
Paradoxical Behavior of Oncogenes Undermines the Somatic Mutation Theory


 , ,
, ,  and
and 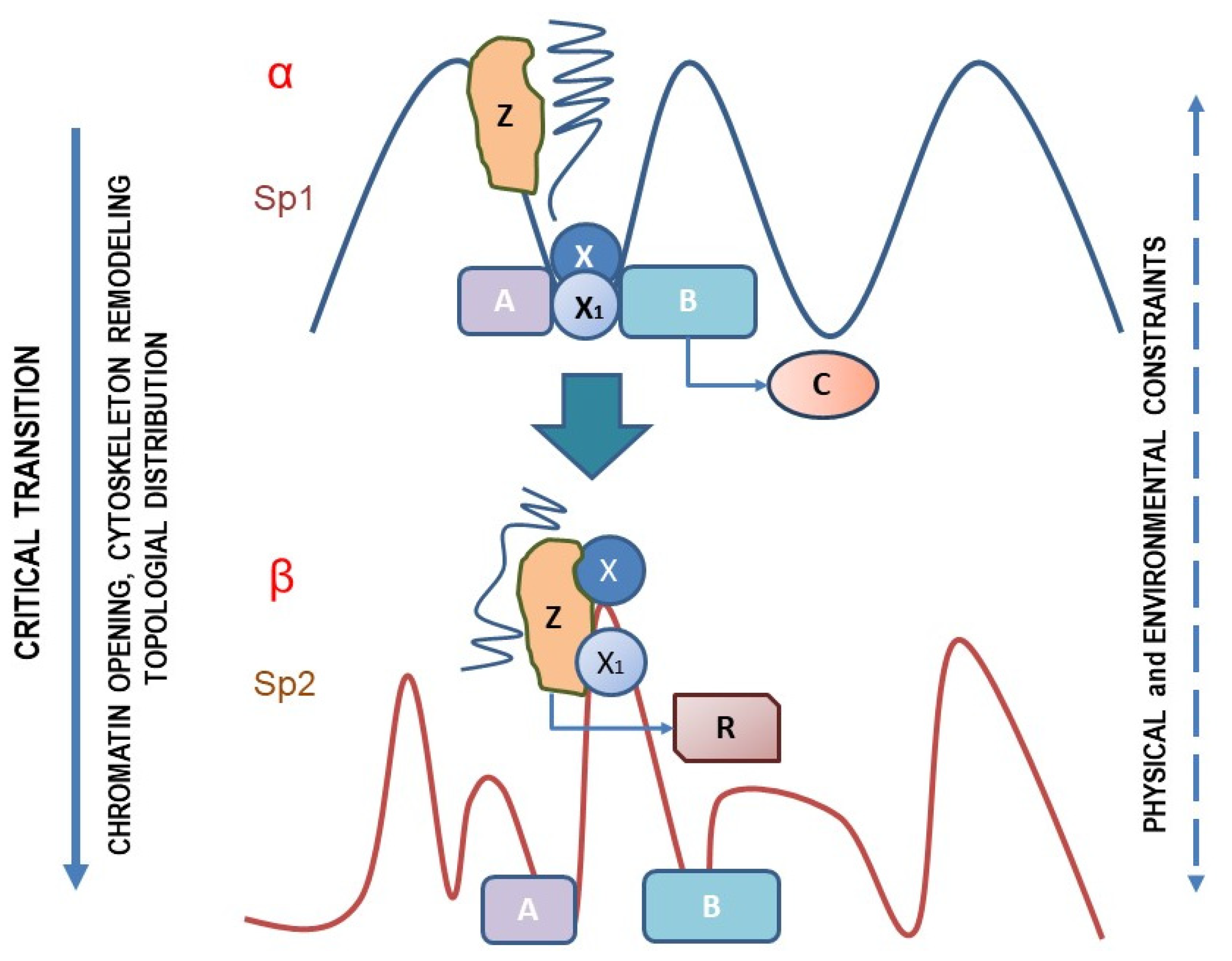{kind=link}
Abstract
:1. Introduction
2. Paradoxes
2.1. Mutations
2.2. Non-Genotoxic Carcinogenic Agents
2.3. Paradoxical Molecular Effects on Cell Signaling
3. A Testable Hypothesis
4. Conclusions
Funding
Conflicts of Interest
References
- Reece, J.B.; Urry, L.A.; Cain, M.L.; Wasserman, S.A.; Minorsky, P.V.; Jackson, R.B. Campbell Biology, 9th ed.; Pearson Benjamin Cummings: San Francisco, CA, USA, 2011; p. 379. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Brücher, B.L.; Jamall, I.S. Somatic Mutation Theory—Why it’s Wrong for Most Cancers. Cell Physiol. Biochem. 2016, 38, 1663–1680. [Google Scholar] [CrossRef] [PubMed]
- Backer, S.G. Recognizing Paradigm Instability in Theories of Carcinogenesis. Br. J. Med. Med. Res. 2014, 4, 1149–1163. [Google Scholar] [CrossRef]
- Autier, P.; Boniol, M.; Gavin, A.; Vatten, L.J. Breast cancer mortality in neighbouring European countries with different levels of screening but similar access to treatment: Trend analysis of WHO mortality database. BMJ 2011, 343, d4411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkes, N. High cost of cancer treatment doesn’t reflect benefits, say specialists. BMJ 2011, 343, d6220. [Google Scholar] [CrossRef]
- Seymour, C.B.; Mothersill, C. Breast cancer causes and treatment: Where are we going wrong? Breast Cancer 2013, 5, 111–119. [Google Scholar] [CrossRef] [Green Version]
- Gawrylewski, A. The Trouble with Animal Models. The Scientist. 2007. Available online: https://www.the-scientist.com/uncategorized/the-trouble-with-animal-models-46344 (accessed on 25 April 2022).
