
Src Family Kinases: A Potential Therapeutic Target for Acute Kidney Injury

 , ,
, ,
Abstract
:1. Introduction
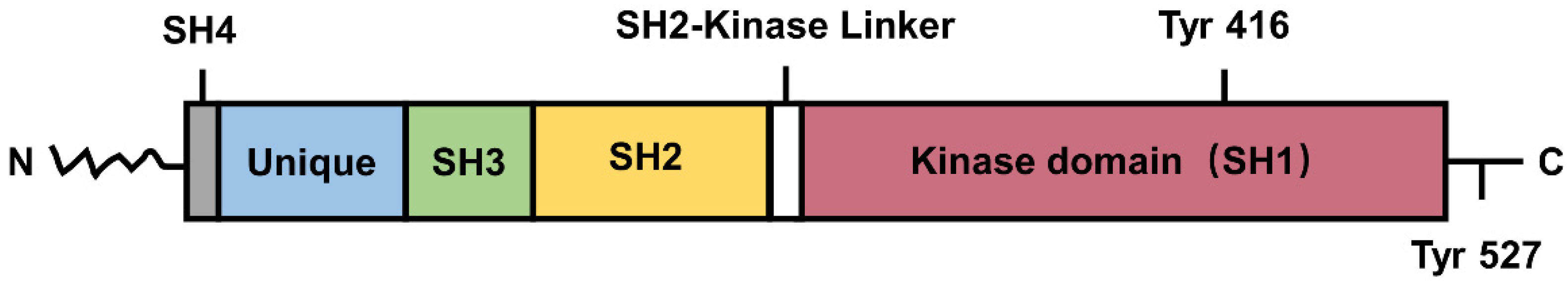
2. Overview of SFKs
2.1. Introduction and Structure of SFKs
2.2. Function of SFKs in Kidney
3. The Pathophysiological Role of SFKs in AKI
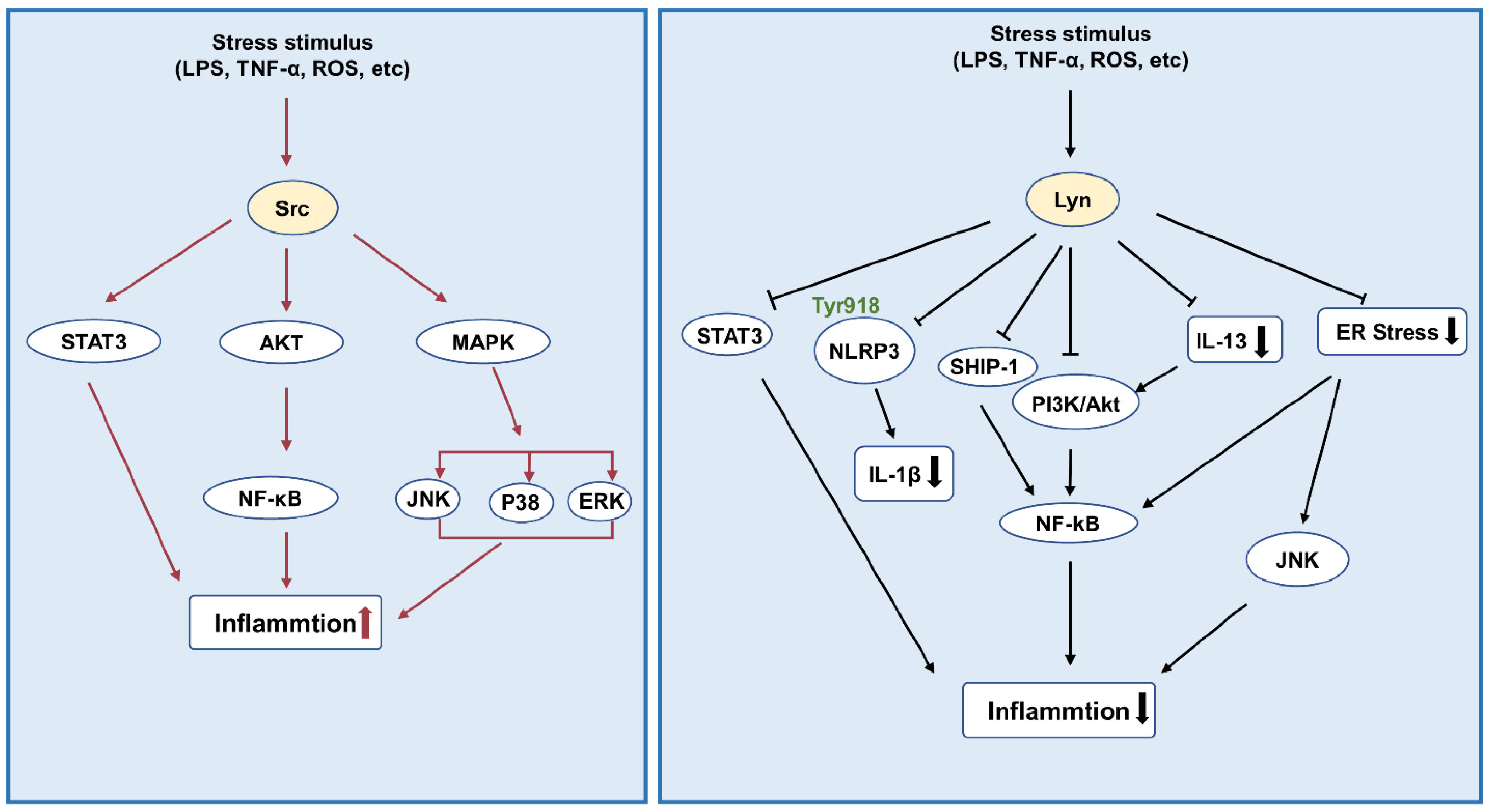
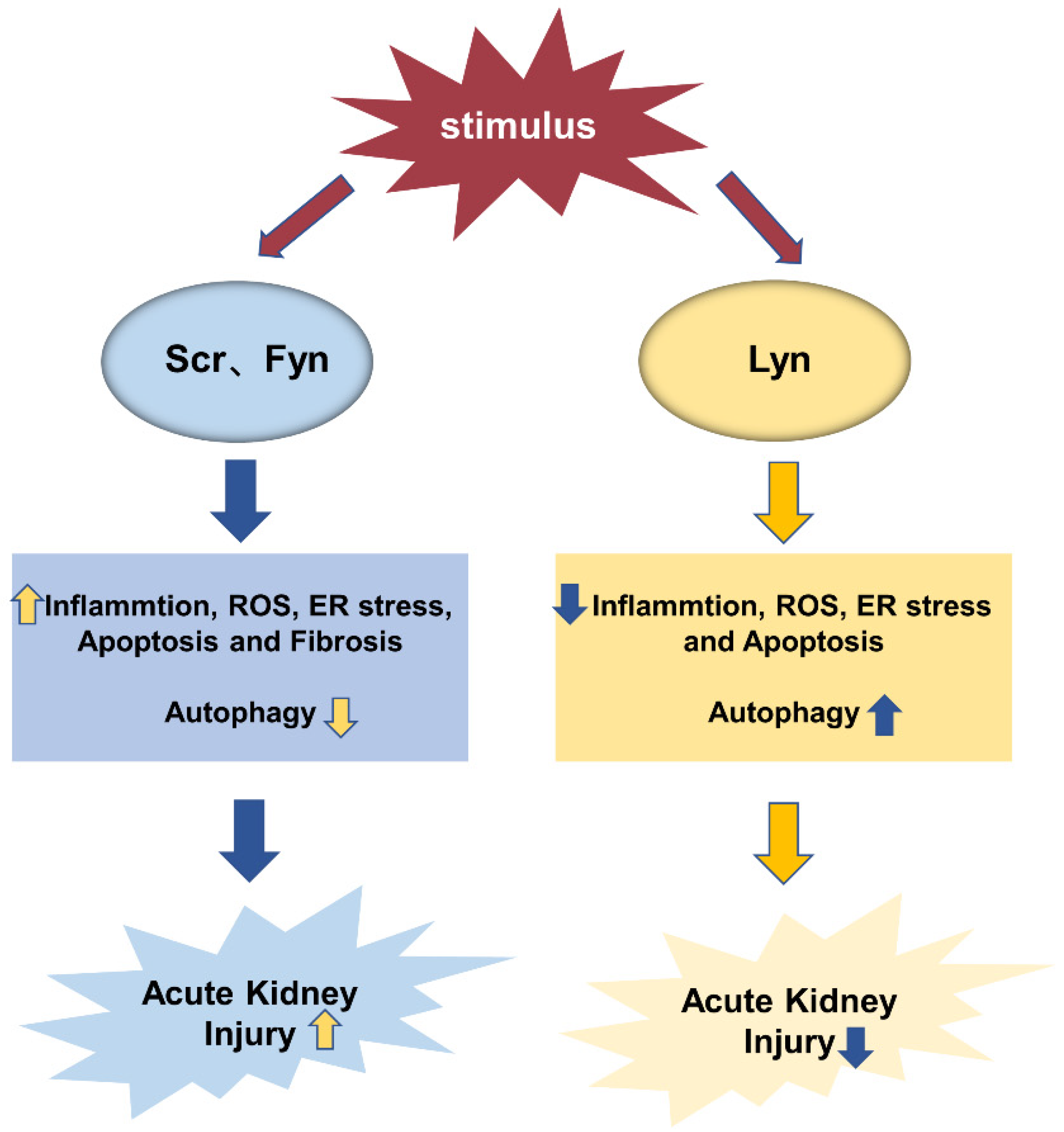
3.1. Inflammation
3.2. Oxidative Stress
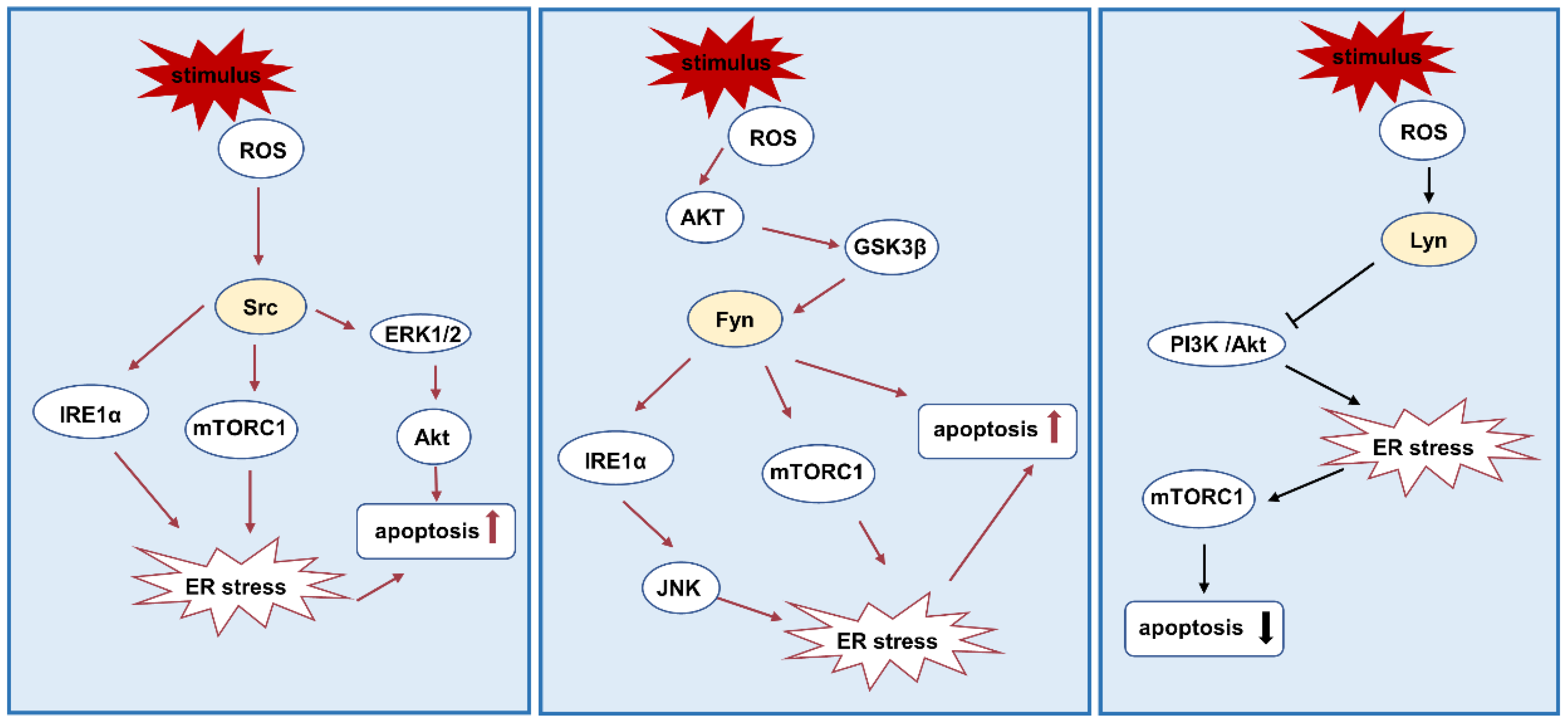
3.3. ER Stress and Apoptosis
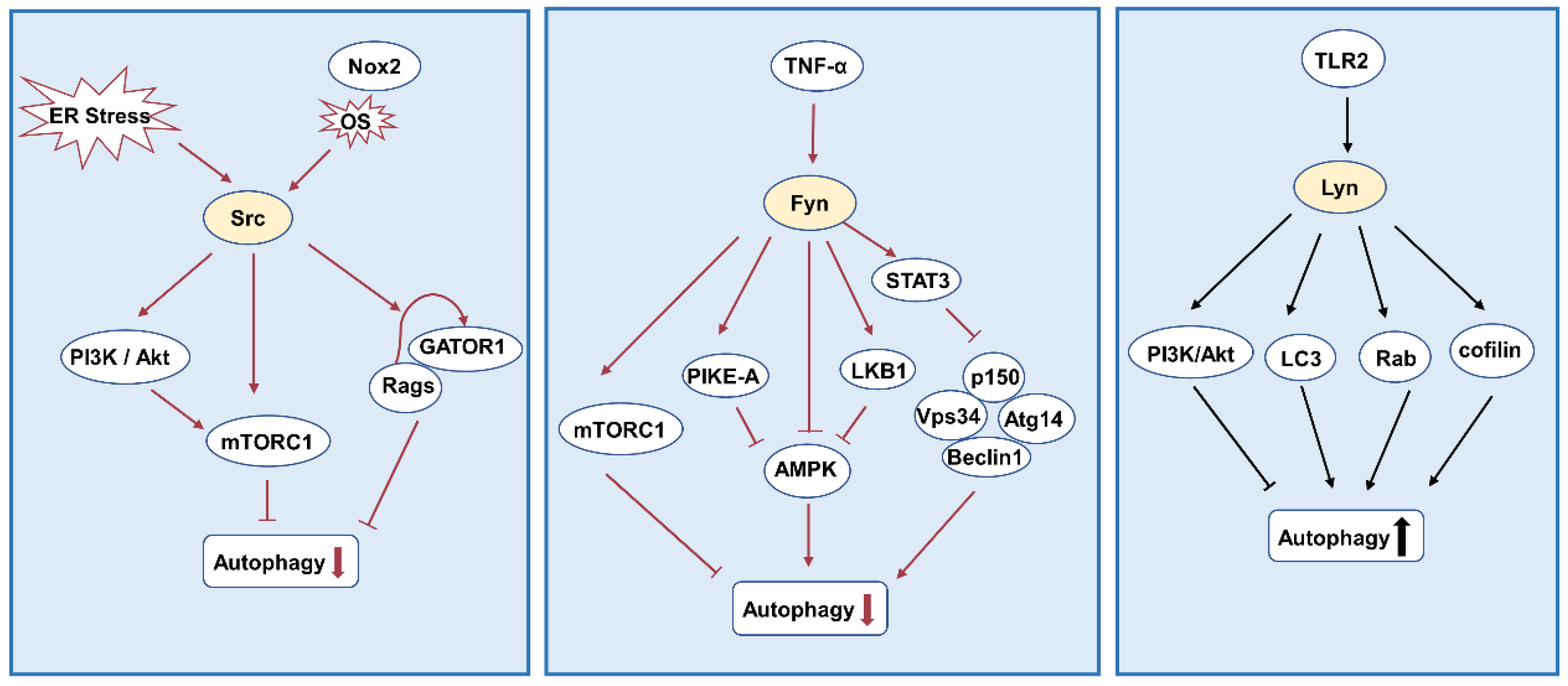
3.4. Autophagy
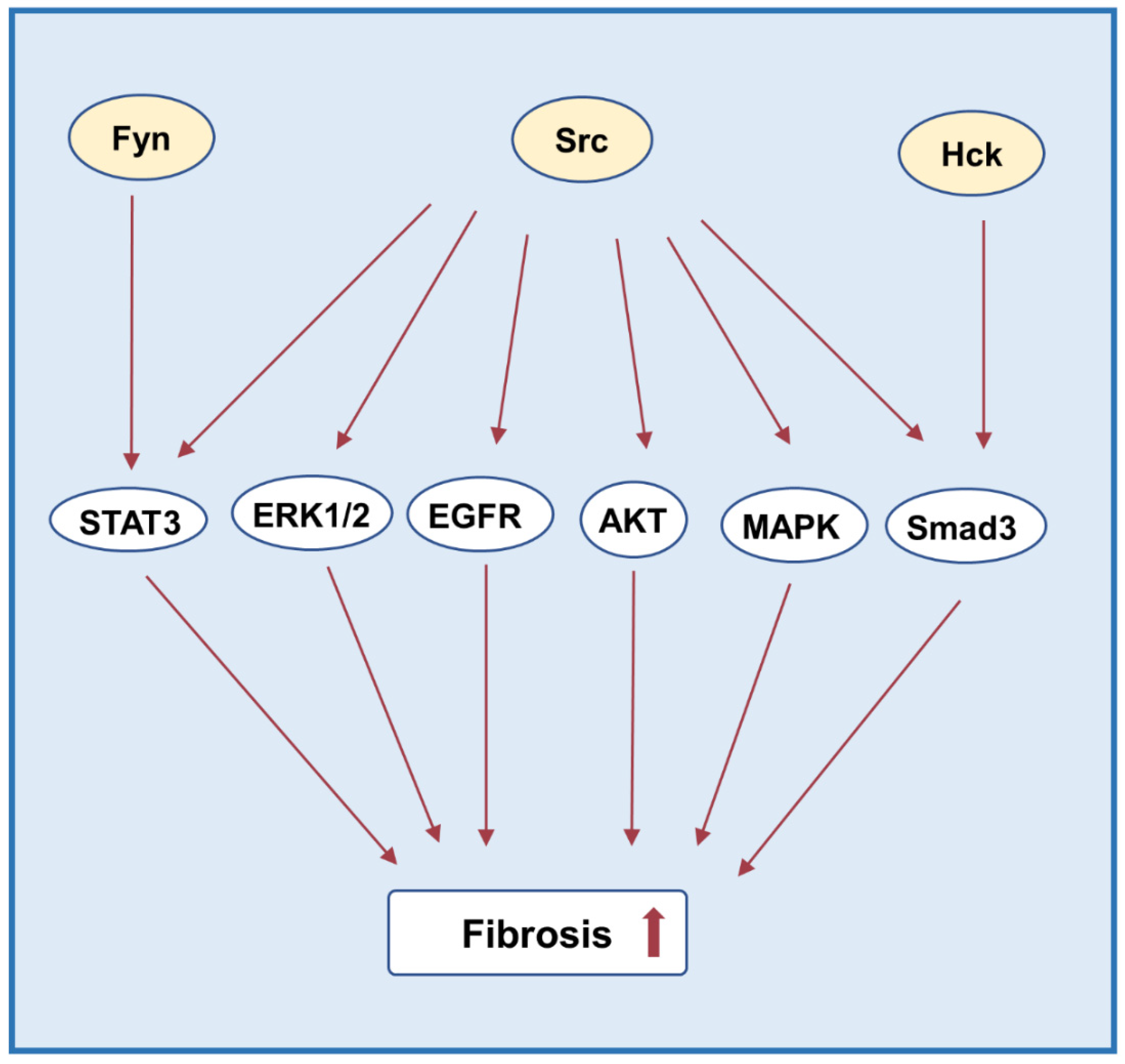
3.5. Fibrosis
4. Targeting SFKs for AKI
5. Conclusions
6. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Levey, A.S.; Eckardt, K.-U.; Dorman, N.M.; Christiansen, S.L.; Hoorn, E.J.; Ingelfinger, J.R.; Inker, L.A.; Levin, A.; Mehrotra, R.; Palevsky, P.M.; et al. Nomenclature for kidney function and disease: Report of a Kidney Disease: Improving Global Outcomes (KDIGO) Consensus Conference. Kidney Int. 2020, 97, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Hoste, E.A.J.; Kellum, J.A.; Selby, N.M.; Zarbock, A.; Palevsky, P.M.; Bagshaw, S.M.; Goldstein, S.L.; Cerdá, J.; Chawla, L.S. Global epidemiology and outcomes of acute kidney injury. Nat. Rev. Nephrol. 2018, 14, 607–625. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wei, Q.; Liu, J.; Yi, M.; Liu, Y.; Liu, H.; Sun, L.; Peng, Y.; Liu, F.; Venkatachalam, M.A.; et al. AKI on CKD: Heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017, 92, 1071–1083. [Google Scholar] [CrossRef]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Duann, P.; Lianos, E.A.; Ma, J.; Lin, P.-H. Autophagy, Innate Immunity and Tissue Repair in Acute Kidney Injury. Int. J. Mol. Sci. 2016, 17, 662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuk, A.; Bonventre, J.V. Acute Kidney Injury. Annu. Rev. Med. 2016, 67, 293–307. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Tanaka, T.; Nangaku, M. Hypoxia as a key player in the AKI-to-CKD transition. Am. J. Physiol. Renal Physiol. 2014, 307, F1187–F1195. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Bai, M.; Lei, J.; Xie, Y.; Xu, S.; Jia, Z.; Zhang, A. Mitochondrial dysfunction and the AKI-to-CKD transition. Am. J. Physiol. Renal Physiol. 2020, 319, F1105–F1116. [Google Scholar] [CrossRef]
- Kurzhagen, J.T.; Dellepiane, S.; Cantaluppi, V.; Rabb, H. AKI: An increasingly recognized risk factor for CKD development and progression. J. Nephrol. 2020, 33, 1171–1187. [Google Scholar] [CrossRef]
- Hosohata, K.; Harnsirikarn, T.; Chokesuwattanaskul, S. Ferroptosis: A Potential Therapeutic Target in Acute Kidney Injury. Int. J. Mol. Sci. 2022, 23, 6583. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Src protein–tyrosine kinase structure and regulation. Biochem. Biophys. Res. Commun. 2004, 324, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; Zang, X.; Zhou, X.; Liu, L.; Masucci, M.V.; Tang, J.; Li, X.; Liu, N.; Bayliss, G.; Zhao, T.C.; et al. Pharmacological inhibition of Src kinase protects against acute kidney injury in a murine model of renal ischemia/reperfusion. Oncotarget 2017, 8, 31238–31253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pak, E.; Uddin, J.; Ha, H. Inhibition of Src Family Kinases Ameliorates LPS-Induced Acute Kidney Injury and Mitochondrial Dysfunction in Mice. Int. J. Mol. Sci. 2020, 21, 8246. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, C.; Tian, Y.; Lu, J.; Zhang, G.; Liang, S.; Chen, D.; Liu, X.; Kuang, W.; Zhu, M. Src family kinases and pulmonary fibrosis: A review. Biomed. Pharmacother. 2020, 127, 110183. [Google Scholar] [CrossRef] [PubMed]
