
Cerebral Iron Deposition in Neurodegeneration




Abstract
:1. Introduction
2. Brain Iron, Neuroinflammation, and Cognitive Decline in Aging

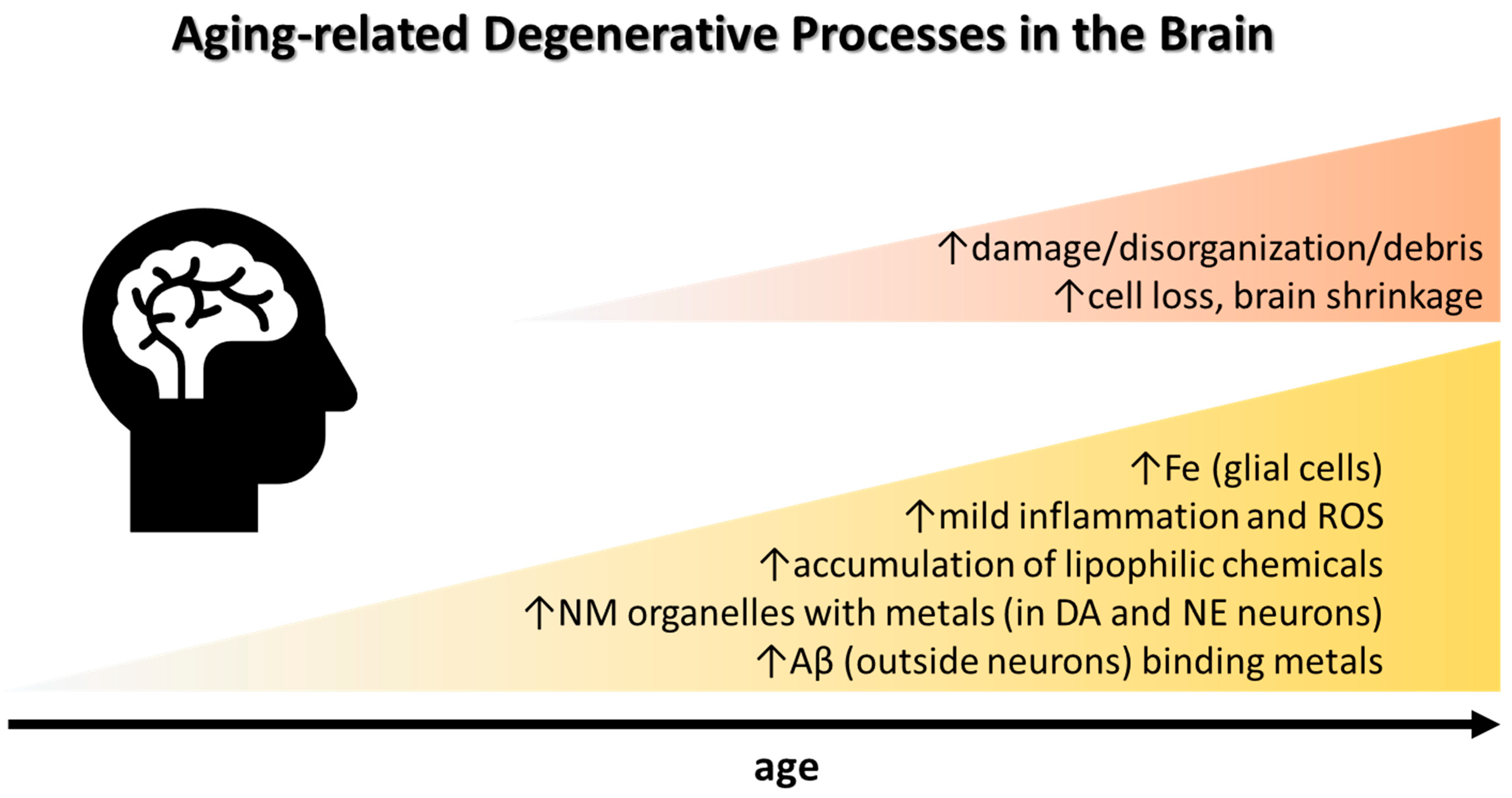{kind=link}
{kind=link}
| Condition | Whole Body Level | Brain Level | ||
|---|---|---|---|---|
| Macroscopic | Cellular | Subcellular | ||
| Aging | ↑ serum ferritin [53] | ↑ Fe in red nucleus, putamen, substantia nigra, dentate nucleus, globus pallidus, caudate nucleus, subthalamic nucleus, cortex [7] | Fe remains stable in oligodendroglia; Fe accumulates in astrocytes and dystrophic microglia in cortex and deep gray matter [12] | Fe bound to ferritin in cytoplasm of microglia and astrocytes and to neuromelanin in neurons [12,28] |
| AceruloplasMinemia | ↑ Fe in liver, pancreas, retina ↑ serum ferritin, ↓ transferrin saturation [54,55] | ↑ Fe in putamen, caudate, lateral, habenular, and pulvinar thalamic nuclei, red nucleus, dentate nucleus, inner cortical layers, hippocampus, mammillary bodies, superior and inferior colliculi [56,57] | ↑ Fe in astrocytes, neurons [58,59,60] | Fe stored in ferritin/ hemosiderin in lysosomal dense bodies and cytoplasmic inclusions [61,62] |
| Hereditary Ferritinopathy | ↑ Fe in liver, kidney, skin, muscle ↓ serum ferritin [63,64,65] | ↑ Fe in globus pallidus, substantia nigra, dentate nucleus, putamen, thalamus, caudate, deep cortical layers [66,67,68] | ↑ Fe in nuclei and cytoplasm of microglia, oligodendroglia, neurons, and also extracellularly [64,69] | Fe stored in inclusion bodies consisting of abnormal ferritin aggregates [69,70] |
| Pantothenate Kinase-Associated Neurodegeneration | - | ↑ Fe in globus pallidus, substantia nigra [71,72,73,74] | ↑ Fe in astrocytes, neurons, perivascular macrophages, iron dust in neuropil [75,76] | Fe stored in cytoplasmic inclusions co-localized with ferritin [75] |
| Mitochondrial Membrane Protein-Associated Neurodegeneration | - | ↑ Fe in globus pallidus, substantia nigra, putamen, caudate [77,78] | ↑ Fe in perivascular macrophages, astrocytes, neurons [79,80] | n.a. |
| Phospholipase A2-Associated Neurodegeneration | - | ↑ Fe in globus pallidus, substantia nigra, dentate nucleus [81,82,83] | ↑ Fe perivascularly in extracellular deposits and in macrophages [82,84,85] | n.a. |
| Beta-Propeller Protein-Associated Neurodegeneration | ↑ serum Tfr/logFerrit ratio [86] | ↑ Fe in substantia nigra, cerebral peduncles, globus pallidus [87] | ↑ Fe in excessive macrophages [88,89] | n.a. |
| Friedreich Ataxia | Fe-positive granules in cardiomyocytes ↓ serum Fe [90,91,92,93] | ↑ Fe in dentate nucleus, red nucleus [94,95] | Fe switched from oligodendroglia to microglia in dentate nucleus [96,97,98] | Fe presumably primarily accumulated in mitochondria [99,100] |
| Wilson Disease | ↑ Fe in liver ↑ serum Fe, ferritin, hepcidin, soluble transferrin receptor [101,102,103] | ↑ Fe in globus pallidus, putamen, caudate, thalamus, substantia nigra, red nucleus, subthalamic nucleus [104,105] | ↑ Fe in excessive macrophages, astrocytes [106] | n.a. |
| Parkinson Disease | ↓ Fe in serum/plasma [107] | ↑ Fe in substantia nigra [108,109] | ↑ Fe in neurons and adjacent neuropil, microglia, perivascularly in extracellular deposits [110,111,112] | Fe bound to neuromelanin in dopaminergic neurons [112,113] |
| Alzheimer Disease | ↓ Fe in serum/plasma [114,115,116] | ↑ Fe in (mostly temporal) cortex, globus pallidus, caudate, putamen [117,118,119,120,121,122] | ↑ Fe in amyloid plaques, microglia, along myelinated fibers [117,123,124,125] | Fe bound to amyloid partially composed of magnetite nanoparticles [126,127] |
| Amyotrophic Lateral Sclerosis | ↑ ferritin, ↓ transferrin in serum [128] ↑ Fe in liver, kidneys [129] ↑ Fe in spinal cord [130,131] | ↑ Fe in motor cortex, caudate, subthalamic nucleus, globus pallidus, substantia nigra, red nucleus [132,133,134] | ↑ Fe in spinal cord neuron nuclei [135] ↑ Fe in microglia in motor cortex [136] | n.a. |
| Multiple Sclerosis | - | ↑ Fe in globus pallidus, putamen, caudate ↓ Fe in normal appearing white matter and thalamus [137,138,139,140,141] | ↑ Fe in macrophages, activated microglia in the rim of lesions; in reactive astrocytes in the inactive centers of lesions; in oligodendroglia, astrocytes, and microglia in the deep gray matter [137,142,143,144,145] | Fe in active lesions stored in ferritin, hemosiderin, and magnetite [144] |
3. Neurodegenerations with Brain Iron Accumulation (NBIA) Group
4. Other Genetic Disorders with Brain Iron Deposits
5. Iron Accumulation and Pathology in Sporadic Neurodegenerative Disorders
6. Demyelinating Disorders and Neuroinflammation
7. Iron Chelation and Other Means to Decrease Cerebral Iron
8. Summary and Future Perspectives
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Urrutia, P.J.; Borquez, D.A.; Nunez, M.T. Inflaming the Brain with Iron. Antioxidants 2021, 10, 61. [Google Scholar] [CrossRef]
- Hinarejos, I.; Machuca-Arellano, C.; Sancho, P.; Espinos, C. Mitochondrial Dysfunction, Oxidative Stress and Neuroinflammation in Neurodegeneration with Brain Iron Accumulation (Nbia). Antioxidants 2020, 9, 1020. [Google Scholar] [CrossRef] [PubMed]
- Huo, T.; Jia, Y.; Yin, C.; Luo, X.; Zhao, J.; Wang, Z.; Lv, P. Iron Dysregulation in Vascular Dementia: Focused on the Ampk/Autophagy Pathway. Brain Res. Bull. 2019, 153, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Garciduenas, L.; Torres-Jardon, R.; Kulesza, R.J.; Mansour, Y.; Gonzalez-Gonzalez, L.O.; Gonzalez-Maciel, A.; Reynoso-Robles, R.; Mukherjee, P.S. Alzheimer Disease Starts in Childhood in Polluted Metropolitan Mexico City. A Major Health Crisis in Progress. Environ. Res. 2020, 183, 109137. [Google Scholar] [CrossRef]
- Drayer, B.; Burger, P.; Darwin, R.; Riederer, S.; Herfkens, R.; Johnson, G.A. Mri of Brain Iron. AJR Am. J. Roentgenol. 1986, 147, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G.; Tishler, T.A.; Lu, P.H.; Villablanca, P.; Altshuler, L.L.; Carter, M.; Huang, D.; Edwards, N.; Mintz, J. Brain Ferritin Iron May Influence Age- and Gender-Related Risks of Neurodegeneration. Neurobiol. Aging 2007, 28, 414–423. [Google Scholar] [CrossRef]
- Burgetova, R.; Dusek, P.; Burgetova, A.; Pudlac, A.; Vaneckova, M.; Horakova, D.; Krasensky, J.; Varga, Z.; Lambert, L. Age-Related Magnetic Susceptibility Changes in Deep Grey Matter and Cerebral Cortex of Normal Young and Middle-Aged Adults Depicted by Whole Brain Analysis. Quant. Imaging Med. Surg. 2021, 11, 3906–3919. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, H.; Cronin, M.J.; He, N.; Yan, F.; Liu, C. Longitudinal Atlas for Normative Human Brain Development and Aging over the Lifespan Using Quantitative Susceptibility Mapping. Neuroimage 2018, 171, 176–189. [Google Scholar] [CrossRef]
- Acosta-Cabronero, J.; Betts, M.J.; Cardenas-Blanco, A.; Yang, S.; Nestor, P.J. In Vivo Mri Mapping of Brain Iron Deposition across the Adult Lifespan. J. Neurosci. 2016, 36, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Reinert, A.; Morawski, M.; Seeger, J.; Arendt, T.; Reinert, T. Iron Concentrations in Neurons and Glial Cells with Estimates on Ferritin Concentrations. BMC Neurosci. 2019, 20, 25. [Google Scholar] [CrossRef]
- Ashraf, A.; Michaelides, C.; Walker, T.A.; Ekonomou, A.; Suessmilch, M.; Sriskanthanathan, A.; Abraha, S.; Parkes, A.; Parkes, H.G.; Geraki, K.; et al. Regional Distributions of Iron, Copper and Zinc and Their Relationships with Glia in a Normal Aging Mouse Model. Front. Aging Neurosci. 2019, 11, 351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, J.R.; Menzies, S.L.; Martin, S.M.S.; Mufson, E.J. Cellular Distribution of Transferrin, Ferritin, and Iron in Normal and Aged Human Brains. J. Neurosci. Res. 1990, 27, 595–611. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The Role of Iron in Brain Ageing and Neurodegenerative Disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef] [Green Version]
- Hofer, T. Oxidation of 2-Deoxyguanosine by H2O2-Ascorbate: Evidence against Free OH• and Thermodynamic Support for Two Electron Reduction of H2O2. J. Chem. Soc. Perkin Trans. 2001, 2, 210–213. [Google Scholar] [CrossRef]
- Hofer, T.; Servais, S.; Seo, A.Y.; Marzetti, E.; Hiona, A.; Upadhyay, J.S.; Wohlgemuth, S.E.; Leeuwenburgh, C. Bioenergetics and Permeability Transition Pore Opening in Heart Subsarcolemmal and Interfibrillar Mitochondria: Effects of Aging and Lifelong Calorie Restriction. Mech. Ageing Dev. 2009, 130, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Aaseth, J.; Skalny, A.V.; Roos, P.M.; Alexander, J.; Aschner, M.; Tinkov, A.A. Copper, Iron, Selenium and Lipo-Glycemic Dysmetabolism in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 9461. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xin, L.; Xiang, M.; Shang, C.; Wang, Y.; Wang, Y.; Cui, X.; Lu, Y. The Molecular Mechanisms of Ferroptosis and Its Role in Cardiovascular Disease. Biomed. Pharmacother. 2022, 145, 112423. [Google Scholar] [CrossRef]
- Mazhar, M.; Din, A.U.; Ali, H.; Yang, G.; Ren, W.; Wang, L.; Fan, X.; Yang, S. Implication of Ferroptosis in Aging. Cell Death Discov. 2021, 7, 149. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Metselaar, B.; Greenough, M.; Bush, A.I.; Ayton, S.J. Ferroptosis and Nrf2: An Emerging Battlefield in the Neurodegeneration of Alzheimer’s Disease. Essays Biochem. 2021, 65, 925–940. [Google Scholar]
- Hofer, T.; Jorgensen, T.O.; Olsen, R.L. Comparison of Food Antioxidants and Iron Chelators in Two Cellular Free Radical Assays: Strong Protection by Luteolin. J. Agric. Food Chem. 2014, 62, 8402–8410. [Google Scholar] [CrossRef]
- Aaseth, J.; Alexander, J.; Alehagen, U. Coenzyme Q10 Supplementation—In Ageing and Disease. Mech. Ageing Dev. 2021, 197, 111521. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Youdim, M.B.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, Brain Ageing and Neurodegenerative Disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Fedorow, H.; Tribl, F.; Halliday, G.; Gerlach, M.; Riederer, P.; Double, K.L. Neuromelanin in Human Dopamine Neurons: Comparison with Peripheral Melanins and Relevance to Parkinson’s Disease. Prog. Neurobiol. 2005, 75, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Zucca, F.A.; Basso, E.; Cupaioli, F.A.; Ferrari, E.; Sulzer, D.; Casella, L.; Zecca, L. Neuromelanin of the Human Substantia Nigra: An Update. Neurotox. Res. 2014, 25, 13–23. [Google Scholar] [CrossRef]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Munoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of Iron, Dopamine and Neuromelanin Pathways in Brain Aging and Parkinson’s Disease. Prog. Neurobiol. 2017, 155, 96–119. [Google Scholar] [CrossRef]
- Zucca, F.A.; Vanna, R.; Cupaioli, F.A.; Bellei, C.; de Palma, A.; di Silvestre, D.; Mauri, P.; Grassi, S.; Prinetti, A.; Casella, L.; et al. Neuromelanin Organelles Are Specialized Autolysosomes That Accumulate Undegraded Proteins and Lipids in Aging Human Brain and Are Likely Involved in Parkinson’s Disease. NPJ Parkinsons Dis. 2018, 4, 17. [Google Scholar] [CrossRef]
- Zecca, L.; Bellei, C.; Costi, P.; Albertini, A.; Monzani, E.; Casella, L.; Gallorini, M.; Bergamaschi, L.; Moscatelli, A.; Turro, N.J.; et al. New Melanic Pigments in the Human Brain That Accumulate in Aging and Block Environmental Toxic Metals. Proc. Natl. Acad. Sci. USA 2008, 105, 17567–17572. [Google Scholar] [CrossRef] [Green Version]
- Halliday, G.M.; Fedorow, H.; Rickert, C.H.; Gerlach, M.; Riederer, P.; Double, K.L. Evidence for Specific Phases in the Development of Human Neuromelanin. J. Neural. Transm. 2006, 113, 721–728. [Google Scholar] [CrossRef]
- Zecca, L.; Tampellini, D.; Gatti, A.; Crippa, R.; Eisner, M.; Sulzer, D.; Ito, S.; Fariello, R.; Gallorini, M. The Neuromelanin of Human Substantia Nigra and Its Interaction with Metals. J. Neural. Transm. 2002, 109, 663–672. [Google Scholar] [CrossRef]
- Ma, Y.; Li, R.; Dong, Y.; You, C.; Huang, S.; Li, X.; Wang, F.; Zhang, Y. Tlyp-1 Peptide Functionalized Human H Chain Ferritin for Targeted Delivery of Paclitaxel. Int. J. Nanomed. 2021, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Billesbolle, C.B.; Azumaya, C.M.; Kretsch, R.C.; Powers, A.S.; Gonen, S.; Schneider, S.; Arvedson, T.; Dror, R.O.; Cheng, Y.; Manglik, A. Structure of Hepcidin-Bound Ferroportin Reveals Iron Homeostatic Mechanisms. Nature 2020, 586, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W. Toxicology of Choroid Plexus: Special Reference to Metal-Induced Neurotoxicities. Microsc. Res. Tech. 2001, 52, 89–103. [Google Scholar] [CrossRef]
- Zheng, W.; Aschner, M.; Ghersi-Egea, J.F. Brain Barrier Systems: A New Frontier in Metal Neurotoxicological Research. Toxicol. Appl. Pharmacol. 2003, 192, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, L.; Corsi, A.; Lauretani, F.; Bandinelli, S.; Bartali, B.; Taub, D.D.; Guralnik, J.M.; Longo, D.L. The Origins of Age-Related Proinflammatory State. Blood 2005, 105, 2294–2299. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and Anti-Inflammaging: A Systemic Perspective on Aging and Longevity Emerged from Studies in Humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef]
- Capucciati, A.; Zucca, F.A.; Monzani, E.; Zecca, L.; Casella, L.; Hofer, T. Interaction of Neuromelanin with Xenobiotics and Consequences for Neurodegeneration; Promising Experimental Models. Antioxidants 2021, 10, 824. [Google Scholar] [CrossRef]
- Myhre, O.; Utkilen, H.; Duale, N.; Brunborg, G.; Hofer, T. Metal Dyshomeostasis and Inflammation in Alzheimer’s and Parkinson’s Diseases: Possible Impact of Environmental Exposures. Oxid. Med. Cell Longev. 2013, 2013, 726954. [Google Scholar] [CrossRef] [Green Version]
- Kell, D.B.; Pretorius, E. Serum Ferritin Is an Important Inflammatory Disease Marker, as It Is Mainly a Leakage Product from Damaged Cells. Metallomics 2014, 6, 748–773. [Google Scholar] [CrossRef] [Green Version]