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar]
- Le Fanu, J. Rise and Fall of Modern Medicine. Lancet 1999, 354, 518. [Google Scholar] [CrossRef] [Green Version]
- McNeal, A.S.; Belote, R.L.; Zeng, H.; Urquijo, M.; Barker, K.; Torres, R.; Curtin, M.; Shain, A.H.; Andtbacka, R.H.; Holmen, S.; et al. BRAFV600E induces reversible mitotic arrest in human melanocytes via microRNA-mediated suppression of AURKB. Elife 2021, 10, e70385. [Google Scholar] [CrossRef]
- Ruiz-Vega, R.; Chen, C.F.; Razzak, E.; Vasudeva, P.; Krasieva, T.B.; Shiu, J.; Caldwell, M.G.; Yan, H.; Lowengrub, J.; Ganesan, A.K.; et al. Dynamics of nevus development implicate cell cooperation in the growth arrest of transformed melanocytes. Elife 2020, 9, e61026. [Google Scholar] [CrossRef] [PubMed]
- Li, J. Context-Dependent Roles of Claudins in Tumorigenesis. Front. Oncol. 2021, 11, 676781. [Google Scholar] [CrossRef] [PubMed]
- Shiu, J.; Lander, A.D. When oncogenes do not cause cancer. Elife 2021, 10, e74912. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. Coming full circle-from endless complexity to simplicity and back again. Cell 2014, 157, 267–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Versteeg, R. Cancer: Tumours outside the mutation box. Nature 2014, 506, 438–439. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.G.; Kramer, B.S. Paradoxes in carcinogenesis: New opportunities for research directions. BMC Cancer 2007, 7, 151. [Google Scholar] [CrossRef] [Green Version]
- Soto, A.M.; Sonnenschein, C. The tissue organization field theory of cancer: A testable replacement for the somatic mutation theory. BioEssays 2011, 33, 332–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, A.M.; Sonnenschein, C. Is systems biology a promising approach to resolve controversies in cancer research? Cancer Cell Int. 2012, 12, 12. [Google Scholar] [CrossRef] [Green Version]
- Bizzarri, M.; Pasqualato, A.; Cucina, A.; Pasta, V. Physical forces and non linear dynamics mould fractal cell shape: Quantitative morphological parameters and cell phenotype. HistolHistopathol 2013, 28, 155–174. [Google Scholar] [CrossRef]
- Sonnenschein, C.; Soto, A.M. And yet another epicycle. Bioessays 2006, 28, 100–101. [Google Scholar] [CrossRef]
- Bizzarri, M.; Cucina, A. SMT and TOFT: Why and How They are Opposite and Incompatible Paradigms. Acta Biotheor. 2016, 64, 221–239. [Google Scholar] [CrossRef]
- Sorsa, M. Somatic mutation theory. J. Toxicol. Environm. Health 1980, 6, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Woodman, S.E.; Mills, G.B. Are oncogenes sufficient to cause human cancer? Proc. Natl. Acad. Sci. USA 2010, 107, 20599–20600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, W.C.; Counter, C.M.; Lundberg, A.S.; Beijersbergen, R.L.; Brooks, M.W.; Weinberg, R.A. Creation of human tumour cells with defined genetic elements. Nature 1999, 400, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Morales, C.P.; Holt, S.E.; Ouellette, M.; Kaur, K.J.; Yan, Y.; Wilson, K.S.; White, M.A.; Wright, W.E.; Shay, J.W. Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nat. Genet. 1999, 21, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Gonda, T.J. The c-Myb oncoprotein. Int. J. Biochem Cell Biol. 1998, 30, 547–551. [Google Scholar] [CrossRef]
- Holliday, R. Neoplastic transformation: The contrasting stability of human and mouse cells. Cancer Surv. 1996, 28, 103–115. [Google Scholar]
- Kacser, H.; Burns, J.A. The molecular basis of dominance. Genetics 1981, 97, 639–665. [Google Scholar] [CrossRef]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Kan, Z.; Jaiswal, B.S.; Stinson, J.; Janakiraman, V.; Bhatt, D.; Stern, H.M.; Yue, P.; Haverty, P.M.; Bourgon, R.; Zheng, J.; et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010, 466, 869–873. [Google Scholar] [CrossRef] [Green Version]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [Green Version]
