | |||
| Requirement | Options |
|---|---|
| Reaction constraining options | Only irreversible reactions All reactions Non—essential gene deletion only Meet minimum growth requirements |
| Gene mapping approach | AND/MIN and OR/MAX AND/MIN and OR/SUM AND/geometric mean and OR/MAX AND/geometric mean and OR/SUM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grausa, K.; Mozga, I.; Pleiko, K.; Pentjuss, A. Integrative Gene Expression and Metabolic Analysis Tool IgemRNA. Biomolecules 2022, 12, 586. https://doi.org/10.3390/biom12040586
Grausa K, Mozga I, Pleiko K, Pentjuss A. Integrative Gene Expression and Metabolic Analysis Tool IgemRNA. Biomolecules. 2022; 12(4):586. https://doi.org/10.3390/biom12040586
Chicago/Turabian StyleGrausa, Kristina, Ivars Mozga, Karlis Pleiko, and Agris Pentjuss. 2022. "Integrative Gene Expression and Metabolic Analysis Tool IgemRNA" Biomolecules 12, no. 4: 586. https://doi.org/10.3390/biom12040586
APA StyleGrausa, K., Mozga, I., Pleiko, K., & Pentjuss, A. (2022). Integrative Gene Expression and Metabolic Analysis Tool IgemRNA. Biomolecules, 12(4), 586. https://doi.org/10.3390/biom12040586