- Kowdley, K.V.; Gochanour, E.M.; Sundaram, V.; Shah, R.A.; Handa, P. Hepcidin Signaling in Health and Disease: Ironing out the Details. Hepatol. Commun. 2021, 5, 723–735. [Google Scholar] [CrossRef]
- Camaschella, C.; Nai, A.; Silvestri, L. Iron Metabolism and Iron Disorders Revisited in the Hepcidin Era. Haematologica 2020, 105, 260–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Du, X.; Xie, J.; Wang, J. Interleukin-6 Regulates Iron-Related Proteins through C-Jun N-Terminal Kinase Activation in Bv2 Microglial Cell Lines. PLoS ONE 2017, 12, e0180464. [Google Scholar] [CrossRef]
- Martin-Bastida, A.; Tilley, B.S.; Bansal, S.; Gentleman, S.M.; Dexter, D.T.; Ward, R.J. Iron and Inflammation: In Vivo and Post-Mortem Studies in Parkinson’s Disease. J. Neural Transm. 2021, 128, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Abu-Rumeileh, S.; Steinacker, P.; Polischi, B.; Mammana, A.; Bartoletti-Stella, A.; Oeckl, P.; Baiardi, S.; Zenesini, C.; Huss, A.; Cortelli, P.; et al. Csf Biomarkers of Neuroinflammation in Distinct Forms and Subtypes of Neurodegenerative Dementia. Alzheimers Res. Ther. 2019, 12, 2. [Google Scholar] [CrossRef] [Green Version]
- Liang, T.; Qian, Z.M.; Mu, M.D.; Yung, W.H.; Ke, Y. Brain Hepcidin Suppresses Major Pathologies in Experimental Parkinsonism. iScience 2020, 23, 101284. [Google Scholar] [CrossRef] [PubMed]
- Yambire, K.F.; Rostosky, C.; Watanabe, T.; Pacheu-Grau, D.; Torres-Odio, S.; Sanchez-Guerrero, A.; Senderovich, O.; Meyron-Holtz, E.G.; Milosevic, I.; Frahm, J.; et al. Impaired Lysosomal Acidification Triggers Iron Deficiency and Inflammation in Vivo. eLife 2019, 8, e51031. [Google Scholar] [CrossRef] [PubMed]
- Borquez, D.A.; Urrutia, P.J.; Nunez, M.T. Iron, the Endolysosomal System and Neuroinflammation: A Matter of Balance. Neural Regen. Res. 2022, 17, 1003–1004. [Google Scholar]
- Sfera, A.; Bullock, K.; Price, A.; Inderias, L.; Osorio, C. Ferrosenescence: The Iron Age of Neurodegeneration? Mech. Ageing Dev. 2018, 174, 63–75. [Google Scholar] [CrossRef]
- Spence, H.; McNeil, C.J.; Waiter, G.D. The Impact of Brain Iron Accumulation on Cognition: A Systematic Review. PLoS ONE 2020, 15, e0240697. [Google Scholar] [CrossRef]
- Salami, A.; Papenberg, G.; Sitnikov, R.; Laukka, E.J.; Persson, J.; Kalpouzos, G. Elevated Neuroinflammation Contributes to the Deleterious Impact of Iron Overload on Brain Function in Aging. Neuroimage 2021, 230, 117792. [Google Scholar] [CrossRef]
- Venkatesh, A.; Daugherty, A.M.; Bennett, I.J. Neuroimaging Measures of Iron and Gliosis Explain Memory Performance in Aging. Hum. Brain Mapp. 2021, 42, 5761–5770. [Google Scholar] [CrossRef] [PubMed]
- Timmers, P.R.H.J.; Wilson, J.F.; Joshi, P.K.; Deelen, J. Multivariate Genomic Scan Implicates Novel Loci and Haem Metabolism in Human Ageing. Nat. Commun. 2020, 11, 3570. [Google Scholar] [CrossRef] [PubMed]
- Casale, G.; Bonora, C.; Migliavacca, A.; Zurita, I.E.; de Nicola, P. Serum Ferritin and Ageing. Age Ageing 1981, 10, 119–122. [Google Scholar] [CrossRef]
- Yoshida, K.; Furihata, K.; Takeda, S.; Nakamura, A.; Yamamoto, K.; Morita, H.; Hiyamuta, S.; Ikeda, S.; Shimizu, N.; Yanagisawa, N. A Mutation in the Ceruloplasmin Gene Is Associated with Systemic Hemosiderosis in Humans. Nat. Genet. 1995, 9, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Pelucchi, S.; Mariani, R.; Ravasi, G.; Pelloni, I.; Marano, M.; Tremolizzo, L.; Alessio, M.; Piperno, A. Phenotypic Heterogeneity in Seven Italian Cases of Aceruloplasminemia. Parkinsonism Relat. Disord. 2018, 51, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chen, Y.; Li, Y.; Gharabaghi, S.; Chen, Y.; Sethi, S.K.; Wu, Y.; Haacke, E.M. Intracranial Iron Distribution and Quantification in Aceruloplasminemia: A Case Study. Magn. Reson. Imaging 2020, 70, 29–35. [Google Scholar] [CrossRef]
- Kim, H.K.; Ki, C.S.; Kim, Y.J.; Lee, M.S. Radiological Findings of Two Sisters with Aceruloplasminemia Presenting with Chorea. Clin. Neuroradiol. 2017, 27, 385–388. [Google Scholar] [CrossRef]
- Kaneko, K.; Hineno, A.; Yoshida, K.; Ohara, S.; Morita, H.; Ikeda, S. Extensive Brain Pathology in a Patient with Aceruloplasminemia with a Prolonged Duration of Illness. Hum. Pathol. 2012, 43, 451–456. [Google Scholar] [CrossRef]
- Gonzalez-Cuyar, L.F.; Perry, G.; Miyajima, H.; Atwood, C.S.; Riveros-Angel, M.; Lyons, P.F.; Siedlak, S.L.; Smith, M.A.; Castellani, R.J. Redox Active Iron Accumulation in Aceruloplasminemia. Neuropathology 2008, 28, 466–471. [Google Scholar] [CrossRef]
- Oide, T.; Yoshida, K.; Kaneko, K.; Ohta, M.; Arima, K. Iron Overload and Antioxidative Role of Perivascular Astrocytes in Aceruloplasminemia. Neuropathol. Appl. Neurobiol. 2006, 32, 170–176. [Google Scholar] [CrossRef]
- Vroegindeweij, L.H.P.; Bossoni, L.; Boon, A.J.W.; Wilson, J.H.P.; Bulk, M.; Labra-Munoz, J.; Huber, M.; Webb, A.; van der Weerd, L.; Langendonk, J.G. Quantification of Different Iron Forms in the Aceruloplasminemia Brain to Explore Iron-Related Neurodegeneration. Neuroimage Clin. 2021, 30, 102657. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, H. Aceruloplasminemia, an Iron Metabolic Disorder. Neuropathology 2003, 23, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Davidzon, G.; Kurlan, R.M.; Tawil, R.; Bonilla, E.; di Mauro, S.; Powers, J.M. Hereditary Ferritinopathy: A Novel Mutation, Its Cellular Pathology, and Pathogenetic Insights. J. Neuropathol. Exp. Neurol. 2005, 64, 280–294. [Google Scholar] [CrossRef] [Green Version]
- Curtis, A.R.; Fey, C.; Morris, C.M.; Bindoff, L.A.; Ince, P.G.; Chinnery, P.F.; Coulthard, A.; Jackson, M.J.; Jackson, A.P.; McHale, D.P.; et al. Mutation in the Gene Encoding Ferritin Light Polypeptide Causes Dominant Adult-Onset Basal Ganglia Disease. Nat. Genet. 2001, 28, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Ory-Magne, F.; Brefel-Courbon, C.; Payoux, P.; Debruxelles, S.; Sibon, I.; Goizet, C.; Labauge, P.; Menegon, P.; Uro-Coste, E.; Ghetti, B.; et al. Clinical Phenotype and Neuroimaging Findings in a French Family with Hereditary Ferritinopathy (Ftl498-499instc). Mov. Disord. 2009, 24, 1676–1683. [Google Scholar] [CrossRef]
- McNeill, A.; Birchall, D.; Hayflick, S.J.; Gregory, A.; Schenk, J.F.; Zimmerman, E.A.; Shang, H.; Miyajima, H.; Chinnery, P.F. T2* and Fse Mri Distinguishes Four Subtypes of Neurodegeneration with Brain Iron Accumulation. Neurology 2008, 70, 1614–1619. [Google Scholar] [CrossRef]
- Keogh, M.J.; Jonas, P.; Coulthard, A.; Chinnery, P.F.; Burn, J. Neuroferritinopathy: A New Inborn Error of Iron Metabolism. Neurogenetics 2012, 13, 93–96. [Google Scholar] [CrossRef]
- McNeill, A.; Gorman, G.; Khan, A.; Horvath, R.; Blamire, A.M.; Chinnery, P.F. Progressive Brain Iron Accumulation in Neuroferritinopathy Measured by the Thalamic T2* Relaxation Rate. AJNR Am. J. Neuroradiol. 2012, 33, 1810–1813. [Google Scholar] [CrossRef] [Green Version]
- Kurzawa-Akanbi, M.; Keogh, M.; Tsefou, E.; Ramsay, L.; Johnson, M.; Keers, S.; Ochieng, L.W.; McNair, A.; Singh, P.; Khan, A.; et al. Neuropathological and Biochemical Investigation of Hereditary Ferritinopathy Cases with Ferritin Light Chain Mutation: Prominent Protein Aggregation in the Absence of Major Mitochondrial or Oxidative Stress. Neuropathol. Appl. Neurobiol. 2021, 47, 26–42. [Google Scholar] [CrossRef]
- Vidal, R.; Delisle, M.B.; Rascol, O.; Ghetti, B. Hereditary Ferritinopathy. J. Neurol. Sci. 2003, 207, 110–111. [Google Scholar] [CrossRef]
- Lee, J.H.; Gregory, A.; Hogarth, P.; Rogers, C.; Hayflick, S.J. Looking Deep into the Eye-of-the-Tiger in Pantothenate Kinase-Associated Neurodegeneration. AJNR Am. J. Neuroradiol. 2018, 39, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dusek, P.; Martinez, E.M.T.; Madai, V.I.; Jech, R.; Sobesky, J.; Paul, F.; Niendorf, T.; Wuerfel, J.; Schneider, S.A. 7-Tesla Magnetic Resonance Imaging for Brain Iron Quantification in Homozygous and Heterozygous Pank2 Mutation Carriers. Mov. Disord. Clin. Pract. 2014, 1, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Xing, W.; Liao, W.; Wang, X. Magnetic Resonance Imaging, Susceptibility Weighted Imaging and Quantitative Susceptibility Mapping Findings of Pantothenate Kinase-Associated Neurodegeneration. J. Clin. Neurosci. 2019, 59, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fermin-Delgado, R.; Roa-Sanchez, P.; Speckter, H.; Perez-Then, E.; Rivera-Mejia, D.; Foerster, B.; Stoeter, P. Involvement of Globus Pallidus and Midbrain Nuclei in Pantothenate Kinase-Associated Neurodegeneration: Measurement of T2 and T2* Time. Clin. Neuroradiol. 2013, 23, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Kruer, M.C.; Hiken, M.; Gregory, A.; Malandrini, A.; Clark, D.; Hogarth, P.; Grafe, M.; Hayflick, S.J.; Woltjer, R.L. Novel Histopathologic Findings in Molecularly-Confirmed Pantothenate Kinase-Associated Neurodegeneration. Brain 2011, 134, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Paudel, R.; Johnson, R.; Courtney, R.; Lees, A.J.; Holton, J.L.; Hardy, J.; Revesz, T.; Houlden, H. Pantothenate Kinase-Associated Neurodegeneration Is Not a Synucleinopathy. Neuropathol. Appl. Neurobiol. 2013, 39, 121–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dusek, P.; Mekle, R.; Skowronska, M.; Acosta-Cabronero, J.; Huelnhagen, T.; Robinson, S.D.; Schubert, F.; Deschauer, M.; Els, A.; Ittermann, B.; et al. Brain Iron and Metabolic Abnormalities in C19orf12 Mutation Carriers: A 7.0 Tesla Mri Study in Mitochondrial Membrane Protein-Associated Neurodegeneration. Mov. Disord. 2020, 35, 142–150. [Google Scholar] [CrossRef]
- Alavi, A.; Mokhtari, M.; Hajati, R.; Davarzani, A.; Fasano, A.; Lang, A.E.; Rohani, M. Late-Onset Mitochondrial Membrane Protein-Associated Neurodegeneration with Extensive Brain Iron Deposition. Mov. Disord. Clin. Pract. 2020, 7, 120–121. [Google Scholar] [CrossRef]
- Hogarth, P.; Gregory, A.; Kruer, M.C.; Sanford, L.; Wagoner, W.; Natowicz, M.R.; Egel, R.T.; Subramony, S.H.; Goldman, J.G.; Berry-Kravis, E.; et al. New Nbia Subtype: Genetic, Clinical, Pathologic, and Radiographic Features of Mpan. Neurology 2013, 80, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Hartig, M.B.; Iuso, A.; Haack, T.; Kmiec, T.; Jurkiewicz, E.; Heim, K.; Roeber, S.; Tarabin, V.; Dusi, S.; Krajewska-Walasek, M.; et al. Absence of an Orphan Mitochondrial Protein, C19orf12, Causes a Distinct Clinical Subtype of Neurodegeneration with Brain Iron Accumulation. Am. J. Hum. Genet. 2011, 89, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Kurian, M.A.; Morgan, N.V.; MacPherson, L.; Foster, K.; Peake, D.; Gupta, R.; Philip, S.G.; Hendriksz, C.; Morton, J.E.; Kingston, H.M.; et al. Phenotypic Spectrum of Neurodegeneration Associated with Mutations in the Pla2g6 Gene (Plan). Neurology 2008, 70, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.; Westaway, S.K.; Holm, I.E.; Kotzbauer, P.T.; Hogarth, P.; Sonek, S.; Coryell, J.C.; Nguyen, T.M.; Nardocci, N.; Zorzi, G.; et al. Neurodegeneration Associated with Genetic Defects in Phospholipase a(2). Neurology 2008, 71, 1402–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, A.; Aguilera-Albesa, S.; Tello, C.A.; Serrano, M.; Tomas, M.; Camino-Leon, R.; Fernandez-Ramos, J.; Jimenez-Escrig, A.; Poo, P.; O’Callaghan, M.; et al. Pla2g6-Associated Neurodegeneration: New Insights into Brain Abnormalities and Disease Progression. Parkinsonism Relat. Disord. 2019, 61, 179–186. [Google Scholar] [CrossRef]
- Paisan-Ruiz, C.; Li, A.; Schneider, S.A.; Holton, J.L.; Johnson, R.; Kidd, D.; Chataway, J.; Bhatia, K.P.; Lees, A.J.; Hardy, J.; et al. Widespread Lewy Body and Tau Accumulation in Childhood and Adult Onset Dystonia-Parkinsonism Cases with Pla2g6 Mutations. Neurobiol. Aging 2012, 33, 814–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riku, Y.; Ikeuchi, T.; Yoshino, H.; Mimuro, M.; Mano, K.; Goto, Y.; Hattori, N.; Sobue, G.; Yoshida, M. Extensive Aggregation of Alpha-Synuclein and Tau in Juvenile-Onset Neuroaxonal Dystrophy: An Autopsied Individual with a Novel Mutation in the Pla2g6 Gene-Splicing Site. Acta Neuropathol. Commun. 2013, 1, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belohlavkova, A.; Sterbova, K.; Betzler, C.; Burkhard, S.; Panzer, A.; Wolff, M.; Lassuthova, P.; Vlckova, M.; Kyncl, M.; Benova, B.; et al. Clinical Features and Blood Iron Metabolism Markers in Children with Beta-Propeller Protein Associated Neurodegeneration. Eur. J. Paediatr. Neurol. 2020, 28, 81–88. [Google Scholar] [CrossRef]
- Kimura, Y.; Sato, N.; Ishiyama, A.; Shigemoto, Y.; Suzuki, F.; Fujii, H.; Maikusa, N.; Matsuda, H.; Nishioka, K.; Hattori, N.; et al. Serial Mri Alterations of Pediatric Patients with Beta-Propeller Protein Associated Neurodegeneration (Bpan). J. Neuroradiol. 2021, 48, 88–93. [Google Scholar] [CrossRef]
- Hayflick, S.J.; Kruer, M.C.; Gregory, A.; Haack, T.B.; Kurian, M.A.; Houlden, H.H.; Anderson, J.; Boddaert, N.; Sanford, L.; Harik, S.I.; et al. Beta-Propeller Protein-Associated Neurodegeneration: A New X-Linked Dominant Disorder with Brain Iron Accumulation. Brain 2013, 136, 1708–1717. [Google Scholar] [CrossRef]
- Paudel, R.; Li, A.; Wiethoff, S.; Bandopadhyay, R.; Bhatia, K.; de Silva, R.; Houlden, H.; Holton, J.L. Neuropathology of Beta-Propeller Protein Associated Neurodegeneration (Bpan): A New Tauopathy. Acta Neuropathol. Commun. 2015, 3, 39. [Google Scholar] [CrossRef] [Green Version]
- Lamarche, J.B.; Cote, M.; Lemieux, B. The Cardiomyopathy of Friedreich’s Ataxia Morphological Observations in 3 Cases. Can. J. Neurol. Sci. 1980, 7, 389–396. [Google Scholar] [CrossRef] [Green Version]