- Konishi, N.; Hiasa, Y.; Matsuda, H.; Tao, M.; Tsuzuki, T.; Hayashi, I.; Kitahori, Y.; Shiraishi, T.; Yatani, R.; Shimazaki, J.; et al. Intratumor cellular heterogeneity and alterations in ras oncogene and p53 tumor suppressor gene in human prostate carcinoma. Am. J. Pathol. 1995, 147, 112–122. [Google Scholar]
- Baisse, B.; Bouzourene, H.; Saraga, E.P.; Bosman, F.T.; Benhattar, J. Intratumor genetic heterogeneity in advanced human colorectal adenocarcinoma. Int. J. Cancer 2001, 93, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Maeda, T.; Mohapatra, G.; Waldman, F.M.; Davis, R.L.; Feuerstein, B.G. Heterogeneity, poliploidy, aneusomy, and 9p deletion in human glioblastoma multiforme. Cancer Genet. Cytogenet. 1995, 83, 127–135. [Google Scholar] [CrossRef]
- Szollosi, J.; Balazs, M.; Fuerstein, B.G.; Benz, C.C.; Waldman, F.M. ERBB-2 (HER2/neu) gene copy number, p185HER-2 overexpression and intratumor heterogeneity in human breast cancer. Cancer Res. 1995, 55, 5400–5407. [Google Scholar] [PubMed]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Plattner, R.; Anderson, M.J.; Sato, K.J.; Fasching, C.L.; Der, C.J.; Stanbridge, E.J. Loss of oncogenic ras expression does not correlate with loss of tumorigenicity in human cells. Proc. Natl. Acad. Sci. USA 1996, 93, 6665–6670. [Google Scholar] [CrossRef] [Green Version]
- Albino, A.P.; Le Strange, R.; Oliff, A.I.; Furth, M.E.; Old, L.J. Transforming ras genes from human melanoma: A manifestation of tumor heterogeneity? Nature 1984, 308, 69–72. [Google Scholar] [CrossRef]
- Washington, C.; Dalbegue, F.; Abreo, F.; Taubenberger, J.K.; Lichy, J.H. Loss of heterozygosity in fibrocystic change of the breast: Genetic relationships between benign proliferative lesions and associated carcinomas. Am. J. Pathol. 2000, 157, 323–329. [Google Scholar] [CrossRef]
- Lupski, J.R. Genetics. Genome mosaicism—One human, multiple genomes. Science 2013, 341, 358–359. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, W.; Velculescu, V.E.; Kern, S.E.; Hruban, R.H.; Hamilton, S.R.; Vogelstein, B.; Kinzler, K.W. Gene expression profiles in normal and cancer cells. Science 1997, 276, 1268–1272. [Google Scholar] [CrossRef]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Jamshidi, N.; Margolis, D.J.; Raman, S.; Huang, J.; Reiter, R.E.; Kuo, M.D. Multiregional Radiogenomic Assessment of Prostate Microenvironments with Multiparametric MR Imaging and DNA Whole-Exome Sequencing of Prostate Glands with Adenocarcinoma. Radiology 2017, 284, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández, J.L.; Rodríguez-Parets, J.O.; Valero, J.M.; Muñoz, M.A.; Benito, M.R.; Hernandez, J.M.; Bullón, A. High resolution genome-wide analysis of chromosomal alterations in elastofibroma. Virchows. Arch. 2010, 456, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, Y.; Boyle, D.L.; Rosengren, S.; Green, D.R.; Zvaifler, N.J.; Firestein, G.S. Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc. Natl. Acad. Sci. USA 2002, 99, 10025–10030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Magalhães, J.P. Every gene can (and possibly will) be associated with cancer. Trends Genet. 2022, 38, 216–217. Available online: https://www.sciencedirect.com/science/article/pii/S0168952521002663 (accessed on 10 February 2022). [CrossRef] [PubMed]
- Schuller, H.M. Mechanisms of smoking-related lung and pancreatic adenocarcinoma development. Nat. Rev. Cancer 2002, 2, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Wellcome Trust, Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, UK. Catalogue Of Somatic Mutations in Cancer (COSMIC). Available online: http://www.sanger.ac.uk/perl/genetics/CGP/cosmic?action=gene&ln=KRAS (accessed on 2 February 2022).