- Brauer, P.M.; Tyner, A.L. Building a better understanding of the intracellular tyrosine kinase PTK6—BRK by BRK. Biochim. et Biophys. Acta 2010, 1806, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by src family kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef] [Green Version]
- Senis, Y.A.; Mazharian, A.; Mori, J. Src family kinases: At the forefront of platelet activation. Blood 2014, 124, 2013–2024. [Google Scholar] [CrossRef] [Green Version]
- Ingley, E. Src family kinases: Regulation of their activities, levels and identification of new pathways. Biochim. et Biophys. Acta (BBA)—Proteins Proteom. 2008, 1784, 56–65. [Google Scholar] [CrossRef]
- McClendon, C.J.; Miller, W.T. Structure, Function, and Regulation of the SRMS Tyrosine Kinase. Int. J. Mol. Sci. 2020, 21, 4233. [Google Scholar] [CrossRef]
- Brown, M.T.; Cooper, J.A. Regulation, substrates and functions of src. Biochim. et Biophys. Acta 1996, 1287, 121–149. [Google Scholar] [CrossRef]
- Berndt, S.; Liebscher, I. New Structural Perspectives in G Protein-Coupled Receptor-Mediated Src Family Kinase Activation. Int. J. Mol. Sci. 2021, 22, 6489. [Google Scholar] [CrossRef]
- Kohmura, N.; Yagi, T.; Tomooka, Y.; Oyanagi, M.; Kominami, R.; Takeda, N.; Chiba, J.; Ikawa, Y.; Aizawa, S. A novel nonreceptor tyrosine kinase, Srm: Cloning and targeted disruption. Mol. Cell. Biol. 1994, 14, 6915–6925. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.J.; Barker, K.T.; Martindale, J.E.; Kamalati, T.; Lowe, P.N.; Page, M.J.; Gusterson, B.A.; Crompton, M.R. Cloning and characterisation of cDNAs encoding a novel non-receptor tyrosine kinase, brk, expressed in human breast tumours. Oncogene 1994, 9, 2383–2390. [Google Scholar] [PubMed]
- Goel, R.K.; Lukong, K.E. Understanding the cellular roles of Fyn-related kinase (FRK): Implications in cancer biology. Cancer Metastasis Rev. 2016, 35, 179–199. [Google Scholar] [CrossRef] [PubMed]
- Brauer, P.M.; Tyner, A.L. RAKing in AKT: A tumor suppressor function for the intracellular tyrosine kinase FRK. Cell Cycle 2009, 8, 2728–2732. [Google Scholar] [CrossRef]
- Martellucci, S.; Clementi, L.; Sabetta, S.; Mattei, V.; Botta, L.; Angelucci, A. Src Family Kinases as Therapeutic Targets in Advanced Solid Tumors: What We Have Learned So Far. Cancers 2020, 12, 1448. [Google Scholar] [CrossRef]
- Wang, J.; Zhuang, S. Src family kinases in chronic kidney disease. Am. J. Physiol. Renal Physiol. 2017, 313, F721–F728. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, S.R.; Till, J.H. Protein Tyrosine Kinase Structure and Function. Annu. Rev. Biochem. 2000, 69, 373–398. [Google Scholar] [CrossRef] [Green Version]
- Tatosyan, A.G.; Mizenina, O.A. Kinases of the Src family: Structure and functions. Biochemistry 2000, 65, 49–58. [Google Scholar]
- Yu, X.-M.; Groveman, B.R. Src family kinases in the nervous system. FEBS J. 2012, 279, 1. [Google Scholar] [CrossRef] [PubMed]
- Ahler, E.; Register, A.C.; Chakraborty, S.; Fang, L.; Dieter, E.M.; Sitko, K.A.; Vidadala, R.S.R.; Trevillian, B.M.; Golkowski, M.; Gelman, H.; et al. A Combined Approach Reveals a Regulatory Mechanism Coupling Src’s Kinase Activity, Localization, and Phosphotransferase-Independent Functions. Mol. Cell 2019, 74, 393–408.e320. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Rosen, M.K.; Shin, T.B.; Seidel-Dugan, C.; Brugge, J.S.; Schreiber, S.L. Solution structure of the SH3 domain of Src and identification of its ligand-binding site. Science 1992, 258, 1665–1668. [Google Scholar] [CrossRef]
- Martin, G.S. The hunting of the Src. Nat. Rev. Mol. Cell Biol. 2001, 2, 467–475. [Google Scholar] [CrossRef]
- Boggon, T.J.; Eck, M.J. Structure and regulation of Src family kinases. Oncogene 2004, 23, 7918–7927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Harrison, S.C.; Eck, M.J. Three-dimensional structure of the tyrosine kinase c-Src. Nature 1997, 385, 595–602. [Google Scholar] [CrossRef]