- Michael, S.; Petrocine, S.V.; Qian, J.; Lamarche, J.B.; Knutson, M.D.; Garrick, M.D.; Koeppen, A.H. Iron and Iron-Responsive Proteins in the Cardiomyopathy of Friedreich’s Ataxia. Cerebellum 2006, 5, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, R.L.; Qian, J.; Santambrogio, P.; Levi, S.; Koeppen, A.H. Relation of Cytosolic Iron Excess to Cardiomyopathy of Friedreich’s Ataxia. Am. J. Cardiol. 2012, 110, 1820–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, D.; Srivastava, A.K.; Gulati, S.; Rajeswari, M.R. Assessment of Cell-Free Levels of Iron and Copper in Patients with Friedreich’s Ataxia. Biometals 2019, 32, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Straub, S.; Mangesius, S.; Emmerich, J.; Indelicato, E.; Nachbauer, W.; Degenhardt, K.S.; Ladd, M.E.; Boesch, S.; Gizewski, E.R. Toward Quantitative Neuroimaging Biomarkers for Friedreich’s Ataxia at 7 Tesla: Susceptibility Mapping, Diffusion Imaging, R2 and R1 Relaxometry. J. Neurosci. Res. 2020, 98, 2219–2231. [Google Scholar] [CrossRef]
- Ward, P.G.D.; Harding, I.H.; Close, T.G.; Corben, L.A.; Delatycki, M.B.; Storey, E.; Georgiou-Karistianis, N.; Egan, G.F. Longitudinal Evaluation of Iron Concentration and Atrophy in the Dentate Nuclei in Friedreich Ataxia. Mov. Disord. 2019, 34, 335–343. [Google Scholar] [CrossRef]
- Koeppen, A.H.; Michael, S.C.; Knutson, M.D.; Haile, D.J.; Qian, J.; Levi, S.; Santambrogio, P.; Garrick, M.D.; Lamarche, J.B. The Dentate Nucleus in Friedreich’s Ataxia: The Role of Iron-Responsive Proteins. Acta Neuropathol. 2007, 114, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Koeppen, A.H.; Ramirez, R.L.; Yu, D.; Collins, S.E.; Qian, J.; Parsons, P.J.; Yang, K.X.; Chen, Z.; Mazurkiewicz, J.E.; Feustel, P.J. Friedreich’s Ataxia Causes Redistribution of Iron, Copper, and Zinc in the Dentate Nucleus. Cerebellum 2012, 11, 845–860. [Google Scholar] [CrossRef] [Green Version]
- Harding, I.H.; Raniga, P.; Delatycki, M.B.; Stagnitti, M.R.; Corben, L.A.; Storey, E.; Georgiou-Karistianis, N.; Egan, G.F. Tissue Atrophy and Elevated Iron Concentration in the Extrapyramidal Motor System in Friedreich Ataxia: The Image-Frda Study. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1261–1263. [Google Scholar] [CrossRef] [Green Version]
- Reelfs, O.; Abbate, V.; Cilibrizzi, A.; Pook, M.A.; Hider, R.C.; Pourzand, C. The Role of Mitochondrial Labile Iron in Friedreich’s Ataxia Skin Fibroblasts Sensitivity to Ultraviolet A. Metallomics 2019, 11, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Bradley, J.L.; Blake, J.C.; Chamberlain, S.; Thomas, P.K.; Cooper, J.M.; Schapira, A.H. Clinical, Biochemical and Molecular Genetic Correlations in Friedreich’s Ataxia. Hum. Mol. Genet. 2000, 9, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Gromadzka, G.; Wierzbicka, D.; Litwin, T.; Przybylkowski, A. Iron Metabolism Is Disturbed and Anti-Copper Treatment Improves but Does Not Normalize Iron Metabolism in Wilson’s Disease. Biometals 2021, 34, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Osterode, W.; Falkenberg, G.; Ferenci, P.; Wrba, F. Quantitative Trace Element Mapping in Liver Tissue from Patients with Wilson’S Disease Determined by Micro X-Ray Fluorescence. J. Trace Elem. Med. Biol. 2019, 51, 42–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hachmoller, O.; Zibert, A.; Zischka, H.; Sperling, M.; Groba, S.R.; Grunewald, I.; Wardelmann, E.; Schmidt, H.H.; Karst, U. Spatial Investigation of the Elemental Distribution in Wilson’s Disease Liver after D-Penicillamine Treatment by La-Icp-Ms. J. Trace Elem. Med. Biol. 2017, 44, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Dusek, P.; Lescinskij, A.; Ruzicka, F.; Acosta-Cabronero, J.; Bruha, R.; Sieger, T.; Hajek, M.; Dezortova, M. Associations of Brain Atrophy and Cerebral Iron Accumulation at Mri with Clinical Severity in Wilson Disease. Radiology 2021, 299, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Dezortova, M.; Lescinskij, A.; Dusek, P.; Herynek, V.; Acosta-Cabronero, J.; Bruha, R.; Jiru, F.; Robinson, S.D.; Hajek, M. Multiparametric Quantitative Brain Mri in Neurological and Hepatic Forms of Wilson’s Disease. J. Magn. Reson. Imaging 2020, 51, 1829–1835. [Google Scholar] [CrossRef]
- Dusek, P.; Bahn, E.; Litwin, T.; Jablonka-Salach, K.; Luciuk, A.; Huelnhagen, T.; Madai, V.I.; Dieringer, M.A.; Bulska, E.; Knauth, M.; et al. Brain Iron Accumulation in Wilson Disease: A Post Mortem 7 Tesla Mri—Histopathological Study. Neuropathol. Appl. Neurobiol. 2017, 43, 514–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Jimenez, F.J.; Alonso-Navarro, H.; Garcia-Martin, E.; Agundez, J.A.G. Biological Fluid Levels of Iron and Iron-Related Proteins in Parkinson’s Disease: Review and Meta-Analysis. Eur. J. Neurol. 2021, 28, 1041–1055. [Google Scholar] [CrossRef]
- Depierreux, F.; Parmentier, E.; Mackels, L.; Baquero, K.; Degueldre, C.; Balteau, E.; Salmon, E.; Phillips, C.; Bahri, M.A.; Maquet, P.; et al. Parkinson’s Disease Multimodal Imaging: F-Dopa Pet, Neuromelanin-Sensitive and Quantitative Iron-Sensitive Mri. NPJ Parkinsons Dis. 2021, 7, 57. [Google Scholar] [CrossRef]
- Biondetti, E.; Santin, M.D.; Valabregue, R.; Mangone, G.; Gaurav, R.; Pyatigorskaya, N.; Hutchison, M.; Yahia-Cherif, L.; Villain, N.; Habert, M.O.; et al. The Spatiotemporal Changes in Dopamine, Neuromelanin and Iron Characterizing Parkinson’s Disease. Brain 2021, 144, 3114–3125. [Google Scholar] [CrossRef]
- Riederer, P.; Monoranu, C.; Strobel, S.; Iordache, T.; Sian-Hulsmann, J. Iron as the Concert Master in the Pathogenic Orchestra Playing in Sporadic Parkinson’s Disease. J. Neural. Transm. 2021, 128, 1577–1598. [Google Scholar] [CrossRef]
- Oakley, A.E.; Collingwood, J.F.; Dobson, J.; Love, G.; Perrott, H.R.; Edwardson, J.A.; Elstner, M.; Morris, C.M. Individual Dopaminergic Neurons Show Raised Iron Levels in Parkinson Disease. Neurology 2007, 68, 1820–1825. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, I.; Reimann, K.; Jankuhn, S.; Kirilina, E.; Stieler, J.; Sonntag, M.; Meijer, J.; Weiskopf, N.; Reinert, T.; Arendt, T.; et al. Cell Specific Quantitative Iron Mapping on Brain Slices by Immuno-Micropixe in Healthy Elderly and Parkinson’s Disease. Acta Neuropathol. Commun. 2021, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.M.; Bohic, S.; Carmona, A.; Ortega, R.; Cottam, V.; Hare, D.J.; Finberg, J.P.; Reyes, S.; Halliday, G.M.; Mercer, J.F.; et al. Copper Pathology in Vulnerable Brain Regions in Parkinson’s Disease. Neurobiol. Aging 2014, 35, 858–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hare, D.J.; Doecke, J.D.; Faux, N.G.; Rembach, A.; Volitakis, I.; Fowler, C.J.; Grimm, R.; Doble, P.A.; Cherny, R.A.; Masters, C.L.; et al. Decreased Plasma Iron in Alzheimer’s Disease Is Due to Transferrin Desaturation. ACS Chem. Neurosci. 2015, 6, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Jouini, N.; Saied, Z.; Sassi, S.B.; Nebli, F.; Messaoud, T.; Hentati, F.; Belal, S. Impacts of Iron Metabolism Dysregulation on Alzheimer’s Disease. J. Alzheimers Dis. 2021, 80, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Kweon, O.J.; Youn, Y.C.; Lim, Y.K.; Lee, M.K.; Kim, H.R. Clinical Utility of Serum Hepcidin and Iron Profile Measurements in Alzheimer’s Disease. J. Neurol. Sci. 2019, 403, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Van Duijn, S.; Bulk, M.; van Duinen, S.G.; Nabuurs, R.J.A.; van Buchem, M.A.; van der Weerd, L.; Natte, R. Cortical Iron Reflects Severity of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 60, 1533–1545. [Google Scholar] [CrossRef] [Green Version]
- Ayton, S.; Wang, Y.; Diouf, I.; Schneider, J.A.; Brockman, J.; Morris, M.C.; Bush, A.I. Brain Iron Is Associated with Accelerated Cognitive Decline in People with Alzheimer Pathology. Mol. Psychiatry 2020, 25, 2932–2941. [Google Scholar] [CrossRef]
- Cogswell, P.M.; Wiste, H.J.; Senjem, M.L.; Gunter, J.L.; Weigand, S.D.; Schwarz, C.G.; Arani, A.; Therneau, T.M.; Lowe, V.J.; Knopman, D.S.; et al. Associations of Quantitative Susceptibility Mapping with Alzheimer’s Disease Clinical and Imaging Markers. Neuroimage 2021, 224, 117433. [Google Scholar] [CrossRef]
- Spotorno, N.; Acosta-Cabronero, J.; Stomrud, E.; Lampinen, B.; Strandberg, O.T.; van Westen, D.; Hansson, O. Relationship between Cortical Iron and Tau Aggregation in Alzheimer’s Disease. Brain 2020, 143, 1341–1349. [Google Scholar] [CrossRef]
- Damulina, A.; Pirpamer, L.; Soellradl, M.; Sackl, M.; Tinauer, C.; Hofer, E.; Enzinger, C.; Gesierich, B.; Duering, M.; Ropele, S.; et al. Cross-Sectional and Longitudinal Assessment of Brain Iron Level in Alzheimer Disease Using 3-T Mri. Radiology 2020, 296, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhao, Z.; Cui, A.; Zhu, Y.; Zhang, L.; Liu, J.; Shi, S.; Fu, C.; Han, X.; Gao, W.; et al. Increased Iron Deposition on Brain Quantitative Susceptibility Mapping Correlates with Decreased Cognitive Function in Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Kenkhuis, B.; Somarakis, A.; de Haan, L.; Dzyubachyk, O.; Ijsselsteijn, M.E.; de Miranda, N.F.C.C.; Lelieveldt, B.P.F.; Dijkstra, J.; van Roon-Mom, W.M.C.; Hollt, T.; et al. Iron Loading Is a Prominent Feature of Activated Microglia in Alzheimer’s Disease Patients. Acta Neuropathol. Commun. 2021, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Madsen, S.J.; DiGiacomo, P.S.; Zeng, Y.; Goubran, M.; Chen, Y.; Rutt, B.K.; Born, D.; Vogel, H.; Sinclair, R.; Zeineh, M.M. Correlative Microscopy to Localize and Characterize Iron Deposition in Alzheimer’s Disease. J. Alzheimers Dis. Rep. 2020, 4, 525–536. [Google Scholar] [CrossRef]
- Zeineh, M.M.; Chen, Y.; Kitzler, H.H.; Hammond, R.; Vogel, H.; Rutt, B.K. Activated Iron-Containing Microglia in the Human Hippocampus Identified by Magnetic Resonance Imaging in Alzheimer Disease. Neurobiol. Aging 2015, 36, 2483–2500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, J.; Collingwood, J.F.; Tjendana-Tjhin, V.; Brooks, J.; Lermyte, F.; Plascencia-Villa, G.; Hands-Portman, I.; Dobson, J.; Perry, G.; Telling, N.D. Nanoscale Synchrotron X-ray Speciation of Iron and Calcium Compounds in Amyloid Plaque Cores from Alzheimer’s Disease Subjects. Nanoscale 2018, 10, 11782–11796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plascencia-Villa, G.; Ponce, A.; Collingwood, J.F.; Arellano-Jimenez, M.J.; Zhu, X.; Rogers, J.T.; Betancourt, I.; Jose-Yacaman, M.; Perry, G. High-Resolution Analytical Imaging and Electron Holography of Magnetite Particles in Amyloid Cores of Alzheimer’s Disease. Sci. Rep. 2016, 6, 24873. [Google Scholar] [CrossRef]
- Wang, L.; Li, C.; Chen, X.; Li, S.; Shang, H. Abnormal Serum Iron-Status Indicator Changes in Amyotrophic Lateral Sclerosis (Als) Patients: A Meta-Analysis. Front. Neurol. 2020, 11, 380. [Google Scholar] [CrossRef]
- Tandon, L.; Kasarskis, E.J.; Ehmann, W.D. Elemental Imbalance Studies by Inaa on Extraneural Tissues from Amyotrophic Lateral Sclerosis Patients. J. Radioanal. Nucl. Chem. 1995, 195, 13–19. [Google Scholar] [CrossRef]
- Ince, P.G.; Shaw, P.J.; Candy, J.M.; Mantle, D.; Tandon, L.; Ehmann, W.D.; Markesbery, W.R. Iron, Selenium and Glutathione Peroxidase Activity Are Elevated in Sporadic Motor Neuron Disease. Neurosci. Lett. 1994, 182, 87–90. [Google Scholar] [CrossRef]
- Roos, P.M. Studies on Metals in Motor Neuron Disease. Ph.D. Thesis, Karolinska Institutet, Stockholm, Sweden, 2013. [Google Scholar]
- Roeben, B.; Wilke, C.; Bender, B.; Ziemann, U.; Synofzik, M. The Motor Band Sign in Als: Presentations and Frequencies in a Consecutive Series of Als Patients. J. Neurol. Sci. 2019, 406, 116440. [Google Scholar] [CrossRef] [PubMed]
- De Reuck, J.; Devos, D.; Moreau, C.; Auger, F.; Durieux, N.; Deramecourt, V.; Pasquier, F.; Maurage, C.A.; Cordonnier, C.; Leys, D.; et al. Topographic Distribution of Brain Iron Deposition and Small Cerebrovascular Lesions in Amyotrophic Lateral Sclerosis and in Frontotemporal Lobar Degeneration: A Post-Mortem 7.0-Tesla Magnetic Resonance Imaging Study with Neuropathological Correlates. Acta Neurol. Belg. 2017, 117, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Cabronero, J.; Machts, J.; Schreiber, S.; Abdulla, S.; Kollewe, K.; Petri, S.; Spotorno, N.; Kaufmann, J.; Heinze, H.J.; Dengler, R.; et al. Quantitative Susceptibility Mri to Detect Brain Iron in Amyotrophic Lateral Sclerosis. Radiology 2018, 289, 195–203. [Google Scholar] [CrossRef]
- Kasarskis, E.J.; Ehmann, W.D.; Markesbery, W.R. Trace Metals in Human Neurodegenerative Diseases. Prog. Clin. Biol. Res. 1993, 380, 299–310. [Google Scholar] [PubMed]
- Kwan, J.Y.; Jeong, S.Y.; van Gelderen, P.; Deng, H.X.; Quezado, M.M.; Danielian, L.E.; Butman, J.A.; Chen, L.; Bayat, E.; Russell, J.; et al. Iron Accumulation in Deep Cortical Layers Accounts for Mri Signal Abnormalities in Als: Correlating 7 Tesla Mri and Pathology. PLoS ONE 2012, 7, e35241. [Google Scholar] [CrossRef]
- Hametner, S.; Wimmer, I.; Haider, L.; Pfeifenbring, S.; Bruck, W.; Lassmann, H. Iron and Neurodegeneration in the Multiple Sclerosis Brain. Ann. Neurol. 2013, 74, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Schweser, F.; Hagemeier, J.; Dwyer, M.G.; Bergsland, N.; Hametner, S.; Weinstock-Guttman, B.; Zivadinov, R. Decreasing Brain Iron in Multiple Sclerosis: The Difference between Concentration and Content in Iron Mri. Hum. Brain Mapp. 2021, 42, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Burgetova, A.; Dusek, P.; Vaneckova, M.; Horakova, D.; Langkammer, C.; Krasensky, J.; Sobisek, L.; Matras, P.; Masek, M.; Seidl, Z. Thalamic Iron Differentiates Primary-Progressive and Relapsing-Remitting Multiple Sclerosis. AJNR Am. J. Neuroradiol. 2017, 38, 1079–1086. [Google Scholar] [CrossRef] [Green Version]
- Khalil, M.; Langkammer, C.; Pichler, A.; Pinter, D.; Gattringer, T.; Bachmaier, G.; Ropele, S.; Fuchs, S.; Enzinger, C.; Fazekas, F. Dynamics of Brain Iron Levels in Multiple Sclerosis: A Longitudinal 3t Mri Study. Neurology 2015, 84, 2396–2402. [Google Scholar] [CrossRef]