- Sasco, A.J.; Secretan, M.B.; Straif, K. Tobacco smoking and cancer: A brief review of recent epidemiological evidence. Lung Cancer 2004, 45 (Suppl. S2), S3–S9. [Google Scholar] [CrossRef]
- Porta, M.; Crous-Bou, M.; Wark, P.A.; Vineis, P.; Real, F.X.; Malats, N.; Kampman, E. Cigarette smoking and K-ras mutations in pancreas, lung and colorectal adenocarcinomas: Etiopathogenic similarities, differences and paradoxes. Mutat. Res. 2009, 682, 83–93. [Google Scholar] [CrossRef]
- Tseng, C.H.; Tsuang, B.J.; Chiang, C.J.; Ku, K.C.; Tseng, J.S.; Yang, T.Y.; Hsu, K.H.; Chen, K.C.; Yu, S.L.; Lee, W.C.; et al. The Relationship Between Air Pollu-tion and Lung Cancer in Nonsmokers in Taiwan. J. Thorac. Oncol. 2019, 14, 784–792. [Google Scholar] [CrossRef]
- Coulombe, L.; Kalousek, D.K.; Eaves, C.J.; Gupta, C.M.; Eaves, A.C. Long-term marrow culture reveals chromosomally normal hematopoietic progenitor cells in patients with Philadelphia chromosome-positive chronic myelogenous leukemia. N. Engl. J. Med. 1983, 308, 1493–1498. [Google Scholar] [CrossRef]
- Gottschalk, S.; Anderson, N.; Hainz, C.; Eckhardt, S.G.; Serkova, N.J. Imatinib (STI571)-mediated changes in glucose metabolism in human leukemia BCR-ABL-positive cells. Clin. Cancer Res. 2004, 10, 6661–6668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisberg, E.; Griffin, J.D. CML cell trafficking: Influence of the stromal microenvironment. Open J. Hematol. 2012, 3, S1–S2. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Li, M.; McDonald, T.; Holyoake, T.L.; Moon, R.T.; Campana, D.; Shultz, L.; Bhatia, R. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-β-catenin signaling. Blood 2013, 121, 1824–1838. [Google Scholar] [CrossRef] [PubMed]
- Pellicano, F.; Mukherjee, L.; Holyoake, T.L. Concise Review: Cancer Cells Escape from Oncogene Addiction: Understanding the Mechanisms Behind Treatment Failure for More Effective Targeting. Stem Cells 2014, 32, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Saw, K.M.; Eaves, A.; Eaves, C. Instability of BCR-ABL gene in primary and cultured chronic myeloid leukemia stem cells. J. Natl. Cancer Inst. 2007, 99, 680–693. [Google Scholar] [CrossRef] [Green Version]
- Erson, A.E.; Petty, E.M. Molecular and genetic events in neoplastic transformation. In Cancer Epidemiology and Prevention, 3rd ed.; Schottenfeld, D., Fraumeni, J.F., Jr., Eds.; Oxford University Press: New York, NY, USA, 2006; Volume 4, pp. 47–64. [Google Scholar] [CrossRef]
- Balmain, A.; Harris, C.C. Carcinogenesis in mouse and human cells: Parallels and paradoxes. Carcinogenesis 2000, 21, 371–377. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Bizzarri, M.; Cucina, A.; Conti, F.; D’Anselmi, F. Beyond the oncogene paradigm: Understanding complexity in cancerogenesis. Acta Biotheor. 2008, 56, 173–196. [Google Scholar] [CrossRef]
- Lorimore, S.A.; Coates, P.J.; Wright, E.G. Radiation-induced genomic instability and bystander effects: Inter-related non targeted effects of exposure to ionizing radiation. Oncogene 2003, 22, 7058–7069. [Google Scholar] [CrossRef] [Green Version]
- Soto, A.M.; Sonnenschein, C. One hundred years of somatic mutation theory of carcinogenesis: Is it time to switch? BioEssays 2014, 36, 118–120. [Google Scholar] [CrossRef] [Green Version]
- Boland, C.R.; Ricciardiello, L. How many mutations does it take to make a tumor? Proc. Natl. Acad. Sci. USA 1999, 96, 14675–14677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, V.Y.; Wang, W.K.; Duesberg, P. Dominant transformation by mutated human ras genes in vitro requires more than 100 times higher expression than is observed in cancers. Proc. Natl. Acad. Sci. USA 1997, 94, 9614–9619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, G.; Carmona, R.; Zakeri, K.; Lee, C.H.; Borgan, S.; Marhoon, Z.; Sharabi, A.; Mell, L.K. Specificity of Genetic Biomarker Studies in Cancer Research: A Systematic Review. PLoS ONE 2016, 11, e0156489. [Google Scholar] [CrossRef] [PubMed]
- Lijinsky, W. Non-genotoxic environmental carcinogens. Environ. Carcinog. Rev. 1990, 8, 45–87. [Google Scholar] [CrossRef]
- Hernández, L.G.; van Steeg, H.; Luijten, M.; van Benthem, J. Mechanisms of non-genotoxic carcinogens and importance of a weight of evidence approach. Mutat. Res. 2009, 682, 94–109. [Google Scholar] [CrossRef]
- Berenblum, I.; Shubik, P. An experimental study of the initiating stage of carcinogenesis, and a re-examination of the somatic cell mutation theory of cancer. Br. J. Cancer 1949, 3, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, I.B. Non-mutagenic mechanism in carcinogenesis: Role of protein kinase C in signal transduction and growth control. Environ. Health Perspect 1991, 93, 175–179. [Google Scholar] [CrossRef]
- Lee, S.J.; Yum, Y.N.; Kim, S.C. Distinguishing between genotoxic and non-genotoxic hepatocarcinogens by gene expression profiling and bioinformatic pathway analysis. Sci. Rep. 2013, 3, 2783. [Google Scholar] [CrossRef] [Green Version]
- Moizhess, T.G. Carcinogenesis induced by foreign bodies. Biochemistry 2008, 73, 763–775. [Google Scholar] [CrossRef]
- Alexander, J.J.; Moawad, J.; Cai, D. Primary intimal sarcoma of the aorta associated with a dacron graft and resulting in arterial rupture. Vasc. Endovasc. Surg. 2006, 40, 509–515. [Google Scholar] [CrossRef]
- IARC Monographs. Volume 74. Surgical Implants and Other Foreign Bodies. Available online: http://monographs.iarc.fr/ENG/Monographs/vol74/volume74.pdf (accessed on 20 January 2022).