- Cheng, Z.; Liu, L.; Wang, Z.; Cai, Y.; Xu, Q.; Chen, P. Hypoxia Activates Src and Promotes Endocytosis Which Decreases MMP-2 Activity and Aggravates Renal Interstitial Fibrosis. Int. J. Mol. Sci. 2018, 19, 581. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Liu, Y. Therapy for kidney fibrosis: Is the Src kinase a potential target? Kidney Int. 2016, 89, 12–14. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, H.; Murata, Y.; Okazawa, H.; Matozaki, T. Src family kinases: Modulators of neurotransmitter receptor function and behavior. Trends Neurosci. 2011, 34, 629–637. [Google Scholar] [CrossRef]
- Satoh, A.; Gukovskaya, A.; Edderkaoui, M.; Daghighian, M.; Reevejr, J.; Shimosegawa, T.; Pandol, S. Tumor Necrosis Factor-α Mediates Pancreatitis Responses in Acinar Cells via Protein Kinase C and Proline-Rich Tyrosine Kinase 2. Gastroenterology 2005, 129, 639–651. [Google Scholar] [CrossRef]
- Luttrell, D.K.; Luttrell, L.M. Not so strange bedfellows: G-protein-coupled receptors and Src family kinases. Oncogene 2004, 23, 7969–7978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, J.T.; Parsons, S.J. Src family protein tyrosine kinases: Cooperating with growth factor and adhesion signaling pathways. Curr. Opin. Cell Biol. 1997, 9, 187–192. [Google Scholar] [CrossRef]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Chen, X.; Chen, X.; Qin, A.; Mao, Y.; Pang, Y.; Yu, S.; Zhang, S. PP121 suppresses RANKL-Induced osteoclast formation in vitro and LPS-Induced bone resorption in vivo. Exp. Cell Res. 2020, 388, 111857. [Google Scholar] [CrossRef]
- Parra-Mercado, G.K.; Fuentes-Gonzalez, A.M.; Hernandez-Aranda, J.; Diaz-Coranguez, M.; Dautzenberg, F.M.; Catt, K.J.; Hauger, R.L.; Olivares-Reyes, J.A.; Olivares-Reyes, J.A. CRF1 Receptor Signaling via the ERK1/2-MAP and Akt Kinase Cascades: Roles of Src, EGF Receptor, and PI3-Kinase Mechanisms. Front. Endocrinol. 2019, 10, 869. [Google Scholar] [CrossRef] [Green Version]
- Calautti, V.; Grossi, M.; Mammucari, C.; Aoyama, Y.; Pirro, M.; Ono, Y.; Li, J.; Dotto, G.P. Fyn tyrosine kinase is a downstream mediator of Rho/PRK2 function in keratinocyte cell–cell adhesion. J. Cell Biol. 2002, 156, 137–148. [Google Scholar] [CrossRef]
- Ishizawar, R.; Parsons, S.J. c-Src and cooperating partners in human cancer. Cancer Cell 2004, 6, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.C.; Yen, T.; Lowell, C.A.; DeFranco, A.L. Lupus-like kidney disease in mice deficient in the Src family tyrosine kinases Lyn and Fyn. Curr. Biol. 2001, 11, 34–38. [Google Scholar] [CrossRef] [Green Version]
- Shang, G.; Tang, X.; Gao, P.; Guo, F.; Liu, H.; Zhao, Z.; Chen, Q.; Jiang, T.; Zhang, N.; Li, H. Sulforaphane attenuation of experimental diabetic nephropathy involves GSK-3 beta/Fyn/Nrf2 signaling pathway. J. Nutr. Biochem. 2015, 26, 596–606. [Google Scholar] [CrossRef]
- Panicker, N.; Saminathan, H.; Jin, H.; Neal, M.; Harischandra, D.S.; Gordon, R.; Kanthasamy, K.; Lawana, V.; Sarkar, S.; Luo, J.; et al. Fyn Kinase Regulates Microglial Neuroinflammatory Responses in Cell Culture and Animal Models of Parkinson’s Disease. J. Neurosci. 2015, 35, 10058–10077. [Google Scholar] [CrossRef]