- Zivadinov, R.; Tavazzi, E.; Bergsland, N.; Hagemeier, J.; Lin, F.; Dwyer, M.G.; Carl, E.; Kolb, C.; Hojnacki, D.; Ramasamy, D.; et al. Brain Iron at Quantitative Mri Is Associated with Disability in Multiple Sclerosis. Radiology 2018, 289, 487–496. [Google Scholar] [CrossRef]
- Kaunzner, U.W.; Kang, Y.; Zhang, S.; Morris, E.; Yao, Y.; Pandya, S.; Rua, S.M.H.; Park, C.; Gillen, K.M.; Nguyen, T.D.; et al. Quantitative Susceptibility Mapping Identifies Inflammation in a Subset of Chronic Multiple Sclerosis Lesions. Brain 2019, 142, 133–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal-Bianco, A.; Grabner, G.; Kronnerwetter, C.; Weber, M.; Hoftberger, R.; Berger, T.; Auff, E.; Leutmezer, F.; Trattnig, S.; Lassmann, H.; et al. Slow Expansion of Multiple Sclerosis Iron Rim Lesions: Pathology and 7 T Magnetic Resonance Imaging. Acta Neuropathol. 2017, 133, 25–42. [Google Scholar] [CrossRef] [Green Version]
- Popescu, B.F.; Frischer, J.M.; Webb, S.M.; Tham, M.; Adiele, R.C.; Robinson, C.A.; Fitz-Gibbon, P.D.; Weigand, S.D.; Metz, I.; Nehzati, S.; et al. Pathogenic Implications of Distinct Patterns of Iron and Zinc in Chronic Ms Lesions. Acta Neuropathol. 2017, 134, 45–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagnato, F.; Hametner, S.; Yao, B.; van Gelderen, P.; Merkle, H.; Cantor, F.K.; Lassmann, H.; Duyn, J.H. Tracking Iron in Multiple Sclerosis: A Combined Imaging and Histopathological Study at 7 Tesla. Brain 2011, 134, 3602–3615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.Q.; Ni, W.; Wang, R.M.; Dong, Y.; Wu, Z.Y. A Novel Ceruloplasmin Mutation Identified in a Chinese Patient and Clinical Spectrum of Aceruloplasminemia Patients. Metab. Brain Dis. 2021, 36, 2273–2281. [Google Scholar] [CrossRef]
- Kenawi, M.; Rouger, E.; Island, M.L.; Leroyer, P.; Robin, F.; Remy, S.; Tesson, L.; Anegon, I.; Nay, K.; Derbre, F.; et al. Ceruloplasmin Deficiency Does Not Induce Macrophagic Iron Overload: Lessons from a New Rat Model of Hereditary Aceruloplasminemia. FASEB J. 2019, 33, 13492–13502. [Google Scholar] [CrossRef] [Green Version]
- Marchi, G.; Busti, F.; Zidanes, A.L.; Castagna, A.; Girelli, D. Aceruloplasminemia: A Severe Neurodegenerative Disorder Deserving an Early Diagnosis. Front. Neurosci. 2019, 13, 325. [Google Scholar] [CrossRef] [Green Version]
- Riboldi, G.M.; Anstett, K.; Jain, R.; Lau, H.; Swope, D. Aceruloplasminemia and Putaminal Cavitation. Parkinsonism Relat. Disord. 2018, 51, 121–123. [Google Scholar] [CrossRef]
- Yoshida, K.; Hayashi, H.; Wakusawa, S.; Shigemasa, R.; Koide, R.; Ishikawa, T.; Tatsumi, Y.; Kato, K.; Ohara, S.; Ikeda, S.I. Coexistence of Copper in the Iron-Rich Particles of Aceruloplasminemia Brain. Biol. Trace Elem. Res. 2017, 175, 79–86. [Google Scholar] [CrossRef]
- Dusek, P.; Schneider, S.A.; Aaseth, J. Iron Chelation in the Treatment of Neurodegenerative Diseases. J. Trace Elem. Med. Biol. 2016, 38, 81–92. [Google Scholar] [CrossRef]
- Miyake, Z.; Nakamagoe, K.; Yoshida, K.; Kondo, T.; Tamaoka, A. Deferasirox Might Be Effective for Microcytic Anemia and Neurological Symptoms Associated with Aceruloplasminemia: A Case Report and Review of the Literature. Intern. Med. 2020, 59, 1755–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vroegindeweij, L.H.P.; Boon, A.J.W.; Wilson, J.H.P.; Langendonk, J.G. Effects of Iron Chelation Therapy on the Clinical Course of Aceruloplasminemia: An Analysis of Aggregated Case Reports. Orphanet J. Rare Dis. 2020, 15, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanardi, A.; Conti, A.; Cremonesi, M.; D’Adamo, P.; Gilberti, E.; Apostoli, P.; Cannistraci, C.V.; Piperno, A.; David, S.; Alessio, M. Ceruloplasmin Replacement Therapy Ameliorates Neurological Symptoms in a Preclinical Model of Aceruloplasminemia. EMBO Mol. Med. 2018, 10, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Piperno, A.; Alessio, M. Aceruloplasminemia: Waiting for an Efficient Therapy. Front. Neurosci. 2018, 12, 903. [Google Scholar] [CrossRef] [Green Version]
- Poli, L.; Alberici, A.; Buzzi, P.; Marchina, E.; Lanari, A.; Arosio, C.; Ciccone, A.; Semeraro, F.; Gasparotti, R.; Padovani, A.; et al. Is Aceruloplasminemia Treatable? Combining Iron Chelation and Fresh-Frozen Plasma Treatment. Neurol. Sci. 2017, 38, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, X.P. Does Ceruloplasmin Defend against Neurodegenerative Diseases? Curr. Neuropharmacol. 2019, 17, 539–549. [Google Scholar] [CrossRef]
- Borges, M.D.; de Albuquerque, D.M.; Lanaro, C.; Costa, F.F.; Fertrin, K.Y. Clinical Relevance of Heterozygosis for Aceruloplasminemia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2019, 180, 266–271. [Google Scholar] [CrossRef]
- Cozzi, A.; Santambrogio, P.; Ripamonti, M.; Rovida, E.; Levi, S. Pathogenic Mechanism and Modeling of Neuroferritinopathy. Cell. Mol. Life Sci. 2021, 78, 3355–3367. [Google Scholar] [CrossRef]
- Muhoberac, B.B.; Vidal, R. Iron, Ferritin, Hereditary Ferritinopathy, and Neurodegeneration. Front. Neurosci. 2019, 13, 1195. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Rizek, P.; Jog, M. Neuroferritinopathy: Pathophysiology, Presentation, Differential Diagnoses and Management. Tremor Other Hyperkinet. Mov. 2016, 6, 355. [Google Scholar] [CrossRef]
- McNally, J.R.; Mehlenbacher, M.R.; Luscieti, S.; Smith, G.L.; Reutovich, A.A.; Maura, P.; Arosio, P.; Bou-Abdallah, F. Mutant L-Chain Ferritins That Cause Neuroferritinopathy Alter Ferritin Functionality and Iron Permeability. Metallomics 2019, 11, 1635–1647. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, T.; Okada, Y.; Yamamoto, T.; Sato, D.; Fujiwara, K.; Fukumura, T.; Ikeguchi, M. Structure, Function, Folding, and Aggregation of a Neuroferritinopathy-Related Ferritin Variant. Biochemistry 2019, 58, 2318–2325. [Google Scholar] [CrossRef] [PubMed]
- Park, C.W.; Kim, N.Y.; Kim, Y.J.; Song, S.K.; Lyoo, C.H. A Patient with Neuroferritinopathy Presenting with Juvenile-Onset Voice Tremor. J. Mov. Disord. 2020, 13, 66–68. [Google Scholar] [CrossRef]
- Cozzi, A.; Orellana, D.I.; Santambrogio, P.; Rubio, A.; Cancellieri, C.; Giannelli, S.; Ripamonti, M.; Taverna, S.; di Lullo, G.; Rovida, E.; et al. Stem Cell Modeling of Neuroferritinopathy Reveals Iron as a Determinant of Senescence and Ferroptosis During Neuronal Aging. Stem Cell Rep. 2019, 13, 832–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garringer, H.J.; Irimia, J.M.; Li, W.; Goodwin, C.B.; Richine, B.; Acton, A.; Chan, R.J.; Peacock, M.; Muhoberac, B.B.; Ghetti, B.; et al. Effect of Systemic Iron Overload and a Chelation Therapy in a Mouse Model of the Neurodegenerative Disease Hereditary Ferritinopathy. PLoS ONE 2016, 11, e0161341. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, S.J.; Kurian, M.A.; Hogarth, P. Neurodegeneration with Brain Iron Accumulation. Handb. Clin. Neurol. 2018, 147, 293–305. [Google Scholar]
- Tello, C.; Darling, A.; Lupo, V.; Perez-Duenas, B.; Espinos, C. On the Complexity of Clinical and Molecular Bases of Neurodegeneration with Brain Iron Accumulation. Clin. Genet. 2018, 93, 731–740. [Google Scholar] [CrossRef]
- Arber, C.E.; Li, A.; Houlden, H.; Wray, S. Review: Insights into Molecular Mechanisms of Disease in Neurodegeneration with Brain Iron Accumulation: Unifying Theories. Neuropathol. Appl. Neurobiol. 2016, 42, 220–241. [Google Scholar] [CrossRef]
- Karin, I.; Buchner, B.; Gauzy, F.; Klucken, A.; Klopstock, T. Treat Iron-Related Childhood-Onset Neurodegeneration (Tircon)-an International Network on Care and Research for Patients with Neurodegeneration with Brain Iron Accumulation (Nbia). Front. Neurol. 2021, 12, 642228. [Google Scholar] [CrossRef]
- Wang, Z.B.; Liu, J.Y.; Xu, X.J.; Mao, X.Y.; Zhang, W.; Zhou, H.H.; Liu, Z.Q. Neurodegeneration with Brain Iron Accumulation: Insights into the Mitochondria Dysregulation. Biomed. Pharmacother. 2019, 118, 109068. [Google Scholar] [CrossRef]
- Kinghorn, K.J.; Castillo-Quan, J.I. Mitochondrial Dysfunction and Defects in Lipid Homeostasis as Therapeutic Targets in Neurodegeneration with Brain Iron Accumulation. Rare Dis. 2016, 4, e1128616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohd Fauzi, N.A.; Ibrahim, N.M.; Mukari, S.A.M.; Jegan, T.; Aziz, Z.A. Amelioration of Dystonic Opisthotonus in Pantothenate Kinase-Associated Neurodegeneration Syndrome with Absent “Eye-of-the-Tiger” Sign Following Bilateral Pallidal Deep Brain Stimulation. Mov. Disord. Clin. Pract. 2019, 6, 332–334. [Google Scholar] [CrossRef] [Green Version]
- Werning, M.; Mullner, E.W.; Mlynek, G.; Dobretzberger, V.; Djinovic-Carugo, K.; Baron, D.M.; Prokisch, H.; Buchner, B.; Klopstock, T.; Salzer, U. Pkan Neurodegeneration and Residual Pank2 Activities in Patient Erythrocytes. Ann. Clin. Transl. Neurol. 2020, 7, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Zhang, J.; Jiang, Y.; Wang, J.; Wu, Y. Natural History and Genotype-Phenotype Correlation of Pantothenate Kinase-Associated Neurodegeneration. CNS Neurosci. Ther. 2020, 26, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Arber, C.; Angelova, P.R.; Wiethoff, S.; Tsuchiya, Y.; Mazzacuva, F.; Preza, E.; Bhatia, K.P.; Mills, K.; Gout, I.; Abramov, A.Y.; et al. Ipsc-Derived Neuronal Models of Pank2-Associated Neurodegeneration Reveal Mitochondrial Dysfunction Contributing to Early Disease. PLoS ONE 2017, 12, e0184104. [Google Scholar] [CrossRef]
- Santambrogio, P.; Ripamonti, M.; Paolizzi, C.; Panteghini, C.; Carecchio, M.; Chiapparini, L.; Raimondi, M.; Rubio, A.; di Meo, I.; Cozzi, A.; et al. Harmful Iron-Calcium Relationship in Pantothenate Kinase Associated Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 3664. [Google Scholar] [CrossRef] [PubMed]
- Orellana, D.I.; Santambrogio, P.; Rubio, A.; Yekhlef, L.; Cancellieri, C.; Dusi, S.; Giannelli, S.G.; Venco, P.; Mazzara, P.G.; Cozzi, A.; et al. Coenzyme a Corrects Pathological Defects in Human Neurons of Pank2-Associated Neurodegeneration. EMBO Mol. Med. 2016, 8, 1197–1211. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, D.; Yang, T. Novel Pank2 Mutation in a Chinese Boy with Pank2-Associated Neurodegeneration: A Case Report and Review of Chinese Cases. Medicine 2019, 98, e14122. [Google Scholar] [CrossRef]
- Shi, X.; Zheng, F.; Ye, X.; Li, X.; Zhao, Q.; Lin, Z.; Hu, Y.; Wang, J. Basal Ganglia Calcification and Novel Compound Heterozygous Mutations in the Pank2 Gene in a Chinese Boy with Classic Pantothenate Kinase-Associated Neurodegeneration: A Case Report. Medicine 2018, 97, e0316. [Google Scholar] [CrossRef]
- Fasano, A.; Shahidi, G.; Lang, A.E.; Rohani, M. Basal Ganglia Calcification in a Case of Pkan. Parkinsonism Relat. Disord. 2017, 36, 98–99. [Google Scholar] [CrossRef]
- Drecourt, A.; Babdor, J.; Dussiot, M.; Petit, F.; Goudin, N.; Garfa-Traore, M.; Habarou, F.; Bole-Feysot, C.; Nitschke, P.; Ottolenghi, C.; et al. Impaired Transferrin Receptor Palmitoylation and Recycling in Neurodegeneration with Brain Iron Accumulation. Am. J. Hum. Genet. 2018, 102, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Petit, F.; Drecourt, A.; Dussiot, M.; Zangarelli, C.; Hermine, O.; Munnich, A.; Rotig, A. Defective Palmitoylation of Transferrin Receptor Triggers Iron Overload in Friedreich Ataxia Fibroblasts. Blood 2021, 137, 2090–2102. [Google Scholar] [CrossRef]
- Klopstock, T.; Tricta, F.; Neumayr, L.; Karin, I.; Zorzi, G.; Fradette, C.; Kmiec, T.; Buchner, B.; Steele, H.E.; Horvath, R.; et al. Safety and Efficacy of Deferiprone for Pantothenate Kinase-Associated Neurodegeneration: A Randomised, Double-Blind, Controlled Trial and an Open-Label Extension Study. Lancet Neurol. 2019, 18, 631–642. [Google Scholar] [CrossRef]
- Zizioli, D.; Tiso, N.; Guglielmi, A.; Saraceno, C.; Busolin, G.; Giuliani, R.; Khatri, D.; Monti, E.; Borsani, G.; Argenton, F.; et al. Knock-Down of Pantothenate Kinase 2 Severely Affects the Development of the Nervous and Vascular System in Zebrafish, Providing New Insights into Pkan Disease. Neurobiol. Dis. 2016, 85, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Khatri, D.; Zizioli, D.; Trivedi, A.; Borsani, G.; Monti, E.; Finazzi, D. Overexpression of Human Mutant Pank2 Proteins Affects Development and Motor Behavior of Zebrafish Embryos. Neuromol. Med. 2019, 21, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Cordoba, M.; Khoury, A.F.; Villanueva-Paz, M.; Gomez-Navarro, C.; Villalon-Garcia, I.; Suarez-Rivero, J.M.; Povea-Cabello, S.; de la Mata, M.; Cotan, D.; Talaveron-Rey, M.; et al. Pantothenate Rescues Iron Accumulation in Pantothenate Kinase-Associated Neurodegeneration Depending on the Type of Mutation. Mol. Neurobiol. 2019, 56, 3638–3656. [Google Scholar] [CrossRef] [Green Version]
- Dusi, S.; Valletta, L.; Haack, T.B.; Tsuchiya, Y.; Venco, P.; Pasqualato, S.; Goffrini, P.; Tigano, M.; Demchenko, N.; Wieland, T.; et al. Exome Sequence Reveals Mutations in Coa Synthase as a Cause of Neurodegeneration with Brain Iron Accumulation. Am. J. Hum. Genet. 2014, 94, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Khatri, D.; Zizioli, D.; Tiso, N.; Facchinello, N.; Vezzoli, S.; Gianoncelli, A.; Memo, M.; Monti, E.; Borsani, G.; Finazzi, D. Down-Regulation of Coasy, the Gene Associated with Nbia-Vi, Reduces Bmp Signaling, Perturbs Dorso-Ventral Patterning and Alters Neuronal Development in Zebrafish. Sci. Rep. 2016, 6, 37660. [Google Scholar] [CrossRef] [Green Version]
- Iankova, V.; Karin, I.; Klopstock, T.; Schneider, S.A. Emerging Disease-Modifying Therapies in Neurodegeneration with Brain Iron Accumulation (Nbia) Disorders. Front. Neurol. 2021, 12, 629414. [Google Scholar] [CrossRef]
- Jackowski, S. Proposed Therapies for Pantothenate-Kinase-Associated Neurodegeneration. J. Exp. Neurosci. 2019, 13, 1179069519851118. [Google Scholar] [CrossRef]
- Dusek, P.; Skoloudik, D.; Roth, J.; Dusek, P. Mitochondrial Membrane Protein-Associated Neurodegeneration: A Case Report and Literature Review. Neurocase 2018, 24, 161–165. [Google Scholar] [CrossRef]
