
CavitySpace: A Database of Potential Ligand Binding Sites in the Human Proteome

 , , , , and
, , , , and 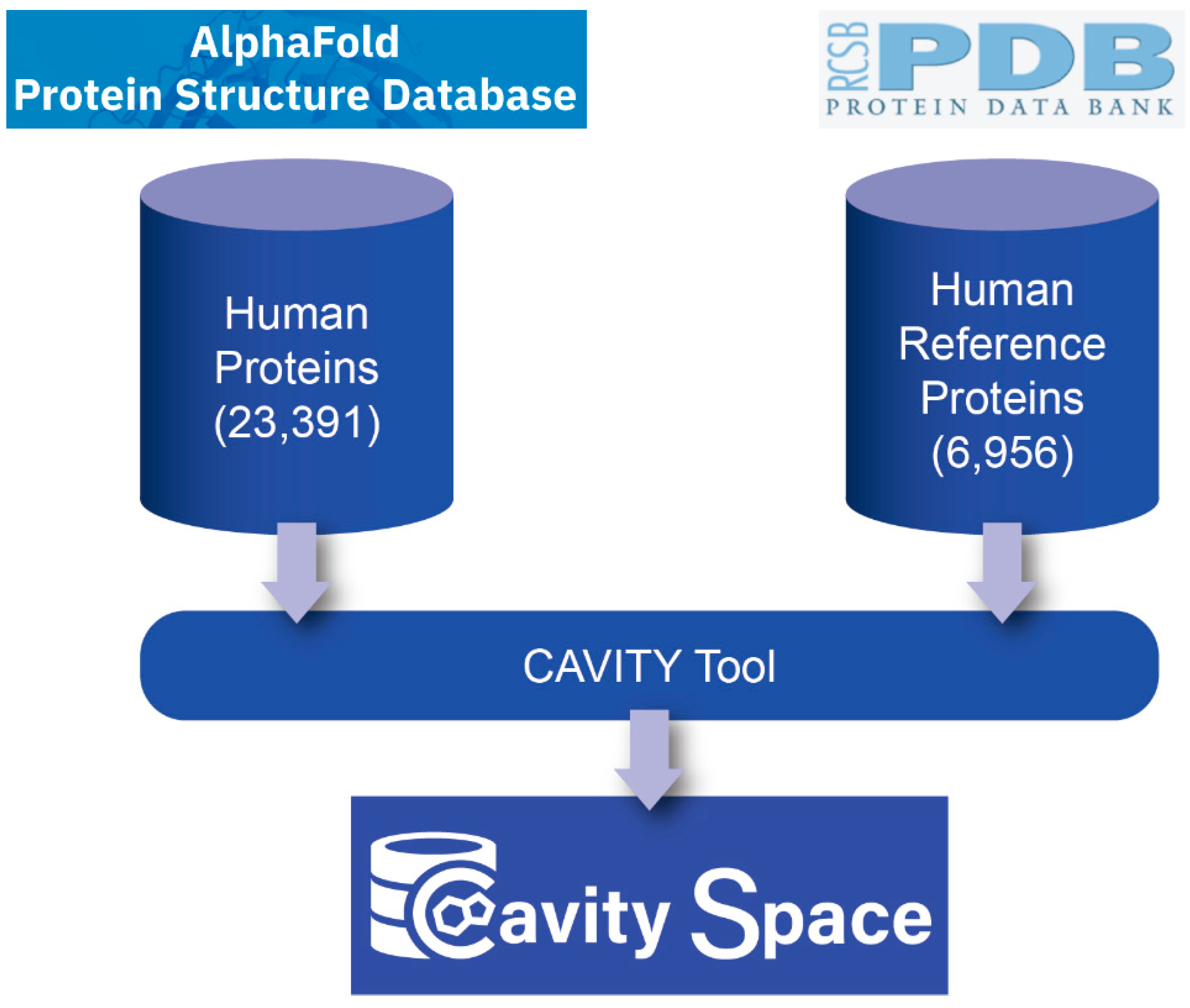{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Cavity Detection
3. Results
3.1. Cavity Library for AlphaFold Structures
3.2. The Quality of Cavity Detection over AlphaFold Structures
3.3. Applications of the Cavity Library
3.4. The Webserver
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pérot, S.; Sperandio, O.; Miteva, M.A.; Camproux, A.-C.; Villoutreix, B.O. Druggable pockets and binding site centric chemical space: A paradigm shift in drug discovery. Drug Discov. Today 2010, 15, 656–667. [Google Scholar] [CrossRef]
- Consortium, W. Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019, 47, D520–D528. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.-R.; Hwang, M.-J. Methods for predicting protein–ligand binding sites. Mol. Modeling Proteins 2015, 1215, 383–398. [Google Scholar]
- Dhakal, A.; McKay, C.; Tanner, J.J.; Cheng, J. Artificial intelligence in the prediction of protein–ligand interactions: Recent advances and future directions. Brief. Bioinform. 2021, 23, bbab476. [Google Scholar] [CrossRef]
- Ito, J.-I.; Tabei, Y.; Shimizu, K.; Tsuda, K.; Tomii, K. PoSSuM: A database of similar protein–ligand binding and putative pockets. Nucleic Acids Res. 2011, 40, D541–D548. [Google Scholar] [CrossRef] [Green Version]
- Kufareva, I.; Ilatovskiy, A.V.; Abagyan, R. Pocketome: An encyclopedia of small-molecule binding sites in 4D. Nucleic Acids Res. 2011, 40, D535–D540. [Google Scholar] [CrossRef] [Green Version]
- Maietta, P.; Lopez, G.; Carro, A.; Pingilley, B.J.; Leon, L.G.; Valencia, A.; Tress, M.L. FireDB: A compendium of biological and pharmacologically relevant ligands. Nucleic Acids Res. 2013, 42, D267–D272. [Google Scholar] [CrossRef] [Green Version]
- Meyer, T.; Knapp, E.-W. Database of protein complexes with multivalent binding ability: Bival-bind. Proteins 2014, 82, 744–751. [Google Scholar] [CrossRef]
- van Linden, O.P.J.; Kooistra, A.J.; Leurs, R.; de Esch, I.J.P.; de Graaf, C. KLIFS: A knowledge-based structural database to navigate Kinase–ligand interaction space. J. Med. Chem. 2014, 57, 249–277. [Google Scholar] [CrossRef]
- Desaphy, J.; Bret, G.; Rognan, D.; Kellenberger, E. sc-PDB: A 3D-database of ligandable binding sites—10 years on. Nucleic Acids Res. 2015, 43, D399–D404. [Google Scholar] [CrossRef]