- Yan, Y.; Ma, L.; Zhou, X.; Ponnusamy, M.; Tang, J.; Zhuang, M.A.; Tolbert, E.; Bayliss, G.; Bai, J.; Zhuang, S. Src inhibition blocks renal interstitial fibroblast activation and ameliorates renal fibrosis. Kidney Int. 2016, 89, 68–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Chen, J.-K.; Nagai, K.; Plieth, D.; Tan, M.; Lee, T.-C.; Threadgill, D.W.; Neilson, E.G.; Harris, R.C. EGFR Signaling Promotes TGFβ-Dependent Renal Fibrosis. J. Am. Soc. Nephrol. 2012, 23, 215–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, N.M.; Shehatou, G.S.; Kenawy, H.I.; Said, E. Dasatinib mitigates renal fibrosis in a rat model of UUO via inhibition of Src/STAT-3/NF-κB signaling. Environ. Toxicol. Pharmacol. 2021, 84, 103625. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Choi, H.-I.; Park, J.S.; Kim, C.S.; Bae, E.H.; Ma, S.K.; Kim, S.W. Src-mediated crosstalk between FXR and YAP protects against renal fibrosis. FASEB J. 2019, 33, 11109–11122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, K.; Xia, L.; Goldberg, H.J.; Lee, K.W.; Shah, A.; Stavar, L.; Masson, E.A.; Momen, A.; Shikatani, E.A.; John, R.; et al. Inhibition of Src Kinase Blocks High Glucose–Induced EGFR Transactivation and Collagen Synthesis in Mesangial Cells and Prevents Diabetic Nephropathy in Mice. Diabetes 2013, 62, 3874–3886. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Shi, Y.; Deng, X.; Su, Y.; Du, C.; Wei, J.; Ren, Y.; Wu, M.; Hou, Y.; Duan, H. Inhibition of c-Src/p38 MAPK pathway ameliorates renal tubular epithelial cells apoptosis in db/db mice. Mol. Cell. Endocrinol. 2015, 417, 27–35. [Google Scholar] [CrossRef]
- Ren, Q.; Guo, F.; Tao, S.; Huang, R.; Ma, L.; Fu, P. Flavonoid fisetin alleviates kidney inflammation and apoptosis via inhibiting Src-mediated NF-κB p65 and MAPK signaling pathways in septic AKI mice. Biomed. Pharmacother. 2020, 122, 109772. [Google Scholar] [CrossRef]
- He, J.C.; Husain, M.; Sunamoto, M.; D’Agati, V.D.; Klotman, M.E.; Iyengar, R.; Klotman, P.E. Nef stimulates proliferation of glomerular podocytes through activation of Src-dependent Stat3 and MAPK1,2 pathways. J. Clin. Investig. 2004, 114, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Chang, J.W.; Yang, W.S.; Kim, S.B.; Kil Park, S.; Park, J.S.; Lee, S.K. Albumin-induced epithelial-mesenchymal transition and ER stress are regulated through a common ROS-c-Src kinase-mTOR pathway: Effect of imatinib mesylate. Am. J. Physiol. Physiol. 2011, 300, F1214–F1222. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Guo, H.; Li, J.; Ma, T.; Zhou, S.; Zhang, Z.; Miao, L.; Cai, L. Sulforaphane prevents type 2 diabetes-induced nephropathy via AMPK-mediated activation of lipid metabolic pathways and Nrf2 antioxidative function. Clin. Sci. 2020, 134, 2469–2487. [Google Scholar] [CrossRef]
- Seo, H.-Y.; Jeon, J.-H.; Jung, Y.-A.; Jung, G.-S.; Lee, E.J.; Choi, Y.-K.; Park, K.-G.; Choe, M.S.; Jang, B.K.; Kim, M.-K.; et al. Fyn deficiency attenuates renal fibrosis by inhibition of phospho-STAT3. Kidney Int. 2016, 90, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Mkaddem, S.B.; Murua, A.; Flament, H.; Titeca-Beauport, D.; Bounaix, C.; Danelli, L.; Launay, P.; Benhamou, M.; Blank, U.; Daugas, E.; et al. Lyn and Fyn function as molecular switches that control immunoreceptors to direct homeostasis or inflammation. Nat. Commun. 2017, 8, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Lin, Q.; Liao, H.; Feng, J.; Dong, X.; Ye, J. TGF-β1 Induces Podocyte Injury Through Smad3-ERK-NF-κB Pathway and Fyn-dependent TRPC6 phosphorylation. Cell. Physiol. Biochem. 2010, 26, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Shen, F.; Tian, K.; Wang, M.; Xi, Y.; Li, J.; Huang, Z. Triptolide induces oxidative damage in NRK-52E cells through facilitating Nrf2 degradation by ubiquitination via the GSK-3β/Fyn pathway. Toxicol. Vitr. 2019, 58, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Yuan, Y.; Zheng, J.; Hu, N. Hydrogen sulfide alleviates uranium-induced kidney cell apoptosis mediated by ER stress via 20S proteasome involving in Akt/GSK-3β/Fyn-Nrf2 signaling. Free Radic. Res. 2018, 52, 1020–1029. [Google Scholar] [CrossRef]