- Sparber, P.; Krylova, T.; Repina, S.; Demina, N.; Rudenskaya, G.; Sharkova, I.; Sharkov, A.; Kadyshev, V.; Kanivets, I.; Korostelev, S.; et al. Retrospective Analysis of 17 Patients with Mitochondrial Membrane Protein-Associated Neurodegeneration Diagnosed in Russia. Parkinsonism Relat. Disord. 2021, 84, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Rickman, O.J.; Salter, C.G.; Gunning, A.C.; Fasham, J.; Voutsina, N.; Leslie, J.S.; McGavin, L.; Cross, H.E.; Posey, J.E.; Akdemir, Z.C.; et al. Dominant Mitochondrial Membrane Protein-Associated Neurodegeneration (Mpan) Variants Cluster within a Specific C19orf12 Isoform. Parkinsonism Relat. Disord. 2021, 82, 84–86. [Google Scholar] [CrossRef]
- Gregory, A.; Lotia, M.; Jeong, S.Y.; Fox, R.; Zhen, D.; Sanford, L.; Hamada, J.; Jahic, A.; Beetz, C.; Freed, A.; et al. Autosomal Dominant Mitochondrial Membrane Protein-Associated Neurodegeneration (Mpan). Mol. Genet. Genom. Med. 2019, 7, e00736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savitt, D.; Jankovic, J. Levodopa-Induced Dyskinesias in Mitochondrial Membrane Protein-Associated Neurodegeneration. Neurol. Clin. Pract. 2019, 9, e7–e9. [Google Scholar] [CrossRef] [PubMed]
- Monfrini, E.; Melzi, V.; Buongarzone, G.; Franco, G.; Ronchi, D.; Dilena, R.; Scola, E.; Vizziello, P.; Bordoni, A.; Bresolin, N.; et al. A De Novo C19orf12 Heterozygous Mutation in a Patient with Mpan. Parkinsonism Relat. Disord. 2018, 48, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Skowronska, M.; Kmiec, T.; Jurkiewicz, E.; Malczyk, K.; Kurkowska-Jastrzebska, I.; Czlonkowska, A. Evolution and Novel Radiological Changes of Neurodegeneration Associated with Mutations in C19orf12. Parkinsonism Relat. Disord. 2017, 39, 71–76. [Google Scholar] [CrossRef]
- Venco, P.; Bonora, M.; Giorgi, C.; Papaleo, E.; Iuso, A.; Prokisch, H.; Pinton, P.; Tiranti, V. Mutations of C19orf12, Coding for a Transmembrane Glycine Zipper Containing Mitochondrial Protein, Cause Mis-Localization of the Protein, Inability to Respond to Oxidative Stress and Increased Mitochondrial Ca(2)(+). Front. Genet. 2015, 6, 185. [Google Scholar] [CrossRef] [Green Version]
- Shao, C.; Zhu, J.; Ma, X.; Siedlak, S.L.; Cohen, M.L.; Lerner, A.; Wang, W. C19orf12 Ablation Causes Ferroptosis in Mitochondrial Membrane Protein-Associated with Neurodegeneration. Free Radic. Biol. Med. 2022, 182, 23–33. [Google Scholar] [CrossRef]
- Skowronska, M.; Buksinska-Lisik, M.; Kmiec, T.; Litwin, T.; Kurkowska-Jastrzebska, I.; Czlonkowska, A. Is There Heart Disease in Cases of Neurodegeneration Associated with Mutations in C19orf12? Parkinsonism Relat. Disord. 2020, 80, 15–18. [Google Scholar] [CrossRef]
- Kasapkara, C.S.; Tumer, L.; Gregory, A.; Ezgu, F.; Inci, A.; Derinkuyu, B.E.; Fox, R.; Rogers, C.; Hayflick, S. A New Nbia Patient from Turkey with Homozygous C19orf12 Mutation. Acta Neurol. Belg. 2019, 119, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.P.; Tang, B.S.; Guo, J.F. Pla2g6-Associated Neurodegeneration (Plan): Review of Clinical Phenotypes and Genotypes. Front. Neurol. 2018, 9, 1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.; Li, Y.; Shi, C.; Gao, Y.; Yang, J.; Liang, D.; Yang, Z.; Xu, Y. Identification of a Novel Mutation in Pla2g6 Gene and Phenotypic Heterogeneity Analysis of Pla2g6-Related Neurodegeneration. Parkinsonism Relat. Disord. 2019, 65, 159–164. [Google Scholar] [CrossRef]
- Gitiaux, C.; Kaminska, A.; Boddaert, N.; Barcia, G.; Gueden, S.; Tich, S.N.T.; de Lonlay, P.; Quijano-Roy, S.; Hully, M.; Pereon, Y.; et al. Pla2g6-Associated Neurodegeneration: Lessons from Neurophysiological Findings. Eur. J. Paediatr. Neurol. 2018, 22, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.T.; Lin, H.Y.; Chen, P.L.; Lin, C.H. Genotype-Phenotype Correlations of Adult-Onset Pla2g6-Associated Neurodegeneration: Case Series and Literature Review. BMC Neurol. 2020, 20, 101. [Google Scholar] [CrossRef] [Green Version]
- Shen, T.; Hu, J.; Jiang, Y.; Zhao, S.; Lin, C.; Yin, X.; Yan, Y.; Pu, J.; Lai, H.Y.; Zhang, B. Early-Onset Parkinson’s Disease Caused by Pla2g6 Compound Heterozygous Mutation, a Case Report and Literature Review. Front. Neurol. 2019, 10, 915. [Google Scholar] [CrossRef] [Green Version]
- Mascalchi, M.; Mari, F.; Berti, B.; Bartolini, E.; Lenge, M.; Bianchi, A.; Antonucci, L.; Santorelli, F.M.; Garavaglia, B.; Guerrini, R. Fast Progression of Cerebellar Atrophy in Pla2g6-Associated Infantile Neuronal Axonal Dystrophy. Cerebellum 2017, 16, 742–745. [Google Scholar] [CrossRef]
- Miki, Y.; Tanji, K.; Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Pla2g6 Accumulates in Lewy Bodies in Park14 and Idiopathic Parkinson’s Disease. Neurosci. Lett. 2017, 645, 40–45. [Google Scholar] [CrossRef]
- Klein, C.; Lochte, T.; Delamonte, S.M.; Braenne, I.; Hicks, A.A.; Zschiedrich-Jansen, K.; Simon, D.K.; Friedman, J.H.; Lohmann, K. Pla2g6 Mutations and Parkinsonism: Long-Term Follow-up of Clinical Features and Neuropathology. Mov. Disord. 2016, 31, 1927–1929. [Google Scholar] [CrossRef]
- Sumi-Akamaru, H.; Beck, G.; Shinzawa, K.; Kato, S.; Riku, Y.; Yoshida, M.; Fujimura, H.; Tsujimoto, Y.; Sakoda, S.; Mochizuki, H. High Expression of Alpha-Synuclein in Damaged Mitochondria with Pla2g6 Dysfunction. Acta Neuropathol. Commun. 2016, 4, 27. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yao, Y.; Liu, H.; Peng, Y.; Ren, J.; Wu, X.; Mao, R.; Zhao, J.; Zhu, Y.; Niu, Z.; et al. Lack of Association between Pla2g6 Genetic Variation and Parkinson’s Disease: A Systematic Review. Neuropsychiatr. Dis. Treat. 2020, 16, 1755–1763. [Google Scholar] [CrossRef] [PubMed]
- Daida, K.; Nishioka, K.; Li, Y.; Yoshino, H.; Shimada, T.; Dougu, N.; Nakatsuji, Y.; Ohara, S.; Hashimoto, T.; Okiyama, R.; et al. Pla2g6 Variants Associated with the Number of Affected Alleles in Parkinson’s Disease in Japan. Neurobiol. Aging 2021, 97, 147.e1–147.e9. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, Y.; Pan, H.; Xu, K.; Jiang, L.; Zhao, Y.; Xu, Q.; Sun, Q.; Tan, J.; Yan, X.; et al. Association of Rare Heterozygous Pla2g6 Variants with the Risk of Parkinson’s Disease. Neurobiol. Aging 2021, 101, 297.e5–297.e8. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Pu, J.; Lai, H.Y.; Xu, L.; Si, X.; Yan, Y.; Jiang, Y.; Zhang, B. Genetic Analysis of Atp13a2, Pla2g6 and Fbxo7 in a Cohort of Chinese Patients with Early-Onset Parkinson’s Disease. Sci. Rep. 2018, 8, 14028. [Google Scholar] [CrossRef]
- Ke, M.; Chong, C.M.; Zeng, H.; Huang, M.; Huang, Z.; Zhang, K.; Cen, X.; Lu, J.H.; Yao, X.; Qin, D.; et al. Azoramide Protects Ipsc-Derived Dopaminergic Neurons with Pla2g6 D331y Mutation through Restoring Er Function and Creb Signaling. Cell Death Dis. 2020, 11, 130. [Google Scholar] [CrossRef] [Green Version]
- Villalon-Garcia, I.; Alvarez-Cordoba, M.; Povea-Cabello, S.; Talaveron-Rey, M.; Villanueva-Paz, M.; Luzon-Hidalgo, R.; Suarez-Rivero, J.M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Salas, J.J.; et al. Vitamin E Prevents Lipid Peroxidation and Iron Accumulation in Pla2g6-Associated Neurodegeneration. Neurobiol. Dis. 2022, 165, 105649. [Google Scholar] [CrossRef]
- Beharier, O.; Tyurin, V.A.; Goff, J.P.; Guerrero-Santoro, J.; Kajiwara, K.; Chu, T.; Tyurina, Y.Y.; Croix, C.M.S.; Wallace, C.T.; Parry, S.; et al. Pla2g6 Guards Placental Trophoblasts against Ferroptotic Injury. Proc. Natl. Acad. Sci. USA 2020, 117, 27319–27328. [Google Scholar] [CrossRef]
- Sanchez, E.; Azcona, L.J.; Paisan-Ruiz, C. Pla2g6 Deficiency in Zebrafish Leads to Dopaminergic Cell Death, Axonal Degeneration, Increased Beta-Synuclein Expression, and Defects in Brain Functions and Pathways. Mol. Neurobiol. 2018, 55, 6734–6754. [Google Scholar] [CrossRef]
- Chiu, C.C.; Lu, C.S.; Weng, Y.H.; Chen, Y.L.; Huang, Y.Z.; Chen, R.S.; Cheng, Y.C.; Huang, Y.C.; Liu, Y.C.; Lai, S.C.; et al. Park14 (D331y) Pla2g6 Causes Early-Onset Degeneration of Substantia Nigra Dopaminergic Neurons by Inducing Mitochondrial Dysfunction, Er Stress, Mitophagy Impairment and Transcriptional Dysregulation in a Knockin Mouse Model. Mol. Neurobiol. 2019, 56, 3835–3853. [Google Scholar] [CrossRef]
- Mori, A.; Hatano, T.; Inoshita, T.; Shiba-Fukushima, K.; Koinuma, T.; Meng, H.; Kubo, S.I.; Spratt, S.; Cui, C.; Yamashita, C.; et al. Parkinson’s Disease-Associated Ipla2-Via/Pla2g6 Regulates Neuronal Functions and Alpha-Synuclein Stability through Membrane Remodeling. Proc. Natl. Acad. Sci. USA 2019, 116, 20689–20699. [Google Scholar] [CrossRef] [Green Version]
- Lin, G.; Lee, P.T.; Chen, K.; Mao, D.; Tan, K.L.; Zuo, Z.; Lin, W.W.; Wang, L.; Bellen, H.J. Phospholipase Pla2g6, a Parkinsonism-Associated Gene, Affects Vps26 and Vps35, Retromer Function, and Ceramide Levels, Similar to Alpha-Synuclein Gain. Cell Metab. 2018, 28, 605–618.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliadi, K.G.; Gluscencova, O.B.; Iliadi, N.; Boulianne, G.L. Mutations in the Drosophila Homolog of Human Pla2g6 Give Rise to Age-Dependent Loss of Psychomotor Activity and Neurodegeneration. Sci. Rep. 2018, 8, 2939. [Google Scholar] [CrossRef] [Green Version]
- Cong, Y.; So, V.; Tijssen, M.A.J.; Verbeek, D.S.; Reggiori, F.; Mauthe, M. Wdr45, One Gene Associated with Multiple Neurodevelopmental Disorders. Autophagy 2021, 17, 3908–3923. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.L.; Gregory, A.; Kurian, M.A.; Bushlin, I.; Mochel, F.; Emrick, L.; Adang, L.; Bpan Guideline Contributing Author Group; Hogarth, P.; Hayflick, S.J. Consensus Clinical Management Guideline for Beta-Propeller Protein-Associated Neurodegeneration. Dev. Med. Child Neurol. 2021, 63, 1402–1409. [Google Scholar] [CrossRef]
- Saffari, A.; Schroter, J.; Garbade, S.F.; Alecu, J.E.; Ebrahimi-Fakhari, D.; Hoffmann, G.F.; Kolker, S.; Ries, M.; Syrbe, S. Quantitative Retrospective Natural History Modeling of Wdr45-Related Developmental and Epileptic Encephalopathy—A Systematic Cross-Sectional Analysis of 160 Published Cases. Autophagy 2021, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Zhao, H.; Chen, D.; Zhang, H.; Zhao, Y.G. Beta-Propeller Proteins Wdr45 and Wdr45b Regulate Autophagosome Maturation into Autolysosomes in Neural Cells. Curr. Biol. 2021, 31, 1666–1677.e6. [Google Scholar] [CrossRef]
- Wan, H.; Wang, Q.; Chen, X.; Zeng, Q.; Shao, Y.; Fang, H.; Liao, X.; Li, H.S.; Liu, M.G.; Xu, T.L.; et al. Wdr45 Contributes to Neurodegeneration through Regulation of Er Homeostasis and Neuronal Death. Autophagy 2020, 16, 531–547. [Google Scholar] [CrossRef]
- Russo, C.; Ardissone, A.; Freri, E.; Gasperini, S.; Moscatelli, M.; Zorzi, G.; Panteghini, C.; Castellotti, B.; Garavaglia, B.; Nardocci, N.; et al. Substantia Nigra Swelling and Dentate Nucleus T2 Hyperintensity May Be Early Magnetic Resonance Imaging Signs of Beta-Propeller Protein-Associated Neurodegeneration. Mov. Disord. Clin. Pract. 2019, 6, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Stige, K.E.; Gjerde, I.O.; Houge, G.; Knappskog, P.M.; Tzoulis, C. Beta-Propeller Protein-Associated Neurodegeneration: A Case Report and Review of the Literature. Clin. Case Rep. 2018, 6, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Rohani, M.; Fasano, A.; Akhoundi, F.H.; Haeri, G.; Lang, A.E.; Bidgoli, M.M.R.; Javanparast, L.; Zamani, B.; Shahidi, G.; Alavi, A. Beta-Propeller Protein Associated Neurodegeneration (Bpan); the First Report of Three Patients from Iran with De Novo Novel Mutations. Parkinsonism Relat. Disord. 2019, 61, 231–233. [Google Scholar] [CrossRef]
- Seibler, P.; Burbulla, L.F.; Dulovic, M.; Zittel, S.; Heine, J.; Schmidt, T.; Rudolph, F.; Westenberger, A.; Rakovic, A.; Munchau, A.; et al. Iron Overload Is Accompanied by Mitochondrial and Lysosomal Dysfunction in Wdr45 Mutant Cells. Brain 2018, 141, 3052–3064. [Google Scholar] [CrossRef] [PubMed]
- Ingrassia, R.; Memo, M.; Garavaglia, B. Ferrous Iron up-Regulation in Fibroblasts of Patients with Beta Propeller Protein-Associated Neurodegeneration (Bpan). Front. Genet. 2017, 8, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aring, L.; Choi, E.K.; Kopera, H.; Lanigan, T.; Iwase, S.; Klionsky, D.J.; Seo, Y.A. A Neurodegeneration Gene, Wdr45, Links Impaired Ferritinophagy to Iron Accumulation. J. Neurochem. 2022, 160, 356–375. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.E.; Jung, M.K.; Noh, S.G.; Choi, H.B.; Chae, S.H.; Lee, J.H.; Mun, J.Y. Iron Accumulation and Changes in Cellular Organelles in Wdr45 Mutant Fibroblasts. Int. J. Mol. Sci. 2021, 22, 11650. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Li, X.; Li, W.; Chen, G.; Xiao, H.; Li, P.; Wu, C. Wdr45 Mutation Impairs the Autophagic Degradation of Transferrin Receptor and Promotes Ferroptosis. Front. Mol. Biosci. 2021, 8, 645831. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Tan, A.H.; Ahmad-Annuar, A.; Schneider, S.A.; Bee, P.C.; Lim, J.L.; Ramli, N.; Idris, M.I. A Patient with Beta-Propeller Protein-Associated Neurodegeneration: Treatment with Iron Chelation Therapy. J. Mov. Disord. 2018, 11, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Fonderico, M.; Laudisi, M.; Andreasi, N.G.; Bigoni, S.; Lamperti, C.; Panteghini, C.; Garavaglia, B.; Carecchio, M.; Emanuele, E.A.; Forni, G.L.; et al. Patient Affected by Beta-Propeller Protein-Associated Neurodegeneration: A Therapeutic Attempt with Iron Chelation Therapy. Front. Neurol. 2017, 8, 385. [Google Scholar] [CrossRef] [Green Version]
- Lehericy, S.; Roze, E.; Goizet, C.; Mochel, F. Mri of Neurodegeneration with Brain Iron Accumulation. Curr. Opin. Neurol. 2020, 33, 462–473. [Google Scholar] [CrossRef]
- Rattay, T.W.; Lindig, T.; Baets, J.; Smets, K.; Deconinck, T.; Sohn, A.S.; Hortnagel, K.; Eckstein, K.N.; Wiethoff, S.; Reichbauer, J.; et al. Fahn/Spg35: A Narrow Phenotypic Spectrum across Disease Classifications. Brain 2019, 142, 1561–1572. [Google Scholar] [CrossRef]