- Tomatis, L. Studies in subcutaneous carcinogenesis with implants of glass and Teflon in mice. Acta Unio. Int. Contra. Cancrum. 1963, 19, 607–611. [Google Scholar] [PubMed]
- Karp, R.D.; Johnson, K.H.; Buoen, L.C.; Ghobrial, H.K.; Brand, I.; Brand, K.G. Tumorigenesis by Millipore filters in mice: Histology and ultrastructure of tissue reactions as related to pore size. J. Natl. Cancer Inst. 1973, 51, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Brand, K.G.; Buoen, L.C.; Johnson, K.H.; Brand, I. Etiological factors, stages, and the role of the foreign body in foreign body tumorigenesis: A review. Cancer Res. 1975, 35, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Sonnenschein, C.; Soto, A.M. Somatic mutation theory of carcinogenesis: Why it should be dropped and replaced. Mol. Carcinog. 2000, 29, 205–211. [Google Scholar] [CrossRef]
- Crick, F.H. On protein synthesis. Symp. Soc. Exp. Biol. 1958, 12, 138–163. [Google Scholar]
- Bailly, F.; Longo, G. Mathematics and Natural Sciences: The Physical Singularity of Life; Imperial College Press: London, UK, 2011. [Google Scholar]
- Longo, G. From exact sciences to life phenomena: Following Schrödinger and Turing on Programs. Inf. Comput. 2009, 207, 545–558. [Google Scholar] [CrossRef] [Green Version]
- Longo, G.; Miquel, P.A.; Sonnenschein, C.; Soto, A.M. Is information a proper observable for biological organization? Prog. Biophys. Mol. Biol. 2012, 109, 108–114. [Google Scholar] [CrossRef] [Green Version]
- LoPiccolo, J.; Granville, C.A.; Gills, J.J.; Dennis, P.A. Targeting Akt in cancer therapy. Anticancer Drugs 2007, 18, 861–874. [Google Scholar] [CrossRef]
- Yoeli-Lerner, M.; You, G.K.; Rabinovitz, I.; Erhardt, P.; Jauliac, S.; Toker, A. Akt blocks breast cancer cell motility and invasion through the transcription factor NFAT. Mol. Cell 2005, 20, 539–550. [Google Scholar] [CrossRef]
- Irie, H.Y.; Pearline, R.V.; Grueneberg, D.; Hsia, M.; Ravichandran, P.; Kothari, N.; Natesan, S.; Brugge, J.S. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial–mesenchymal transition. J. Cell Biol. 2005, 171, 1023–1034. [Google Scholar] [CrossRef]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, D.A.; Wang, X.J.; Donehower, L.A.; Roop, D.R. Paradoxical tumor inhibitory effect of p53 loss in transgenic mice expressing epidermal-targeted v-ras, Ha, v-fos, or human transforming growth factor alpha. Cancer Res. 1996, 56, 4413–4423. [Google Scholar] [PubMed]
- Wang, X.J.; Greenhalgh, D.A.; Jiang, A.; He, D.; Zhong, L.; Medina, D.; Brinkley, B.R.; Roop, D.R. Expression of a p53 mutant in the epidermis of transgenic mice accelerates chemical carcinogenesis. Oncogene 1998, 17, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, G.S. Conflicting roles of molecules in hepatocarcinogenesis: Paradigm or paradox. Cancer Cell 2012, 21, 150–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satgé, D.; Bénard, J. Carcinogenesis in Down syndrome: What can be learned from trisomy 21? Semin. Cancer Biol. 2008, 18, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P.; Benchimol, S. p53: Oncogene or anti-oncogene? Genes Dev. 1990, 4, 1–8. [Google Scholar] [CrossRef] [Green Version]
- de Keizer, P.L.; Laberge, R.M.; Campisi, J. p53: Pro-aging or pro-longevity? Aging 2010, 2, 377–379. [Google Scholar] [CrossRef] [Green Version]
- Bizzarri, M.; Palombo, A.; Cucina, A. Theoretical aspects of systems biology. Prog. Biophys. Mol. Biol. 2013, 112, 33–43. [Google Scholar] [CrossRef]
- Stern, R. Hyaluronan metabolism: A major paradox in cancer biology. Pathol. Biol. 2005, 53, 372–382. [Google Scholar] [CrossRef]
- Anttila, M.A.; Tammi, R.H.; Tammi, M.I.; Syrjänen, K.J.; Saarikoski, S.V.; Kosma, V.M. High levels of stromal hyaluronan predict poor disease outcome in epithelial ovarian cancer. Cancer Res. 2000, 60, 150–155. [Google Scholar]