- Andrade-Oliveira, V.; Foresto-Neto, O.; Watanabe, I.K.M.; Zatz, R.; Câmara, N.O.S. Inflammation in Renal Diseases: New and Old Players. Front. Pharmacol. 2019, 10, 1192. [Google Scholar] [CrossRef]
- Uddin, J.; Dorotea, D.; Pak, E.S.; Ha, H. Fyn Kinase: A Potential Therapeutic Target in Acute Kidney Injury. Biomol. Ther. 2020, 28, 213–221. [Google Scholar] [CrossRef]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of Acute Kidney Injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-H.; Zhang, Y.-G. Kidney and innate immunity. Immunol. Lett. 2017, 183, 73–78. [Google Scholar] [CrossRef]
- Dellepiane, S.; Marengo, M.; Cantaluppi, V. Detrimental cross-talk between sepsis and acute kidney injury: New pathogenic mechanisms, early biomarkers and targeted therapies. Crit. Care 2016, 20, 61. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.R.; Rabb, H. The innate immune response in ischemic acute kidney injury. Clin. Immunol. 2009, 130, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Piskol, R.; Ybarra, R.; Chen, Y.-J.J.; Li, J.; Slaga, D.; Hristopoulos, M.; Clark, R.; Modrusan, Z.; Totpal, K.; et al. CD3 bispecific antibody–induced cytokine release is dispensable for cytotoxic T cell activity. Sci. Transl. Med. 2019, 11, eaax8861. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, M.-Y.; Kim, J.-H.; Cho, J.Y. Fisetin Suppresses Macrophage-Mediated Inflammatory Responses by Blockade of Src and Syk. Biomol. Ther. 2015, 23, 414–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, A.B.; Sanchez-Niño, M.D.; Ramos, A.M.; Moreno, J.A.; Santamaria, B.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. NF-κB in Renal Inflammation. J. Am. Soc. Nephrol. 2010, 21, 1254–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajay, A.K.; Kim, T.-M.; Ramirez-Gonzalez, V.; Park, P.J.; Frank, D.A.; Vaidya, V.S. A Bioinformatics Approach Identifies Signal Transducer and Activator of Transcription-3 and Checkpoint Kinase 1 as Upstream Regulators of Kidney Injury Molecule-1 after Kidney Injury. J. Am. Soc. Nephrol. 2014, 25, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Ramnath, R.D.; Sun, J.; Bhatia, M. Involvement of Src Family Kinases in Substance P-Induced Chemokine Production in Mouse Pancreatic Acinar Cells and Its Significance in Acute Pancreatitis. J. Pharmacol. Exp. Ther. 2009, 329, 418–428. [Google Scholar] [CrossRef]
- Tang, J.; Xiao, Y.; Lin, G.; Guo, H.; Deng, H.-X.; Tu, S.; Langdon, W.Y.; Yang, H.; Tao, L.; Li, Y.; et al. Tyrosine phosphorylation of NLRP3 by the Src family kinase Lyn suppresses the activity of the NLRP3 inflammasome. Sci. Signal. 2021, 14, eabe3410. [Google Scholar] [CrossRef]
- Gao, R.; Ma, Z.; Ma, M.; Yu, J.; Chen, J.; Li, Z.; Shetty, S.; Fu, J. Deletion of Src family kinase Lyn aggravates endotoxin-induced lung inflammation. Am. J. Physiol. Cell. Mol. Physiol. 2015, 309, L1376–L1381. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yang, X.; Li, Y.; Wang, X.; Zhang, Y.; Dai, X.; Niu, B.; Wu, J.; Yuan, X.; Xiong, A.; et al. Lyn kinase represses mucus hypersecretion by regulating IL-13-induced endoplasmic reticulum stress in asthma. eBioMedicine 2017, 15, 137–149. [Google Scholar] [CrossRef]