- Bhagavat, R.; Sankar, S.; Srinivasan, N.; Chandra, N. An Augmented Pocketome: Detection and Analysis of Small-Molecule Binding Pockets in Proteins of Known 3D Structure. Structure 2018, 26, 499–512.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Qiu, J.; Liu, H.; Xu, Y.; Jia, Y.; Zhao, Y. HKPocket: Human kinase pocket database for drug design. BMC Bioinform. 2019, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Konc, J.; Lešnik, S.; Škrlj, B.; Janežič, D. ProBiS-Dock database: A web server and interactive web repository of small ligand–protein binding sites for drug design. J. Chem. Inf. Model. 2021, 61, 4097–4107. [Google Scholar] [CrossRef]
- Radusky, L.; Defelipe, L.A.; Lanzarotti, E. TuberQ: A Mycobacterium tuberculosis protein druggability database. Database 2014, 2014, bau035. [Google Scholar] [CrossRef] [Green Version]
- Tseng, Y.Y.; Chen, Z.J. and Li, W-H. fPOP: Footprinting functional pockets of proteins by comparative spatial patterns. Nucleic Acids Res. 2009, 38, D288–D295. [Google Scholar] [CrossRef] [Green Version]
- Schreyer, A.; Blundell, T. CREDO: A protein–ligand interaction database for drug discovery. Chem. Biol. Drug. Des. 2009, 73, 157–167. [Google Scholar] [CrossRef]
- Bauer, R.A.; Günther, S.; Jansen, D.; Heeger, C.; Thaben, P.F.; Preissner, R. SuperSite: Dictionary of metabolite and drug binding sites in proteins. Nucleic Acids Res. 2008, 37, D195–D200. [Google Scholar] [CrossRef] [Green Version]
- Dessailly, B.H.; Lensink, M.F.; Orengo, C.A.; Wodak, S.J. LigASite—a database of biologically relevant binding sites in proteins with known apo -structures. Nucleic Acids Res. 2007, 36, D667–D673. [Google Scholar] [CrossRef]
- Gold, N.D.; Jackson, R.M. SitesBase: A database for structure-based protein–ligand binding site comparisons. Nucleic Acids Res. 2006, 34, D231–D234. [Google Scholar] [CrossRef] [Green Version]
- Ivanisenko, V.A.; Pintus, S.S.; Grigorovich, D.A.; Kolchanov, N.A. PDBSite: A database of the 3D structure of protein functional sites. Nucleic Acids Res. 2005, 33 (Suppl. 1), D183–D187. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, A.; Iida, K.; Matsui, N.; Tomoda, S.; Yura, K.; Go, M. Het-PDB Navi.: A database for protein–small molecule interactions. J. Biochem. 2004, 135, 79–84. [Google Scholar] [CrossRef]
- Hedderich, J.B.; Persechino, M.; Becker, K.; Heydenreich, F.M.; Gutermuth, T.; Bouvier, M.; Bünemann, M.; Kolb, P. The pocketome of G-protein-coupled receptors reveals previously untargeted allosteric sites. Nat. Commun. 2022, 13, 2567. [Google Scholar] [CrossRef]
- Consortium, U. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, R.; Baker, D. Ab initio protein structure prediction: Progress and prospects. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 173–189. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y. Progress and challenges in protein structure prediction. Curr. Opin. Struct. Biol. 2008, 18, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Kuhlman, B.; Bradley, P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.X.; Pei, J.F.; Lai, L.H. Binding site detection and druggability prediction of protein targets for structure-based drug design. Curr. Pharm. Des. 2013, 19, 2326–2333. [Google Scholar] [CrossRef]