- Posey, J.T.; Soloway, M.S.; Ekici, S.; Sofer, M.; Civantos, F.; Duncan, R.C.; Lokeshwar, V.B. Evaluation of the prognostic potential of hyaluronic acid and hyaluronidase (HYAL1) for prostate cancer. Cancer Res. 2003, 63, 2638–2644. [Google Scholar] [PubMed]
- Powell, A.E.; Wang, Y.; Li, Y.; Poulin, E.J.; Means, A.L.; Washington, M.K.; Higginbotham, J.N.; Juchheim, A.; Prasad, N.; Levy, S.E.; et al. The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell 2012, 149, 146–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomasson, M.; Wang, B.; Hammarsten, P.; Dahlman, A.; Persson, J.L.; Josefsson, A.; Stattin, P.; Granfors, T.; Egevad, L.; Henriksson, R.; et al. LRIG1 and the liar paradox in prostate cancer: A study of the expression and clinical significance of LRIG1 in prostate cancer. Int. J. Cancer 2011, 128, 2843–2852. [Google Scholar] [CrossRef] [PubMed]
- Isalan, M.; Morrison, M. This title is false. Nature 2009, 458, 969. [Google Scholar] [CrossRef]
- de La Coste, A.; Mignon, A.; Fabre, M. Paradoxical inhibition of c-myc-induced carcinogenesis by Bcl-2 in transgenic mice. Cancer Res. 1999, 59, 5017–5022. [Google Scholar]
- Sumi, T.; Tsuneyoshi, N.; Nakatsuji, N.; Suemori, H. Apoptosis and differentiation of human embryonic stem cells induced by sustained activation of c-Myc. Oncogene 2007, 26, 5564–5576. [Google Scholar] [CrossRef] [Green Version]
- Amati, B.; Land, H. Myc-Max-Mad: A transcription factor network controlling cell cycle progression, differentiation and death. Curr. Opin. Genet. Dev. 1994, 4, 102–108. [Google Scholar] [CrossRef]
- You, Z.; Saims, D.; Chen, S.; Zhang, Z.; Guttridge, D.C.; Guan, K.L.; MacDougald, O.A.; Brown, A.M.; Evan, G.; Kitajewski, J.; et al. Wnt signaling promotes oncogenic transformation by inhibiting c-Myc–induced apoptosis. J. Cell Biol. 2002, 157, 429–440. [Google Scholar] [CrossRef]
- Lee, L.A.; Resar, L.M.; Dang, C.V. Cell Density and Paradoxical Transcriptional Properties of c-Myc and Max in Cultured Mouse Fibroblasts. J. Clin. Investig. 1995, 95, 900–904. [Google Scholar] [CrossRef] [Green Version]
- Cucina, A.; Biava, P.M.; D’Anselmi, F.; Coluccia, P.; Conti, F.; di Clemente, R.; Miccheli, A.; Frati, L.; Gulino, A.; Bizzarri, M. Zebrafish embryo proteins induce apoptosis in human colon cancer cells (Caco2). Apoptosis 2006, 11, 1617–1628. [Google Scholar] [CrossRef]
- Payne, S.L.; Hendrix, M.J.; Kirschmann, D.A. Paradoxical roles for lysyl oxidases in cancer—A prospect. J. Cell Biochem. 2007, 101, 1338–1354. [Google Scholar] [CrossRef] [PubMed]
- Moses, H.L.; Yang, E.Y.; Pietenpol, J.A. TGF-𝛽 stimulation and inhibition of cell proliferation: New mechanistic insights. Cell 1990, 63, 245–247. [Google Scholar] [CrossRef]
- Tian, M.; Schiemann, W.P. The TGF-β paradox in human cancer: An update. Future Oncol. 2009, 5, 259–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierie, B.; Moses, H.L. Tumour microenvironment: TGF-β: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef]
- Grinnell, F.; Ho, C.H. Transforming growth factor 𝛽 stimulates fibroblast-collagen matrix contraction by different mechanisms in mechanically loaded and unloaded matrices. Exp. Cell Res. 2002, 273, 248–255. [Google Scholar] [CrossRef]
- Uhler, C.; Shivashankar, G.V. Geometric control and modeling of genome reprogramming. Bioarchitecture 2016, 6, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Jain, N.; Iyer, K.V.; Kumar, A.; Shivashankar, G.V. Cell geometric constraints induce modular gene-expression patterns via redistribution of HDAC3 regulated by actomyosin contractility. Proc. Natl. Acad. Sci. USA 2013, 110, 11349–11354. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Ingber, D.E. A non-genetic basis for cancer progression and metastasis: Self-organizing attractors in cell regulatory networks. Breast Dis. 2007, 26, 27–54. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Wang, S.C.; Wei, Y.; Luo, X.; Jia, Y.; Li, L.; Gopal, P.; Zhu, M.; Nassour, I.; Chuang, J.C.; et al. Arid1a Has Context-Dependent Oncogenic and Tumor Suppressor Functions in Liver Cancer. Cancer Cell 2017, 33, 151–152. [Google Scholar] [CrossRef] [Green Version]