- Li, R.; Fang, L.; Pu, Q.; Lin, P.; Hoggarth, A.; Huang, H.; Li, X.; Li, G.; Wu, M. Lyn prevents aberrant inflammatory responses to Pseudomonas infection in mammalian systems by repressing a SHIP-1-associated signaling cluster. Signal Transduct. Target. Ther. 2016, 1, 16032. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Bian, Z.; Kidder, K.; Liang, H.; Liu, Y. Non-Lyn Src Family Kinases Activate SIRPα–SHP-1 to Inhibit PI3K–Akt2 and Dampen Proinflammatory Macrophage Polarization. J. Immunol. 2021, 207, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aranda-Rivera, A.; Cruz-Gregorio, A.; Aparicio-Trejo, O.; Pedraza-Chaverri, J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules 2021, 11, 1144. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, E.; Buricchi, F.; Raugei, G.; Ramponi, G.; Chiarugi, P. Intracellular Reactive Oxygen Species Activate Src Tyrosine Kinase during Cell Adhesion and Anchorage-Dependent Cell Growth. Mol. Cell. Biol. 2005, 25, 6391–6403. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.-T.; Shih, R.-H.; Lin, C.-C.; Chen, J.-T.; Yang, C.-M. Role of TLR4/NADPH oxidase/ROS-activated p38 MAPK in VCAM-1 expression induced by lipopolysaccharide in human renal mesangial cells. Cell Commun. Signal. 2012, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.-F.; Chang, C.-C.; Lee, I.-T.; Lee, C.-W.; Lin, W.-N.; Lin, C.-C.; Yang, C.-M. Activation of ROS/NF-κB and Ca2+/CaM kinase II are necessary for VCAM-1 induction in IL-1β-treated human tracheal smooth muscle cells. Toxicol. Appl. Pharmacol. 2009, 237, 8–21. [Google Scholar] [CrossRef]
- Sun, J.; Chen, L.; Jiang, P.; Duan, B.; Wang, R.; Xu, J.; Liu, W.; Xu, Y.; Xie, Z.; Feng, F.; et al. Phenylethanoid glycosides of Callicarpa kwangtungensis Chun exert cardioprotective effect by weakening Na+-K+-ATPase/Src/ERK1/2 pathway and inhibiting apoptosis mediated by oxidative stress and inflammation. J. Ethnopharmacol. 2020, 258, 112881. [Google Scholar] [CrossRef]
- Qi, S.; Feng, Z.; Li, Q.; Qi, Z.; Zhang, Y. Inhibition of ROS-mediated activation Src-MAPK/AKT signaling by orientin alleviates H2O2-induced apoptosis in PC12 cells. Drug Des. Dev. Ther. 2018, 12, 3973–3984. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Wu, X.; Xian, X.; Wang, L.; Zhu, L.; Sun, H.; Yang, L.; Liu, W. Calcitonin gene-related peptide inhibits angiotensin II-induced NADPH oxidase-dependent ROS via the Src/STAT3 signalling pathway. J. Cell. Mol. Med. 2020, 24, 6426–6437. [Google Scholar] [CrossRef]
- Huang, Y.; Li, W.; Su, Z.-Y.; Kong, A.-N.T. The complexity of the Nrf2 pathway: Beyond the antioxidant response. J. Nutr. Biochem. 2015, 26, 1401–1413. [Google Scholar] [CrossRef]
- Abhinav, K.; Jain, A.K.J. Show footnotes. GSK-3β Acts Upstream of Fyn Kinase in Regulation of Nuclear Export and Degradation of NF-E2 Related Factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef] [Green Version]
- Kaspar, J.W.; Jaiswal, A.K. Tyrosine phosphorylation controls nuclear export of Fyn, allowing Nrf2 activation of cytoprotective gene expression. FASEB J. 2011, 25, 1076–1087. [Google Scholar] [CrossRef] [Green Version]
- Culbreth, M.; Zhang, Z.; Aschner, M. Methylmercury augments Nrf2 activity by downregulation of the Src family kinase Fyn. NeuroToxicology 2017, 62, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Medina, B.E.; Lerma, D.; Hwang, M.; Ross, J.A.; Skouta, R.; Aguilera, R.J.; Kirken, R.A.; Varela-Ramirez, A.; Robles-Escajeda, E. Green barley mitigates cytotoxicity in human lymphocytes undergoing aggressive oxidative stress, via activation of both the Lyn/PI3K/Akt and MAPK/ERK pathways. Sci. Rep. 2019, 9, 6005. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R. Endoplasmic Reticulum Stress in the Kidney as a Novel Mediator of Kidney Injury. Nephron Exp. Nephrol. 2009, 112, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, A.; Zirak, M.R.; Hayes, A.W.; Reiter, R.; Karimi, G. Curcumin and its analogues protect from endoplasmic reticulum stress: Mechanisms and pathways. Pharmacol. Res. 2019, 146, 104335. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef]