- Krasowski, A.; Muthas, D.; Sarkar, A.; Schmitt, S.; Brenk, R. DrugPred: A structure-based approach to predict protein druggability developed using an extensive nonredundant data set. J. Chem. Inf. Model. 2011, 51, 2829–2842. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, J.E.A.; Pinotsis, N.; Ghisleni, A.; Salmazo, A.; Konarev, P.V.; Kostan, J.; Sjöblom, B.; Schreiner, C.; Polyansky, A.A.; Gkougkoulia, E.A.; et al. The structure and regulation of human muscle α-actinin. Cell 2014, 159, 1447–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeturu, K.; Chandra, N. PocketMatch: A new algorithm to compare binding sites in protein structures. BMC Bioinform. 2008, 9, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butina, D. Unsupervised data base clustering based on Daylight’s fingerprint and Tanimoto similarity: A fast and automated way To cluster small and large data sets. J. Chem. Inf. Comput. Sci. 1999, 39, 747–750. [Google Scholar] [CrossRef]
- Luginina, A.; Gusach, A.; Marin, E.; Mishin, A.; Brouillette, R.; Popov, P.; Shiriaeva, A.; Borshchevskiy, V.; Longpré, J.-M.; Lyapina, E.; et al. Structure-based mechanism of cysteinyl leukotriene receptor inhibition by antiasthmatic drugs. Sci. Adv. 2019, 5, eaax2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F. AutoDock Vina 1.2.0: New docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, S.; Hu, Q.; Gao, S.; Ma, X.; Zhang, W.; Shen, Y.; Chen, F.; Lai, L.; Pei, J. CavityPlus: A web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 2018, 46, W374–W379. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Lin, H.; Huang, Z.; He, Y.; Deng, X.; Xu, Y.; Pei, J.; Lai, L. CavitySpace: A Database of Potential Ligand Binding Sites in the Human Proteome. Biomolecules 2022, 12, 967. https://doi.org/10.3390/biom12070967
Wang S, Lin H, Huang Z, He Y, Deng X, Xu Y, Pei J, Lai L. CavitySpace: A Database of Potential Ligand Binding Sites in the Human Proteome. Biomolecules. 2022; 12(7):967. https://doi.org/10.3390/biom12070967
Chicago/Turabian StyleWang, Shiwei, Haoyu Lin, Zhixian Huang, Yufeng He, Xiaobing Deng, Youjun Xu, Jianfeng Pei, and Luhua Lai. 2022. "CavitySpace: A Database of Potential Ligand Binding Sites in the Human Proteome" Biomolecules 12, no. 7: 967. https://doi.org/10.3390/biom12070967
APA StyleWang, S., Lin, H., Huang, Z., He, Y., Deng, X., Xu, Y., Pei, J., & Lai, L. (2022). CavitySpace: A Database of Potential Ligand Binding Sites in the Human Proteome. Biomolecules, 12(7), 967. https://doi.org/10.3390/biom12070967