- Blanco Calvo, M.; Bolós Fernández, V.; Medina Villaamil, V.; Aparicio Gallego, G.; Díaz Prado, S.; Grande Pulido, E. Biology of BMP signalling and cancer. Clin. Transl. Oncol. 2009, 11, 126–137. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Ye, Y.; Long, X.; Xiao, P.; Ren, X.; Yu, J. BMP signaling and its paradoxical effects in tumorigenesis and dissemination. Oncotarget 2016, 7, 78206–78218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, N.; Malik, S.; Villanueva, K.E.; Urano, A.; Lu, X.; Von Figura, G.; Seeley, E.S.; Dawson, D.W.; Collisson, E.A.; Hebrok, M. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev. 2015, 29, 658–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, K.; Yamada, Y.; Semi, K.; Yagi, M.; Tanaka, A.; Itakura, F.; Aoki, H.; Kunisada, T.; Woltjen, K.; Haga, H.; et al. Cellular context-dependent consequences of Apc mutations on gene regulation and cellular behavior. Proc. Natl. Acad. Sci. USA 2017, 114, 758–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kage, H.; Flodby, P.; Zhou, B.; Borok, Z. Dichotomous roles of claudins as tumor promoters or suppressors: Lessons from knockout mice. Cell Mol. Life Sci. 2019, 76, 4663–4672. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Hosono, Y.; Yanagisawa, K.; Takahashi, T. NKX2-1/TTF-1: An enigmatic oncogene that functions as a double-edged sword for cancer cell survival and progression. Cancer Cell 2013, 23, 718–723. [Google Scholar] [CrossRef] [Green Version]
- Otálora-Otálora, B.A.; Henríquez, B.; López-Kleine, L.; Rojas, A. RUNX family: Oncogenes or tumor suppressors (Review). Oncol. Rep. 2019, 42, 3–19. [Google Scholar] [CrossRef]
- Hruschka, N.; Kalisz, M.; Subijana, M.; Graña-Castro, O.; Del Cano-Ochoa, F.; Brunet, L.P.; Chernukhin, I.; Sagrera, A.; De Reynies, A.; Kloesch, B.; et al. The GATA3 X308_Splice breast cancer mutation is a hormone context-dependent oncogenic driver. Oncogene 2020, 39, 5455–5467. [Google Scholar] [CrossRef]
- Goel, R.K.; Lukong, K.E. Understanding the cellular roles of Fyn-related kinase (FRK): Implications in cancer biology. Cancer Metastasis. Rev. 2016, 35, 179–199. [Google Scholar] [CrossRef]
- Ferreirós, A.; Pedrosa, P.; Da Silva-Álvarez, S.; Triana-Martínez, F.; Vilas, J.M.; Picallos-Rabina, P.; González, P.; Gómez, M.; Li, H.; García-Caballero, T.; et al. Context-Dependent Impact of RAS Oncogene Expression on Cellular Reprogramming to Pluripotency. Stem Cell Rep. 2019, 12, 1099–1112. [Google Scholar] [CrossRef] [Green Version]
- Lambert, M.W. Cytoskeletal and nucleoskeletal interacting protein networks play critical roles in cellular function and dysfunction. Exp. Biol. Med. 2019, 244, 1233–1239. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. Systems biology—How far has it come? Biochemist 2011, 33, 16–18. [Google Scholar] [CrossRef] [Green Version]
- Bizzarri, M.; Giuliani, A.; Minini, M.; Monti, N.; Cucina, A. Constraints Shape Cell Function and Morphology by Canalizing the Developmental Path along the Waddington’s Landscape. Bioessays 2020, 42, e1900108. [Google Scholar] [CrossRef] [PubMed]
- Tatematsu, M.; Nagamine, Y.; Farber, E. Redifferentiation as a basis for remodeling of carcinogen-induced hepatocyte nodules to normal appearing liver. Cancer Res. 1983, 43, 5049–5058. [Google Scholar] [PubMed]
- Horii, R.; Akiyama, F.; Kasumi, F.; Koike, M.; Sakamoto, G. Spontaneous “ healing” of breast cancer. Breast Cancer 2005, 12, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Papac, R.J. Spontaneous regression of cancer: Possible mechanisms. In Vivo 1998, 12, 571–578. [Google Scholar]
- Challis, G.B.; Stam, H.J. The spontaneous regression of cancer. A review of cases from 1900 to 1987. Acta Oncol. 1990, 29, 545–550. [Google Scholar] [CrossRef]