- Rashid, H.-O.; Yadav, R.K.; Kim, H.-R.; Chae, H.-J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [Green Version]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Sovolyova, N.; Healy, S.; Samali, A.; Logue, S.E. Stressed to death—mechanisms of ER stress-induced cell death. Biol. Chem. 2014, 395, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R. Endoplasmic reticulum stress as a progression factor for kidney injury. Curr. Opin. Pharmacol. 2010, 10, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Nakajima, S.; Saito, Y.; Takahashi, S.; Katoh, R.; Kitamura, M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1–JNK pathway. Cell Death Differ. 2012, 19, 310–320. [Google Scholar] [CrossRef] [Green Version]
- Ying, R.; Wang, X.-Q.; Yang, Y.; Gu, Z.-J.; Mai, J.-T.; Qiu, Q.; Chen, Y.-X.; Wang, J.-F. Hydrogen sulfide suppresses endoplasmic reticulum stress-induced endothelial-to-mesenchymal transition through Src pathway. Life Sci. 2016, 144, 208–217. [Google Scholar] [CrossRef]
- Tsai, Y.-L.; Ha, D.P.; Zhao, H.; Carlos, A.J.; Wei, S.; Pun, T.K.; Wu, K.; Zandi, E.; Kelly, K.; Lee, A.S. Endoplasmic reticulum stress activates SRC, relocating chaperones to the cell surface where GRP78/CD109 blocks TGF-β signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E4245–E4254. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.-K.; Cho, W.Y.; Sung, S.A.; Kim, H.K.; Won, N.H. MEK inhibitor, U0126, attenuates cisplatin-induced renal injury by decreasing inflammation and apoptosis. Kidney Int. 2005, 67, 458–466. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yamada, E.; Zong, H.; Pessin, J.E. Fyn Activation of mTORC1 Stimulates the IRE1α-JNK Pathway, Leading to Cell Death. J. Biol. Chem. 2015, 290, 24772–24783. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, Z.; Hill, J.A.; Lin, F. New Autophagy Reporter Mice Reveal Dynamics of Proximal Tubular Autophagy. J. Am. Soc. Nephrol. 2014, 25, 305–315. [Google Scholar] [CrossRef]
- Anding, A.L.; Baehrecke, E.H. Autophagy in Cell Life and Cell Death. Curr. Top. Dev. Biol. 2015, 114, 67–91. [Google Scholar] [CrossRef]
- Takabatake, Y.; Kimura, T.; Takahashi, A.; Isaka, Y. Autophagy and the kidney: Health and disease. Nephrol. Dial. Transplant. 2014, 29, 1639–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Nartiss, Y.; Steipe, B.; McQuibban, G.A.; Kim, P.K. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 2012, 8, 1462–1476. [Google Scholar] [CrossRef] [Green Version]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, R.; Palmieri, M.; Chaudhury, A.; Klisch, T.J.; Di Ronza, A.; Neilson, J.R.; Rodney, G.; Sardiello, M. Src regulates amino acid-mediated mTORC1 activation by disrupting GATOR1-Rag GTPase interaction. Nat. Commun. 2018, 9, 4351. [Google Scholar] [CrossRef]
- Pal, R.; Palmieri, M.; Loehr, J.A.; Li, S.; Abo-Zahrah, R.; Monroe, T.; Thakur, P.B.; Sardiello, M.; Rodney, G.G. Src-dependent impairment of autophagy by oxidative stress in a mouse model of Duchenne muscular dystrophy. Nat. Commun. 2014, 5, 4425. [Google Scholar] [CrossRef] [Green Version]
- Moon, S.Y.; Kim, H.S.; Nho, K.W.; Jang, Y.J.; Lee, S.K. Endoplasmic Reticulum Stress Induces Epithelial-Mesenchymal Transition through Autophagy via Activation of c-Src Kinase. Nephron Exp. Nephrol. 2014, 126, 127–140. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Yamada, E.; Pessin, J.E.; Kurland, I.J.; Schwartz, G.J.; Bastie, C.C. Fyn-Dependent Regulation of Energy Expenditure and Body Weight Is Mediated by Tyrosine Phosphorylation of LKB1. Cell Metab. 2010, 11, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Bastie, C.C.; Zong, H.; Xu, J.; Busa, B.; Judex, S.; Kurland, I.J.; Pessin, J.E. Integrative Metabolic Regulation of Peripheral Tissue Fatty Acid Oxidation by the Src Kinase Family Member Fyn. Cell Metab. 