- Haeri, G.; Akhoundi, F.H.; Alavi, A.; Abdi, S.; Rohani, M. Endocrine Abnormalities in a Case of Neurodegeneration with Brain Iron Accumulation. Mov. Disord. Clin. Pract. 2020, 7, 706–707. [Google Scholar] [CrossRef]
- Abusrair, A.H.; Bohlega, S.; Al-Semari, A.; Al-Ajlan, F.S.; Al-Ahmadi, K.; Mohamed, B.; AlDakheel, A. Brain Mr Imaging Findings in Woodhouse-Sakati Syndrome. AJNR Am. J. Neuroradiol. 2018, 39, 2256–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, S.A.; Paisan-Ruiz, C.; Quinn, N.P.; Lees, A.J.; Houlden, H.; Hardy, J.; Bhatia, K.P. Atp13a2 Mutations (Park9) Cause Neurodegeneration with Brain Iron Accumulation. Mov. Disord. 2010, 25, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Bruggemann, N.; Hagenah, J.; Reetz, K.; Schmidt, A.; Kasten, M.; Buchmann, I.; Eckerle, S.; Bahre, M.; Munchau, A.; Djarmati, A.; et al. Recessively Inherited Parkinsonism: Effect of Atp13a2 Mutations on the Clinical and Neuroimaging Phenotype. Arch. Neurol. 2010, 67, 1357–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roubertie, A.; Hieu, N.; Roux, C.J.; Leboucq, N.; Manes, G.; Charif, M.; Echenne, B.; Goizet, C.; Guissart, C.; Meyer, P.; et al. Ap4 Deficiency: A Novel Form of Neurodegeneration with Brain Iron Accumulation? Neurol. Genet. 2018, 4, e217. [Google Scholar] [CrossRef] [Green Version]
- Horvath, R.; Lewis-Smith, D.; Douroudis, K.; Duff, J.; Keogh, M.; Pyle, A.; Fletcher, N.; Chinnery, P.F. Scp2 Mutations and Neurodegeneration with Brain Iron Accumulation. Neurology 2015, 85, 1909–1911. [Google Scholar] [CrossRef] [Green Version]
- Dard, R.; Meyniel, C.; Touitou, V.; Stevanin, G.; Lamari, F.; Durr, A.; Ewenczyk, C.; Mochel, F. Mutations in Ddhd1, Encoding a Phospholipase A1, Is a Novel Cause of Retinopathy and Neurodegeneration with Brain Iron Accumulation. Eur. J. Med. Genet. 2017, 60, 639–642. [Google Scholar] [CrossRef]
- Correa-Vela, M.; Lupo, V.; Montpeyo, M.; Sancho, P.; Marce-Grau, A.; Hernandez-Vara, J.; Darling, A.; Jenkins, A.; Fernandez-Rodriguez, S.; Tello, C.; et al. Impaired Proteasome Activity and Neurodegeneration with Brain Iron Accumulation in Fbxo7 Defect. Ann. Clin. Transl. Neurol. 2020, 7, 1436–1442. [Google Scholar] [CrossRef]
- Jaberi, E.; Rohani, M.; Shahidi, G.A.; Nafissi, S.; Arefian, E.; Soleimani, M.; Rasooli, P.; Ahmadieh, H.; Daftarian, N.; KaramiNejadRanjbar, M.; et al. Identification of Mutation in Gtpbp2 in Patients of a Family with Neurodegeneration Accompanied by Iron Deposition in the Brain. Neurobiol. Aging 2016, 38, 216.e11–216.e18. [Google Scholar] [CrossRef] [Green Version]
- Carter, M.T.; Venkateswaran, S.; Shapira-Zaltsberg, G.; Davila, J.; Humphreys, P.; Canada, C.C.; Kernohan, K.D.; Boycott, K.M. Clinical Delineation of Gtpbp2-Associated Neuro-Ectodermal Syndrome: Report of Two New Families and Review of the Literature. Clin. Genet. 2019, 95, 601–606. [Google Scholar] [CrossRef]
- Ediz, S.S.; Aralasmak, A.; Yilmaz, T.F.; Toprak, H.; Yesil, G.; Alkan, A. Mri and Mrs Findings in Fucosidosis; a Rare Lysosomal Storage Disease. Brain Dev. 2016, 38, 435–438. [Google Scholar] [CrossRef]
- Vieira, J.P.; Conceicao, C.; Scortenschi, E. Gm1 Gangliosidosis, Late Infantile Onset Dystonia, and T2 Hypointensity in the Globus Pallidus and Substantia Nigra. Pediatr. Neurol. 2013, 49, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Lyon, G.J.; Marchi, E.; Ekstein, J.; Meiner, V.; Hirsch, Y.; Scher, S.; Yang, E.; de Vivo, D.C.; Madrid, R.; Li, Q.; et al. Vac14 Syndrome in Two Siblings with Retinitis Pigmentosa and Neurodegeneration with Brain Iron Accumulation. Cold Spring Harb. Mol. Case Stud. 2019, 5, a003715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sferra, A.; Baillat, G.; Rizza, T.; Barresi, S.; Flex, E.; Tasca, G.; D’Amico, A.; Bellacchio, E.; Ciolfi, A.; Caputo, V.; et al. Tbce Mutations Cause Early-Onset Progressive Encephalopathy with Distal Spinal Muscular Atrophy. Am. J. Hum. Genet. 2016, 99, 974–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, E.; Carss, K.J.; Rankin, J.; Nichols, J.M.; Grozeva, D.; Joseph, A.P.; Mencacci, N.E.; Papandreou, A.; Ng, J.; Barral, S.; et al. Mutations in the Histone Methyltransferase Gene Kmt2b Cause Complex Early-Onset Dystonia. Nat. Genet. 2017, 49, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Skorvanek, M.; Dusek, P.; Rydzanicz, M.; Walczak, A.; Kosinska, J.; Kostrzewa, G.; Brzozowska, M.; Han, V.; Dosekova, P.; Gdovinova, Z.; et al. Neurodevelopmental Disorder Associated with Irf2bpl Gene Mutation: Expanding the Phenotype? Parkinsonism Relat. Disord. 2019, 62, 239–241. [Google Scholar] [CrossRef]
- Zoons, E.; de Koning, T.J.; Abeling, N.G.; Tijssen, M.A. Neurodegeneration with Brain Iron Accumulation on Mri: An Adult Case of Alpha-Mannosidosis. JIMD Rep. 2012, 4, 99–102. [Google Scholar]
- Majovska, J.; Nestrasil, I.; Paulson, A.; Nascene, D.; Jurickova, K.; Hlavata, A.; Lund, T.; Orchard, P.J.; Vaneckova, M.; Zeman, J.; et al. White Matter Alteration and Cerebellar Atrophy Are Hallmarks of Brain Mri in Alpha-Mannosidosis. Mol. Genet. Metab. 2021, 132, 189–197. [Google Scholar] [CrossRef]
- Stepanova, A.; Magrane, J. Mitochondrial Dysfunction in Neurons in Friedreich’s Ataxia. Mol. Cell. Neurosci. 2020, 102, 103419. [Google Scholar] [CrossRef]
- Llorens, J.V.; Soriano, S.; Calap-Quintana, P.; Gonzalez-Cabo, P.; Molto, M.D. The Role of Iron in Friedreich’s Ataxia: Insights from Studies in Human Tissues and Cellular and Animal Models. Front. Neurosci. 2019, 13, 75. [Google Scholar] [CrossRef] [Green Version]
- Li, K. Iron Pathophysiology in Friedreich’s Ataxia. Adv. Exp. Med. Biol. 2019, 1173, 125–143. [Google Scholar]
- Turchi, R.; Faraonio, R.; Lettieri-Barbato, D.; Aquilano, K. An Overview of the Ferroptosis Hallmarks in Friedreich’s Ataxia. Biomolecules 2020, 10, 1489. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, P.; Petrillo, S.; Turchi, R.; Berardinelli, F.; Schirinzi, T.; Vasco, G.; Lettieri-Barbato, D.; Fiorenza, M.T.; Bertini, E.S.; Aquilano, K.; et al. The Nrf2 Induction Prevents Ferroptosis in Friedreich’s Ataxia. Redox Biol. 2021, 38, 101791. [Google Scholar] [CrossRef] [PubMed]
- Tamarit, J.; Britti, E.; Delaspre, F.; Medina-Carbonero, M.; Sanz-Alcazar, A.; Cabiscol, E.; Ros, J. Mitochondrial Iron and Calcium Homeostasis in Friedreich Ataxia. IUBMB Life 2021, 73, 543–553. [Google Scholar] [CrossRef]
- Elincx-Benizri, S.; Glik, A.; Merkel, D.; Arad, M.; Freimark, D.; Kozlova, E.; Cabantchik, I.; Hassin-Baer, S. Clinical Experience with Deferiprone Treatment for Friedreich Ataxia. J. Child Neurol. 2016, 31, 1036–1040. [Google Scholar] [CrossRef]
- Dusek, P.; Litwin, T.; Czlonkowska, A. Neurologic Impairment in Wilson Disease. Ann. Transl. Med. 2019, 7 (Suppl. 2), S64. [Google Scholar] [CrossRef] [PubMed]
- Czlonkowska, A.; Litwin, T.; Dusek, P.; Ferenci, P.; Lutsenko, S.; Medici, V.; Rybakowski, J.K.; Weiss, K.H.; Schilsky, M.L. Wilson Disease. Nat. Rev. Dis. Primers 2018, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Gromadzka, G.; Wierzbicka, D.; Litwin, T.; Przybylkowski, A. Difference in Iron Metabolism May Partly Explain Sex-Related Variability in the Manifestation of Wilson’s Disease. J. Trace Elem. Med. Biol. 2020, 62, 126637. [Google Scholar] [CrossRef]
- Azbukina, N.V.; Lopachev, A.V.; Chistyakov, D.V.; Goriainov, S.V.; Astakhova, A.A.; Poleshuk, V.V.; Kazanskaya, R.B.; Fedorova, T.N.; Sergeeva, M.G. Oxylipin Profiles in Plasma of Patients with Wilson’s Disease. Metabolites 2020, 10, 222. [Google Scholar] [CrossRef]
- Zhou, X.X.; Xiao, X.; Qin, H.; Chen, D.; Wu, C. Study on Different Pathogenic Factors in Different Disease Stages of Patients with Wilson Disease. Neurol. Sci. 2021, 42, 3749–3756. [Google Scholar] [CrossRef]
- Hachmoller, O.; Buzanich, A.G.; Aichler, M.; Radtke, M.; Dietrich, D.; Schwamborn, K.; Lutz, L.; Werner, M.; Sperling, M.; Walch, A.; et al. Elemental Bioimaging and Speciation Analysis for the Investigation of Wilson’s Disease Using Muxrf and Xanes. Metallomics 2016, 8, 648–653. [Google Scholar] [CrossRef] [Green Version]
- Hachmoller, O.; Aichler, M.; Schwamborn, K.; Lutz, L.; Werner, M.; Sperling, M.; Walch, A.; Karst, U. Element Bioimaging of Liver Needle Biopsy Specimens from Patients with Wilson’s Disease by Laser Ablation-Inductively Coupled Plasma-Mass Spectrometry. J. Trace Elem. Med. Biol. 2016, 35, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Gungor, S.; Selimoglu, M.A.; Varol, F.I.; Gungor, S.; Uremis, M.M. The Effects of Iron and Zinc Status on Prognosis in Pediatric Wilson’s Disease. J. Trace Elem. Med. Biol. 2019, 55, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Dusek, P.; Skoloudik, D.; Maskova, J.; Huelnhagen, T.; Bruha, R.; Zahorakova, D.; Niendorf, T.; Ruzicka, E.; Schneider, S.A.; Wuerfel, J. Brain Iron Accumulation in Wilson’s Disease: A Longitudinal Imaging Case Study During Anticopper Treatment Using 7.0t Mri and Transcranial Sonography. J. Magn. Reson. Imaging 2018, 47, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Gromadzka, G.; Wierzbicka, D.W.; Przybylkowski, A.; Litwin, T. Effect of Homeostatic Iron Regulator Protein Gene Mutation on Wilson’s Disease Clinical Manifestation: Original Data and Literature Review. Int. J. Neurosci. 2020, 1–7. [Google Scholar] [CrossRef]
- Delatycki, M.B.; Tai, G.; Corben, L.; Yiu, E.M.; Evans-Galea, M.V.; Stephenson, S.E.; Gurrin, L.; Allen, K.J.; Lynch, D.; Lockhart, P.J. Hfe P.C282y Heterozygosity Is Associated with Earlier Disease Onset in Friedreich Ataxia. Mov. Disord. 2014, 29, 940–943. [Google Scholar] [CrossRef]
- Pak, K.; Ordway, S.; Sadowski, B.; Canevari, M.; Torres, D. Wilson’s Disease and Iron Overload: Pathophysiology and Therapeutic Implications. Clin. Liver Dis. 2021, 17, 61–66. [Google Scholar] [CrossRef]
- Jonczy, A.; Lipinski, P.; Ogorek, M.; Starzynski, R.R.; Krzysztofik, D.; Bednarz, A.; Krzeptowski, W.; Szudzik, M.; Haberkiewicz, O.; Milon, A.; et al. Functional Iron Deficiency in Toxic Milk Mutant Mice (Tx-J) Despite High Hepatic Ferroportin: A Critical Role of Decreased Gpi-Ceruloplasmin Expression in Liver Macrophages. Metallomics 2019, 11, 1079–1092. [Google Scholar] [CrossRef]
- Tisdall, M.D.; Ohm, D.T.; Lobrovich, R.; Das, S.R.; Mizsei, G.; Prabhakaran, K.; Ittyerah, R.; Lim, S.; McMillan, C.T.; Wolk, D.A.; et al. Ex Vivo Mri and Histopathology Detect Novel Iron-Rich Cortical Inflammation in Frontotemporal Lobar Degeneration with Tau Versus Tdp-43 Pathology. Neuroimage Clin. 2022, 33, 102913. [Google Scholar] [CrossRef]
- Bhattarai, A.; Egan, G.F.; Talman, P.; Chua, P.; Chen, Z. Magnetic Resonance Iron Imaging in Amyotrophic Lateral Sclerosis. J. Magn. Reson. Imaging 2021, 55, 1283–1300. [Google Scholar] [CrossRef]
- Zarow, C.; Lyness, S.A.; Mortimer, J.A.; Chui, H.C. Neuronal Loss Is Greater in the Locus Coeruleus Than Nucleus Basalis and Substantia Nigra in Alzheimer and Parkinson Diseases. Arch. Neurol. 2003, 60, 337–341. [Google Scholar] [CrossRef]
- Fu, H.; Hardy, J.; Duff, K.E. Selective Vulnerability in Neurodegenerative Diseases. Nat. Neurosci. 2018, 21, 1350–1358. [Google Scholar] [CrossRef]
- Ehrenberg, A.J.; Nguy, A.K.; Theofilas, P.; Dunlop, S.; Suemoto, C.K.; Alho, A.T.d.; Leite, R.P.; Rodriguez, R.D.; Mejia, M.B.; Rub, U.; et al. Quantifying the Accretion of Hyperphosphorylated Tau in the Locus Coeruleus and Dorsal Raphe Nucleus: The Pathological Building Blocks of Early Alzheimer’s Disease. Neuropathol. Appl. Neurobiol. 2017, 43, 393–408. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Oda, M.; Tanabe, H. Diminution of Dopaminergic Neurons in the Substantia Nigra of Sporadic Amyotrophic Lateral Sclerosis. Neuropathol. Appl. Neurobiol. 1993, 19, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Hofer, T.; Perry, G. Nucleic Acid Oxidative Damage in Alzheimer’s Disease-Explained by the Hepcidin-Ferroportin Neuronal Iron Overload Hypothesis? J. Trace Elem. Med. Biol. 2016, 38, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Azad, M.G.; Dharmasivam, M.; Richardson, V.; Quinn, R.J.; Feng, Y.; Pountney, D.L.; Tonissen, K.F.; Mellick, G.D.; Yanatori, I.; et al. Parkinson’s Disease: Alterations in Iron and Redox Biology as a Key to Unlock Therapeutic Strategies. Redox Biol. 2021, 41, 101896. [Google Scholar] [CrossRef]
- Zecca, L.; Stroppolo, A.; Gatti, A.; Tampellini, D.; Toscani, M.; Gallorini, M.; Giaveri, G.; Arosio, P.; Santambrogio, P.; Fariello, R.G.; et al. The Role of Iron and Copper Molecules in the Neuronal Vulnerability of Locus Coeruleus and Substantia Nigra During Aging. Proc. Natl. Acad. Sci. USA 2004, 101, 9843–9848. [Google Scholar] [CrossRef] [Green Version]
- Biesemeier, A.; Eibl, O.; Eswara, S.; Audinot, J.N.; Wirtz, T.; Pezzoli, G.; Zucca, F.A.; Zecca, L.; Schraermeyer, U. Elemental Mapping of Neuromelanin Organelles of Human Substantia Nigra: Correlative Ultrastructural and Chemical Analysis by Analytical Transmission Electron Microscopy and Nano-Secondary Ion Mass Spectrometry. J. Neurochem. 2016, 138, 339–353. [Google Scholar] [CrossRef]
- Aaseth, J.; Dusek, P.; Roos, P.M. Prevention of Progression in Parkinson’s Disease. Biometals 2018, 31, 737–747. [Google Scholar] [CrossRef] [Green Version]
- D’Amato, R.J.; Lipman, Z.P.; Snyder, S.H. Selectivity of the Parkinsonian Neurotoxin Mptp: Toxic Metabolite Mpp+ Binds to Neuromelanin. Science 1986, 231, 987–989. [Google Scholar] [CrossRef]
- Goldman, S.M.; Musgrove, R.E.; Jewell, S.A.; Di Monte, D.A. Chapter Three—Pesticides and Parkinson’s Disease: Current experimental and epidemiological evidence. In Advances in Neurotoxicology; Costa, L.G., Aschner, M., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 83–117. [Google Scholar]
- Bjorklund, G.; Hofer, T.; Nurchi, V.M.; Aaseth, J. Iron and Other Metals in the Pathogenesis of Parkinson’s Disease: Toxic Effects and Possible Detoxification. J. Inorg. Biochem. 2019, 199, 110717. [Google Scholar] [CrossRef]