- Proietti, S.; Cucina, A.; Pensotti, A.; Biava, P.M.; Minini, M.; Monti, N.; Catizone, A.; Ricci, G.; Leonetti, E.; Harrath, A.H.; et al. Active Fraction from Embryo Fish Extracts Induces Reversion of the Malignant Invasive Phenotype in Breast Cancer through Down-regulation of TCTP and Modulation of E-cadherin/β-catenin Pathway. Int. J. Mol. Sci. 2019, 20, 2151. [Google Scholar] [CrossRef] [Green Version]
- Pierce, G.B.; Wallace, C. Differentiation of malignant to benign cells. Cancer Res. 1971, 31, 127–134. [Google Scholar]
- Proietti, S.; Cucina, A.; Pensotti, A.; Fuso, A.; Marchese, C.; Nicolini, A.; Bizzarri, M. Tumor reversion and embryo morphogenetic factors. Semin Cancer Biol. 2020, 79, 83–90. [Google Scholar] [CrossRef]
- Willhauck, M.J.; Mirancea, N.; Vosseler, S. Reversion of tumor phenotype in surface transplants of skin SCC cells by scaffold-induced stroma modulation. Carcinogenesis 2007, 28, 595–610. [Google Scholar] [CrossRef]
- Krause, S.; Maffini, M.V.; Soto, A.M.; Sonnenschein, C. The microenvironment determines the breast cancer cells’ phenotype: Organization of MCF7 cells in 3D cultures. BMC Cancer 2010, 10, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bissell, M.; Kenny, P.A.; Radisky, D.C. Microenvironmental regulators of tissue structure and function also regulate tumor induction and progression: The role of extracellular matrix and its degrading enzymes. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Kenny, P.A.; Bissell, M.J. Tumor reversion: Correction of malignant behavior by microenvironmental cues. Int. J. Cancer 2003, 107, 688–695. [Google Scholar] [CrossRef] [Green Version]
- Hendrix, M.J.; Seftor, E.A.; Seftor, R.E.; Kasemeier-Kulesa, J.; Kulesa, P.M.; Postovit, L.M. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat. Rev. Cancer 2007, 7, 246–255. [Google Scholar] [CrossRef] [PubMed]
- D’Anselmi, F.; Valerio, M.; Cucina, A.; Galli, L.; Proietti, S.; Dinicola, S.; Pasqualato, A.; Manetti, C.; Ricci, G.; Giuliani, A.; et al. Metabolism and cell shape in cancer: A fractal analysis. Int. J. Biochem. Cell Biol. 2011, 43, 1052–1058. [Google Scholar] [CrossRef]
- Bizzarri, M.; Cucina, A. Tumor and the microenvironment: A chance to reframe the paradigm of carcinogenesis? Biomed. Res. Int. 2014, 2014, 934038. [Google Scholar] [CrossRef] [Green Version]
- Pape, J.; Emberton, M.; Cheema, U. 3D Cancer Models: The Need for a Complex Stroma, Compartmentalization and Stiffness. Front. Bioeng. Biotechnol. 2021, 9, 660502. [Google Scholar] [CrossRef]
- Veenstra, V.L.; Garcia-Garijo, A.; van Laarhoven, H.W.; Bijlsma, M.F. Extracellular Influences: Molecular Subclasses and the Microenvironment in Pancreatic Cancer. Cancers 2018, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- Sung, K.E.; Beebe, D.J. Microfluidic 3D models of cancer. Adv. Drug Deliv. Rev. 2014, 79–80, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Sonnenschein, C.; Soto, A.M. Over a century of cancer research: Inconvenient truths and promising leads. PLoS Biol. 2020, 18, e3000670. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monti, N.; Verna, R.; Piombarolo, A.; Querqui, A.; Bizzarri, M.; Fedeli, V. Paradoxical Behavior of Oncogenes Undermines the Somatic Mutation Theory. Biomolecules 2022, 12, 662. https://doi.org/10.3390/biom12050662
Monti N, Verna R, Piombarolo A, Querqui A, Bizzarri M, Fedeli V. Paradoxical Behavior of Oncogenes Undermines the Somatic Mutation Theory. Biomolecules. 2022; 12(5):662. https://doi.org/10.3390/biom12050662
Chicago/Turabian StyleMonti, Noemi, Roberto Verna, Aurora Piombarolo, Alessandro Querqui, Mariano Bizzarri, and Valeria Fedeli. 2022. "Paradoxical Behavior of Oncogenes Undermines the Somatic Mutation Theory" Biomolecules 12, no. 5: 662. https://doi.org/10.3390/biom12050662
APA StyleMonti, N., Verna, R., Piombarolo, A., Querqui, A., Bizzarri, M., & Fedeli, V. (2022). Paradoxical Behavior of Oncogenes Undermines the Somatic Mutation Theory. Biomolecules, 12(5), 662. https://doi.org/10.3390/biom12050662