- Genoud, S.; Senior, A.M.; Hare, D.J.; Double, K.L. Meta-Analysis of Copper and Iron in Parkinson’s Disease Brain and Biofluids. Mov. Disord. 2020, 35, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.Y.; Shin, D.W.; Roh, Y.; Jang, W.; Cho, J.W.; Lee, E.A.; Ko, H.; Han, K.; Yoo, J.H. Anemia and the Risk of Parkinson’s Disease in Korean Older Adults: A Nationwide Population-Based Study. Sci. Rep. 2020, 10, 4268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joppe, K.; Nicolas, J.D.; Grunewald, T.A.; Eckermann, M.; Salditt, T.; Lingor, P. Elemental Quantification and Analysis of Structural Abnormalities in Neurons from Parkinson’s-Diseased Brains by X-ray Fluorescence Microscopy and Diffraction. Biomed. Opt. Express. 2020, 11, 3423–3443. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Guo, P.; Lian, T.H.; Zuo, L.J.; Yu, S.Y.; Liu, L.; Jin, Z.; Yu, Q.J.; Wang, R.D.; Li, L.X.; et al. Clinical Characteristics, Iron Metabolism and Neuroinflammation: New Insight into Excessive Daytime Sleepiness in Parkinson’s Disease. Neuropsychiatr. Dis. Treat. 2021, 17, 2041–2051. [Google Scholar] [CrossRef]
- Kwiatek-Majkusiak, J.; Geremek, M.; Koziorowski, D.; Tomasiuk, R.; Szlufik, S.; Friedman, A. Serum Levels of Hepcidin and Interleukin 6 in Parkinson’s Disease. Acta Neurobiol. Exp. 2020, 80, 297–304. [Google Scholar] [CrossRef]
- Huang, C.; Ma, W.; Luo, Q.; Shi, L.; Xia, Y.; Lao, C.; Liu, W.; Zou, Y.; Cheng, A.; Shi, R.; et al. Iron Overload Resulting from the Chronic Oral Administration of Ferric Citrate Induces Parkinsonism Phenotypes in Middle-Aged Mice. Aging 2019, 11, 9846–9861. [Google Scholar] [CrossRef]
- Cabantchik, Z.I.; Munnich, A.; Youdim, M.B.; Devos, D. Regional Siderosis: A New Challenge for Iron Chelation Therapy. Front. Pharmacol. 2013, 4, 167. [Google Scholar] [CrossRef] [Green Version]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garcon, G.; Rouaix, N.; et al. Targeting Chelatable Iron as a Therapeutic Modality in Parkinson’s Disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef] [Green Version]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing Research Diagnostic Criteria for Alzheimer’s Disease: The Iwg-2 Criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; del Tredici, K. Stages of the Pathologic Process in Alzheimer Disease: Age Categories from 1 to 100 Years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef]
- Goodman, L. Alzheimer’s Disease: A Clinico-Pathologic Analysis of Twenty-Three Cases with a Theory on Pathogenesis. J. Nerv. Ment. Dis. 1953, 118, 97–130. [Google Scholar] [CrossRef] [PubMed]
- Gleason, A.; Bush, A.I. Iron and Ferroptosis as Therapeutic Targets in Alzheimer’s Disease. Neurotherapeutics 2021, 18, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Jakaria, M.; Belaidi, A.A.; Bush, A.I.; Ayton, S. Ferroptosis as a Mechanism of Neurodegeneration in Alzheimer’s Disease. J. Neurochem. 2021, 159, 804–825. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Bush, A.I. The Essential Elements of Alzheimer’s Disease. J. Biol. Chem. 2021, 296, 100105. [Google Scholar] [CrossRef] [PubMed]
- Bulk, M.; Abdelmoula, W.M.; Geut, H.; Wiarda, W.; Ronen, I.; Dijkstra, J.; van der Weerd, L. Quantitative Mri and Laser Ablation-Inductively Coupled Plasma-Mass Spectrometry Imaging of Iron in the Frontal Cortex of Healthy Controls and Alzheimer’s Disease Patients. Neuroimage 2020, 215, 116808. [Google Scholar] [CrossRef]
- Cruz-Alonso, M.; Fernandez, B.; Navarro, A.; Junceda, S.; Astudillo, A.; Pereiro, R. Laser Ablation Icp-Ms for Simultaneous Quantitative Imaging of Iron and Ferroportin in Hippocampus of Human Brain Tissues with Alzheimer’s Disease. Talanta 2019, 197, 413–421. [Google Scholar] [CrossRef]
- Exley, C.; House, E.; Polwart, A.; Esiri, M.M. Brain Burdens of Aluminum, Iron, and Copper and Their Relationships with Amyloid-Beta Pathology in 60 Human Brains. J. Alzheimers Dis. 2012, 31, 725–730. [Google Scholar] [CrossRef]
- Hare, D.J.; Raven, E.P.; Roberts, B.R.; Bogeski, M.; Portbury, S.D.; McLean, C.A.; Masters, C.L.; Connor, J.R.; Bush, A.I.; Crouch, P.J.; et al. Laser Ablation-Inductively Coupled Plasma-Mass Spectrometry Imaging of White and Gray Matter Iron Distribution in Alzheimer’s Disease Frontal Cortex. Neuroimage 2016, 137, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Gong, N.J.; Dibb, R.; Bulk, M.; van der Weerd, L.; Liu, C. Imaging Beta Amyloid Aggregation and Iron Accumulation in Alzheimer’s Disease Using Quantitative Susceptibility Mapping Mri. Neuroimage 2019, 191, 176–185. [Google Scholar] [CrossRef]
- Bulk, M.; Abdelmoula, W.M.; Nabuurs, R.J.A.; van der Graaf, L.M.; Mulders, C.W.H.; Mulder, A.A.; Jost, C.R.; Koster, A.J.; van Buchem, M.A.; Natte, R.; et al. Postmortem Mri and Histology Demonstrate Differential Iron Accumulation and Cortical Myelin Organization in Early- and Late-Onset Alzheimer’s Disease. Neurobiol. Aging 2018, 62, 231–242. [Google Scholar] [CrossRef]
- Zeng, Y.; DiGiacomo, P.S.; Madsen, S.J.; Zeineh, M.M.; Sinclair, R. Exploring Valence States of Abnormal Mineral Deposits in Biological Tissues Using Correlative Microscopy and Spectroscopy Techniques: A Case Study on Ferritin and Iron Deposits from Alzheimer’s Disease Patients. Ultramicroscopy 2021, 231, 113254. [Google Scholar] [CrossRef] [PubMed]
- Everett, J.; Lermyte, F.; Brooks, J.; Tjendana-Tjhin, V.; Plascencia-Villa, G.; Hands-Portman, I.; Donnelly, J.M.; Billimoria, K.; Perry, G.; Zhu, X.; et al. Biogenic Metallic Elements in the Human Brain? Sci. Adv. 2021, 7, eabf6707. [Google Scholar] [CrossRef]
- Ayton, S.; Faux, N.G.; Bush, A.I.; Alzheimer’s Disease Neuroimaging Initiative. Ferritin Levels in the Cerebrospinal Fluid Predict Alzheimer’s Disease Outcomes and Are Regulated by Apoe. Nat. Commun. 2015, 6, 6760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayton, S.; Faux, N.G.; Bush, A.I. Association of Cerebrospinal Fluid Ferritin Level with Preclinical Cognitive Decline in Apoe-Epsilon4 Carriers. JAMA Neurol. 2017, 74, 122–125. [Google Scholar] [CrossRef] [Green Version]
- Ahmadi, S.; Zhu, S.; Sharma, R.; Wilson, D.J.; Kraatz, H.B. Interaction of Metal Ions with Tau Protein. The Case for a Metal-Mediated Tau Aggregation. J. Inorg. Biochem. 2019, 194, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Bourassa, M.W.; Leskovjan, A.C.; Tappero, R.V.; Farquhar, E.R.; Colton, C.A.; van Nostrand, W.E.; Miller, L.M. Elevated Copper in the Amyloid Plaques and Iron in the Cortex Are Observed in Mouse Models of Alzheimer’s Disease That Exhibit Neurodegeneration. Biomed. Spectrosc. Imaging 2013, 2, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative Stress and the Amyloid Beta Peptide in Alzheimer’s Disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Bu, G. The Low-Density Lipoprotein Receptor-Related Protein 1 and Amyloid-Beta Clearance in Alzheimer’s Disease. Front. Aging Neurosci. 2014, 6, 93. [Google Scholar] [CrossRef] [Green Version]
- Angelova, D.M.; Brown, D.R. Microglia and the Aging Brain: Are Senescent Microglia the Key to Neurodegeneration? J. Neurochem. 2019, 151, 676–688. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, S.; Ashok, A.; McDonald, D.; Wise, A.S.; Kritikos, A.E.; Rana, N.A.; Harding, C.V.; Singh, N. Upregulation of Local Hepcidin Contributes to Iron Accumulation in Alzheimer’s Disease Brains. J. Alzheimers Dis. 2021, 82, 1487–1497. [Google Scholar] [CrossRef]
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking While the Iron Is Hot: Iron Metabolism and Ferroptosis in Neurodegeneration. Free Radic. Biol. Med. 2019, 133, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Zhang, J. Iron Metabolism, Ferroptosis, and the Links with Alzheimer’s Disease. Front. Neurosci. 2019, 13, 1443. [Google Scholar] [CrossRef] [PubMed]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of Ferroptosis Regulator Glutathione Peroxidase 4 in Forebrain Neurons Promotes Cognitive Impairment and Neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef]
- Chen, L.; Dar, N.J.; Na, R.; McLane, K.D.; Yoo, K.; Han, X.; Ran, Q. Enhanced Defense against Ferroptosis Ameliorates Cognitive Impairment and Reduces Neurodegeneration in 5xfad Mice. Free Radic. Biol. Med. 2022, 180, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Karuppagounder, S.S.; Alin, L.; Chen, Y.; Brand, D.; Bourassa, M.W.; Dietrich, K.; Wilkinson, C.M.; Nadeau, C.A.; Kumar, A.; Perry, S.; et al. N-Acetylcysteine Targets 5 Lipoxygenase-Derived, Toxic Lipids and Can Synergize with Prostaglandin E2 to Inhibit Ferroptosis and Improve Outcomes Following Hemorrhagic Stroke in Mice. Ann. Neurol. 2018, 84, 854–872. [Google Scholar] [CrossRef] [Green Version]
- Adair, J.C.; Knoefel, J.E.; Morgan, N. Controlled Trial of N-Acetylcysteine for Patients with Probable Alzheimer’s Disease. Neurology 2001, 57, 1515–1517. [Google Scholar] [CrossRef]
- Ratan, R.R. The Chemical Biology of Ferroptosis in the Central Nervous System. Cell Chem. Biol. 2020, 27, 479–498. [Google Scholar] [CrossRef]
- Fasae, K.D.; Abolaji, A.O.; Faloye, T.R.; Odunsi, A.Y.; Oyetayo, B.O.; Enya, J.I.; Rotimi, J.A.; Akinyemi, R.O.; Whitworth, A.J.; Aschner, M. Metallobiology and Therapeutic Chelation of Biometals (Copper, Zinc and Iron) in Alzheimer’s Disease: Limitations, and Current and Future Perspectives. J. Trace Elem. Med. Biol. 2021, 67, 126779. [Google Scholar] [CrossRef]
- McLachlan, D.R.; Smith, W.L.; Kruck, T.P. Desferrioxamine and Alzheimer’s Disease: Video Home Behavior Assessment of Clinical Course and Measures of Brain Aluminum. Ther. Drug Monit. 1993, 15, 602–607. [Google Scholar] [CrossRef]
- Rao, S.S.; Portbury, S.D.; Lago, L.; McColl, G.; Finkelstein, D.I.; Bush, A.I.; Adlard, P.A. The Iron Chelator Deferiprone Improves the Phenotype in a Mouse Model of Tauopathy. J. Alzheimers Dis. 2020, 77, 753–771. [Google Scholar] [CrossRef]
- Prasanthi, J.R.; Schrag, M.; Dasari, B.; Marwarha, G.; Dickson, A.; Kirsch, W.M.; Ghribi, O. Deferiprone Reduces Amyloid-Beta and Tau Phosphorylation Levels but Not Reactive Oxygen Species Generation in Hippocampus of Rabbits Fed a Cholesterol-Enriched Diet. J. Alzheimers Dis. 2012, 30, 167–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faux, N.G.; Ritchie, C.W.; Gunn, A.; Rembach, A.; Tsatsanis, A.; Bedo, J.; Harrison, J.; Lannfelt, L.; Blennow, K.; Zetterberg, H.; et al. Pbt2 Rapidly Improves Cognition in Alzheimer’s Disease: Additional Phase Ii Analyses. J. Alzheimers Dis. 2010, 20, 509–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shefner, J.M.; Al-Chalabi, A.; Baker, M.R.; Cui, L.Y.; de Carvalho, M.; Eisen, A.; Grosskreutz, J.; Hardiman, O.; Henderson, R.; Matamala, J.M.; et al. A Proposal for New Diagnostic Criteria for Als. Clin. Neurophysiol. 2020, 131, 1975–1978. [Google Scholar] [CrossRef]
- Jankovska, N.; Matej, R. Molecular Pathology of Als: What We Currently Know and What Important Information Is Still Missing. Diagnostics 2021, 11, 1365. [Google Scholar] [CrossRef] [PubMed]
- Sheelakumari, R.; Madhusoodanan, M.; Radhakrishnan, A.; Ranjith, G.; Thomas, B. A Potential Biomarker in Amyotrophic Lateral Sclerosis: Can Assessment of Brain Iron Deposition with Swi and Corticospinal Tract Degeneration with Dti Help? AJNR Am. J. Neuroradiol. 2016, 37, 252–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, X.L.; Xiang, Y.; Guo, Y. The Role of Iron in Amyotrophic Lateral Sclerosis. Adv. Exp. Med. Biol. 2019, 1173, 145–152. [Google Scholar] [PubMed]
- Kurlander, H.M.; Patten, B.M. Metals in Spinal Cord Tissue of Patients Dying of Motor Neuron Disease. Ann. Neurol. 1979, 6, 21–24. [Google Scholar] [CrossRef]
- Veyrat-Durebex, C.; Corcia, P.; Mucha, A.; Benzimra, S.; Mallet, C.; Gendrot, C.; Moreau, C.; Devos, D.; Piver, E.; Pages, J.C.; et al. Iron Metabolism Disturbance in a French Cohort of Als Patients. Biomed. Res. Int. 2014, 2014, 485723. [Google Scholar] [CrossRef]
- Mitchell, R.M.; Simmons, Z.; Beard, J.L.; Stephens, H.E.; Connor, J.R. Plasma Biomarkers Associated with Als and Their Relationship to Iron Homeostasis. Muscle Nerve 2010, 42, 95–103. [Google Scholar] [CrossRef]
- Nadjar, Y.; Gordon, P.; Corcia, P.; Bensimon, G.; Pieroni, L.; Meininger, V.; Salachas, F. Elevated Serum Ferritin Is Associated with Reduced Survival in Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e45034. [Google Scholar] [CrossRef]
- Orts-Del’Immagine, A.; Wyart, C. Cerebrospinal-Fluid-Contacting Neurons. Curr. Biol. 2017, 27, R1198–R1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, P.M.; Vesterberg, O.; Syversen, T.; Flaten, T.P.; Nordberg, M. Metal Concentrations in Cerebrospinal Fluid and Blood Plasma from Patients with Amyotrophic Lateral Sclerosis. Biol. Trace Elem. Res. 2013, 151, 159–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patti, F.; Fiore, M.; Chisari, C.G.; D’Amico, E.; Fermo, S.L.; Toscano, S.; Copat, C.; Ferrante, M.; Zappia, M. Csf Neurotoxic Metals/Metalloids Levels in Amyotrophic Lateral Sclerosis Patients: Comparison between Bulbar and Spinal Onset. Environ. Res. 2020, 188, 109820. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Raivich, G.; Kreutzberg, G.W. Increase of Transferrin Receptors and Iron Uptake in Regenerating Motor Neurons. J. Neurosci. Res. 1989, 23, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Stuber, C.; Pitt, D.; Wang, Y. Iron in Multiple Sclerosis and Its Noninvasive Imaging with Quantitative Susceptibility Mapping. Int. J. Mol. Sci. 2016, 17, 100. [Google Scholar] [CrossRef] [Green Version]
- Al-Radaideh, A.; Athamneh, I.; Alabadi, H.; Hbahbih, M. Cortical and Subcortical Morphometric and Iron Changes in Relapsing-Remitting Multiple Sclerosis and Their Association with White Matter T2 Lesion Load: A 3-Tesla Magnetic Resonance Imaging Study. Clin. Neuroradiol. 2019, 29, 51–64. [Google Scholar] [CrossRef]
- Taege, Y.; Hagemeier, J.; Bergsland, N.; Dwyer, M.G.; Weinstock-Guttman, B.; Zivadinov, R.; Schweser, F. Assessment of Mesoscopic Properties of Deep Gray Matter Iron through a Model-Based Simultaneous Analysis of Magnetic Susceptibility and R2*—A Pilot Study in Patients with Multiple Sclerosis and Normal Controls. Neuroimage 2019, 186, 308–320. [Google Scholar] [CrossRef]
- Elkady, A.M.; Cobzas, D.; Sun, H.; Blevins, G.; Wilman, A.H. Progressive Iron Accumulation across Multiple Sclerosis Phenotypes Revealed by Sparse Classification of Deep Gray Matter. J. Magn. Reson. Imaging 2017, 46, 1464–1473. [Google Scholar] [CrossRef]
- Bergsland, N.; Tavazzi, E.; Lagana, M.M.; Baglio, F.; Cecconi, P.; Viotti, S.; Zivadinov, R.; Baselli, G.; Rovaris, M. White Matter Tract Injury Is Associated with Deep Gray Matter Iron Deposition in Multiple Sclerosis. J. Neuroimaging 2017, 27, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Yarnykh, V.L.; Krutenkova, E.P.; Aitmagambetova, G.; Repovic, P.; Mayadev, A.; Qian, P.; Henson, L.K.J.; Gangadharan, B.; Bowen, J.D. Iron-Insensitive Quantitative Assessment of Subcortical Gray Matter Demyelination in Multiple Sclerosis Using the Macromolecular Proton Fraction. AJNR Am. J. Neuroradiol. 2018, 39, 618–625. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, E.; Kmech, J.A.; Cobzas, D.; Sun, H.; Seres, P.; Blevins, G.; Wilman, A.H. Cognitive Implications of Deep Gray Matter Iron in Multiple Sclerosis. AJNR Am. J. Neuroradiol. 2017, 38, 942–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmalbrock, P.; Prakash, R.S.; Schirda, B.; Janssen, A.; Yang, G.K.; Russell, M.; Knopp, M.V.; Boster, A.; Nicholas, J.A.; Racke, M.; et al. Basal Ganglia Iron in Patients with Multiple Sclerosis Measured with 7 T Quantitative Susceptibility Mapping Correlates with Inhibitory Control. AJNR Am. J. Neuroradiol. 2016, 37, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Elkady, A.M.; Cobzas, D.; Sun, H.; Blevins, G.; Wilman, A.H. Discriminative Analysis of Regional Evolution of Iron and Myelin/Calcium in Deep Gray Matter of Multiple Sclerosis and Healthy Subjects. J. Magn. Reson. Imaging 2018, 48, 652–668. [Google Scholar] [CrossRef] [PubMed]
- Elkady, A.M.; Cobzas, D.; Sun, H.; Seres, P.; Blevins, G.; Wilman, A.H. Five Year Iron Changes in Relapsing-Remitting Multiple Sclerosis Deep Gray Matter Compared to Healthy Controls. Mult. Scler. Relat. Disord. 2019, 33, 107–115. [Google Scholar] [CrossRef]
- Hernandez-Torres, E.; Wiggermann, V.; Machan, L.; Sadovnick, A.D.; Li, D.K.B.; Traboulsee, A.; Hametner, S.; Rauscher, A. Increased Mean R2* in the Deep Gray Matter of Multiple Sclerosis Patients: Have We Been Measuring Atrophy? J. Magn. Reson. Imaging 2019, 50, 201–208. [Google Scholar] [CrossRef]
- Pontillo, G.; Petracca, M.; Monti, S.; Quarantelli, M.; Criscuolo, C.; Lanzillo, R.; Tedeschi, E.; Elefante, A.; Morra, V.B.; Brunetti, A.; et al. Unraveling Deep Gray Matter Atrophy and Iron and Myelin Changes in Multiple Sclerosis. AJNR Am. J. Neuroradiol. 2021, 42, 1223–1230. [Google Scholar] [CrossRef]
- Pudlac, A.; Burgetova, A.; Dusek, P.; Nytrova, P.; Vaneckova, M.; Horakova, D.; Krasensky, J.; Lambert, L. Deep Gray Matter Iron Content in Neuromyelitis Optica and Multiple Sclerosis. Biomed. Res. Int. 2020, 2020, 6492786. [Google Scholar] [CrossRef] [PubMed]
- Schweser, F.; Martins, A.L.R.D.; Hagemeier, J.; Lin, F.; Hanspach, J.; Weinstock-Guttman, B.; Hametner, S.; Bergsland, N.; Dwyer, M.G.; Zivadinov, R. Mapping of Thalamic Magnetic Susceptibility in Multiple Sclerosis Indicates Decreasing Iron with Disease Duration: A Proposed Mechanistic Relationship between Inflammation and Oligodendrocyte Vitality. Neuroimage 2018, 167, 438–452. [Google Scholar] [CrossRef]
- Castellaro, M.; Magliozzi, R.; Palombit, A.; Pitteri, M.; Silvestri, E.; Camera, V.; Montemezzi, S.; Pizzini, F.B.; Bertoldo, A.; Reynolds, R.; et al. Heterogeneity of Cortical Lesion Susceptibility Mapping in Multiple Sclerosis. AJNR Am. J. Neuroradiol. 2017, 38, 1087–1095. [Google Scholar] [CrossRef] [Green Version]
- Tham, M.; Frischer, J.M.; Weigand, S.D.; Fitz-Gibbon, P.D.; Webb, S.M.; Guo, Y.; Adiele, R.C.; Robinson, C.A.; Bruck, W.; Lassmann, H.; et al. Iron Heterogeneity in Early Active Multiple Sclerosis Lesions. Ann. Neurol. 2021, 89, 498–510. [Google Scholar] [CrossRef]
- Chawla, S.; Kister, I.; Wuerfel, J.; Brisset, J.C.; Liu, S.; Sinnecker, T.; Dusek, P.; Haacke, E.M.; Paul, F.; Ge, Y. Iron and Non-Iron-Related Characteristics of Multiple Sclerosis and Neuromyelitis Optica Lesions at 7 T Mri. AJNR Am. J. Neuroradiol. 2016, 37, 1223–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggi, P.; Sati, P.; Nair, G.; Cortese, I.C.M.; Jacobson, S.; Smith, B.R.; Nath, A.; Ohayon, J.; van Pesch, V.; Perrotta, G.; et al. Paramagnetic Rim Lesions Are Specific to Multiple Sclerosis: An International Multicenter 3t Mri Study. Ann. Neurol. 2020, 88, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Preziosa, P.; Rocca, M.A.; Filippi, M. Central Vein Sign and Iron Rim in Multiple Sclerosis: Ready for Clinical Use? Curr. Opin. Neurol. 2021, 34, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Sinnecker, T.; Schumacher, S.; Mueller, K.; Pache, F.; Dusek, P.; Harms, L.; Ruprecht, K.; Nytrova, P.; Chawla, S.; Niendorf, T.; et al. Mri Phase Changes in Multiple Sclerosis vs. Neuromyelitis Optica Lesions at 7 T. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e259. [Google Scholar] [CrossRef] [Green Version]
- Dal-Bianco, A.; Grabner, G.; Kronnerwetter, C.; Weber, M.; Kornek, B.; Kasprian, G.; Berger, T.; Leutmezer, F.; Rommer, P.S.; Trattnig, S.; et al. Long-Term Evolution of Multiple Sclerosis Iron Rim Lesions in 7 T Mri. Brain 2021, 144, 833–847. [Google Scholar] [CrossRef]
- Zhang, Y.; Gauthier, S.A.; Gupta, A.; Chen, W.; Comunale, J.; Chiang, G.C.; Zhou, D.; Askin, G.; Zhu, W.; Pitt, D.; et al. Quantitative Susceptibility Mapping and R2* Measured Changes During White Matter Lesion Development in Multiple Sclerosis: Myelin Breakdown, Myelin Debris Degradation and Removal, and Iron Accumulation. AJNR Am. J. Neuroradiol. 2016, 37, 1629–1635. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.A.; Pareto, D.; Pessini-Ferreira, L.; Arrambide, G.; Alberich, M.; Crescenzo, F.; Cappelle, S.; Tintore, M.; Sastre-Garriga, J.; Auger, C.; et al. Value of 3t Susceptibility-Weighted Imaging in the Diagnosis of Multiple Sclerosis. AJNR Am. J. Neuroradiol. 2020, 41, 1001–1008. [Google Scholar] [CrossRef]
- Absinta, M.; Sati, P.; Masuzzo, F.; Nair, G.; Sethi, V.; Kolb, H.; Ohayon, J.; Wu, T.; Cortese, I.C.M.; Reich, D.S. Association of Chronic Active Multiple Sclerosis Lesions with Disability in Vivo. JAMA Neurol. 2019, 76, 1474–1483. [Google Scholar] [CrossRef]
- Gillen, K.M.; Mubarak, M.; Nguyen, T.D.; Pitt, D. Significance and in Vivo Detection of Iron-Laden Microglia in White Matter Multiple Sclerosis Lesions. Front. Immunol. 2018, 9, 255. [Google Scholar] [CrossRef] [Green Version]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of ‘Homeostatic’ Microglia and Patterns of Their Activation in Active Multiple Sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef]
- Ding, D.; Valdivia, A.O.; Bhattacharya, S.K. Nuclear Prelamin a Recognition Factor and Iron Dysregulation in Multiple Sclerosis. Metab. Brain Dis. 2020, 35, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Van Rensburg, S.J.; Peeters, A.V.; van Toorn, R.; Schoeman, J.; Moremi, K.E.; van Heerden, C.J.; Kotze, M.J. Identification of an Iron-Responsive Subtype in Two Children Diagnosed with Relapsing-Remitting Multiple Sclerosis Using Whole Exome Sequencing. Mol. Genet. Metab. Rep. 2019, 19, 100465. [Google Scholar] [CrossRef] [PubMed]
- Herbert, E.; Engel-Hills, P.; Hattingh, C.; Fouche, J.P.; Kidd, M.; Lochner, C.; Kotze, M.J.; van Rensburg, S.J. Fractional Anisotropy of White Matter, Disability and Blood Iron Parameters in Multiple Sclerosis. Metab. Brain Dis. 2018, 33, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Siotto, M.; Filippi, M.M.; Simonelli, I.; Landi, D.; Ghazaryan, A.; Vollaro, S.; Ventriglia, M.; Pasqualetti, P.; Rongioletti, M.C.A.; Squitti, R.; et al. Oxidative Stress Related to Iron Metabolism in Relapsing Remitting Multiple Sclerosis Patients with Low Disability. Front. Neurosci. 2019, 13, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogan, H.O.; Yildiz, O.K. Serum Nadph Oxidase Concentrations and the Associations with Iron Metabolism in Relapsing Remitting Multiple Sclerosis. J. Trace Elem. Med. Biol. 2019, 55, 39–43. [Google Scholar] [CrossRef]
- Bergsland, N.; Agostini, S.; Lagana, M.M.; Mancuso, R.; Mendozzi, L.; Tavazzi, E.; Cecconi, P.; Clerici, M.; Baglio, F. Serum Iron Concentration Is Associated with Subcortical Deep Gray Matter Iron Levels in Multiple Sclerosis Patients. Neuroreport 2017, 28, 645–648. [Google Scholar] [CrossRef]
- Khalil, M.; Renner, A.; Langkammer, C.; Enzinger, C.; Ropele, S.; Stojakovic, T.; Scharnagl, H.; Bachmaier, G.; Pichler, A.; Archelos, J.J.; et al. Cerebrospinal Fluid Lipocalin 2 in Patients with Clinically Isolated Syndromes and Early Multiple Sclerosis. Mult. Scler. 2016, 22, 1560–1568. [Google Scholar] [CrossRef]
- Dekens, D.W.; Eisel, U.L.M.; Gouweleeuw, L.; Schoemaker, R.G.; de Deyn, P.P.; Naude, P.J.W. Lipocalin 2 as a Link between Ageing, Risk Factor Conditions and Age-Related Brain Diseases. Ageing Res. Rev. 2021, 70, 101414. [Google Scholar] [CrossRef]
- Magliozzi, R.; Hametner, S.; Facchiano, F.; Marastoni, D.; Rossi, S.; Castellaro, M.; Poli, A.; Lattanzi, F.; Visconti, A.; Nicholas, R.; et al. Iron Homeostasis, Complement, and Coagulation Cascade as Csf Signature of Cortical Lesions in Early Multiple Sclerosis. Ann. Clin. Transl. Neurol. 2019, 6, 2150–2163. [Google Scholar] [CrossRef] [Green Version]
- Aspli, K.T.; Flaten, T.P.; Roos, P.M.; Holmoy, T.; Skogholt, J.H.; Aaseth, J. Iron and Copper in Progressive Demyelination—New Lessons from Skogholt’s Disease. J. Trace Elem. Med. Biol. 2015, 31, 183–187. [Google Scholar] [CrossRef]
- Hider, R.C.; Hoffbrand, A.V. The Role of Deferiprone in Iron Chelation. N. Engl. J. Med. 2018, 379, 2140–2150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aaseth, J.; Skaug, M.A.; Cao, Y.; Andersen, O. Chelation in Metal Intoxication—Principles and Paradigms. J. Trace Elem. Med. Biol. 2015, 31, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Piga, A.; Gaglioti, C.; Fogliacco, E.; Tricta, F. Comparative Effects of Deferiprone and Deferoxamine on Survival and Cardiac Disease in Patients with Thalassemia Major: A Retrospective Analysis. Haematologica 2003, 88, 489–496. [Google Scholar]
- Rao, S.S.; Lago, L.; Volitakis, I.; Shukla, J.J.; McColl, G.; Finkelstein, D.I.; Adlard, P.A. Deferiprone Treatment in Aged Transgenic Tau Mice Improves Y-Maze Performance and Alters Tau Pathology. Neurotherapeutics 2021, 18, 1081–1094. [Google Scholar] [CrossRef]
- Flaten, T.P.; Aaseth, J.; Andersen, O.; Kontoghiorghes, G.J. Iron Mobilization Using Chelation and Phlebotomy. J. Trace Elem. Med. Biol. 2012, 26, 127–130. [Google Scholar] [CrossRef]
- Mourad, F.H.; Hoffbrand, A.V.; Sheikh-Taha, M.; Koussa, S.; Khoriaty, A.I.; Taher, A. Comparison between Desferrioxamine and Combined Therapy with Desferrioxamine and Deferiprone in Iron Overloaded Thalassaemia Patients. Br. J. Haematol. 2003, 121, 187–189. [Google Scholar] [CrossRef] [Green Version]
- Sedgwick, A.C.; Yan, K.C.; Mangel, D.N.; Shang, Y.; Steinbrueck, A.; Han, H.H.; Brewster, J.T., II; Hu, X.L.; Snelson, D.W.; Lynch, V.M.; et al. Deferasirox (Exjade): An Fda-Approved Aiegen Platform with Unique Photophysical Properties. J. Am. Chem. Soc. 2021, 143, 1278–1283. [Google Scholar] [CrossRef]
- Dou, H.; Qin, Y.; Chen, G.; Zhao, Y. Effectiveness and Safety of Deferasirox in Thalassemia with Iron Overload: A Meta-Analysis. Acta Haematol. 2019, 141, 32–42. [Google Scholar] [CrossRef]
- Totadri, S.; Bansal, D.; Bhatia, P.; Attri, S.V.; Trehan, A.; Marwaha, R.K. The Deferiprone and Deferasirox Combination Is Efficacious in Iron Overloaded Patients with Beta-Thalassemia Major: A Prospective, Single Center, Open-Label Study. Pediatr. Blood Cancer 2015, 62, 1592–1596. [Google Scholar] [CrossRef]
- DivakarJose, R.R.; Delhikumar, C.G.; Kumar, G.R. Efficacy and Safety of Combined Oral Chelation with Deferiprone and Deferasirox on Iron Overload in Transfusion Dependent Children with Thalassemia—A Prospective Observational Study. Indian J. Pediatr. 2021, 88, 330–335. [Google Scholar] [CrossRef]
- Mu, M.D.; Qian, Z.M.; Yang, S.X.; Rong, K.L.; Yung, W.H.; Ke, Y. Therapeutic Effect of a Histone Demethylase Inhibitor in Parkinson’s Disease. Cell Death Dis. 2020, 11, 927. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.L.; Wu, J.; Shah, B.N.; Greutelaers, K.C.; Ghosh, M.C.; Ollivierre, H.; Su, X.Z.; Thuma, P.E.; Bedu-Addo, G.; Mockenhaupt, F.P.; et al. Erythrocytic Ferroportin Reduces Intracellular Iron Accumulation, Hemolysis, and Malaria Risk. Science 2018, 359, 1520–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dusek, P.; Hofer, T.; Alexander, J.; Roos, P.M.; Aaseth, J.O. Cerebral Iron Deposition in Neurodegeneration. Biomolecules 2022, 12, 714. https://doi.org/10.3390/biom12050714
Dusek P, Hofer T, Alexander J, Roos PM, Aaseth JO. Cerebral Iron Deposition in Neurodegeneration. Biomolecules. 2022; 12(5):714. https://doi.org/10.3390/biom12050714
Chicago/Turabian StyleDusek, Petr, Tim Hofer, Jan Alexander, Per M. Roos, and Jan O. Aaseth. 2022. "Cerebral Iron Deposition in Neurodegeneration" Biomolecules 12, no. 5: 714. https://doi.org/10.3390/biom12050714
APA StyleDusek, P., Hofer, T., Alexander, J., Roos, P. M., & Aaseth, J. O. (2022). Cerebral Iron Deposition in Neurodegeneration. Biomolecules, 12(5), 714. https://doi.org/10.3390/biom12050714