2007, 5, 371–381. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Qi, Q.; Chan, C.B.; Zhou, W.; Chen, J.; Luo, H.R.; Appin, C.; Brat, D.J.; Ye, K. Fyn-phosphorylated PIKE-A binds and inhibits AMPK signaling, blocking its tumor suppressive activity. Cell Death Differ. 2016, 23, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Yamada, E.; Okada, S.; Bastie, C.C.; Vatish, M.; Nakajima, Y.; Shibusawa, R.; Ozawa, A.; Pessin, J.E.; Yamada, M. Fyn phosphorylates AMPK to inhibit AMPK activity and AMP-dependent activation of autophagy. Oncotarget 2016, 7, 74612–74629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, E.; Bastie, C.C.; Koga, H.; Wang, Y.; Cuervo, A.M.; Pessin, J.E. Mouse Skeletal Muscle Fiber-Type-Specific Macroautophagy and Muscle Wasting Are Regulated by a Fyn/STAT3/Vps34 Signaling Pathway. Cell Rep. 2012, 1, 557–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, L.; Mourner, R.; Leclerc, J.; Pende, M.; Foretz, M.; Viollet, B. Coordinated maintenance of muscle cell size control by AMP-activated protein kinase. FASEB J. 2010, 24, 3555–3561. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.M.; Huang, P.; Kar, N.; Burgett, M.; Muller-Greven, G.; Nowacki, A.S.; Distelhorst, C.W.; Lathia, J.D.; Rich, J.N.; Kappes, J.C.; et al. Lyn Facilitates Glioblastoma Cell Survival under Conditions of Nutrient Deprivation by Promoting Autophagy. PLoS ONE 2013, 8, e70804. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, S.; Zhou, X.; Ye, Y.; Tan, S.; Zhang, S.; Li, R.; Yu, M.; Jundt, M.C.; Hidebrand, A.; et al. Lyn Delivers Bacteria to Lysosomes for Eradication through TLR2-Initiated Autophagy Related Phagocytosis. PLOS Pathog. 2016, 12, e1005363. [Google Scholar] [CrossRef]
- Zhang, Q.; Meng, X.; Qin, G.; Xue, X.; Dang, N. Lyn Kinase Promotes the Proliferation of Malignant Melanoma Cells through Inhibition of Apoptosis and Autophagy via the PI3K/Akt Signaling Pathway. J. Cancer 2019, 10, 1197–1208. [Google Scholar] [CrossRef]
- Yang, L. How Acute Kidney Injury Contributes to Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 117–142. [Google Scholar] [CrossRef]
- Coca, S.G.; Singanamala, S.; Parikh, C.R. Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney Int. 2012, 81, 442–448. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Solbes, A.S.; Youker, K. Epithelial to Mesenchymal Transition (EMT) and Endothelial to Mesenchymal Transition (EndMT): Role and Implications in Kidney Fibrosis. Results Probl. Cell Differ. 2017, 60, 345–372. [Google Scholar] [CrossRef]
- Tan, Y.; Wang, B.; Keum, J.-S.; Jaffa, A.A. Mechanisms through which bradykinin promotes glomerular injury in diabetes. Am. J. Physiol. Renal Physiol. 2005, 288, F483–F492. [Google Scholar] [CrossRef] [Green Version]
- Moonen, L.; D’Haese, P.C.; Vervaet, B.A. Epithelial Cell Cycle Behaviour in the Injured Kidney. Int. J. Mol. Sci. 2018, 19, 2038. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.-M.; Tang, P.M.K.; Li, J.; Lan, H.Y.; Meng, X.-M.; Tang, P.M.K.; Li, J.; Lan, H.Y. TGF-β/Smad signaling in renal fibrosis. Front. Physiol. 2015, 6, 82. [Google Scholar] [CrossRef] [Green Version]
- Gewin, L.; Vadivelu, S.; Neelisetty, S.; Srichai, M.B.; Paueksakon, P.; Pozzi, A.; Harris, R.C.; Zent, R. Deleting the TGF-βReceptor Attenuates Acute Proximal Tubule Injury. J. Am. Soc. Nephrol. 2012, 23, 2001–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronco, P.; Lelongt, B.; Piedagnel, R.; Chatziantoniou, C. Matrix Metalloproteinases in Kidney Disease Progression and Repair: A Case of Flipping the Coin. Semin. Nephrol. 2007, 27, 352–362. [Google Scholar] [CrossRef]
- Cheng, Z.; Limbu, M.H.; Wang, Z.; Liu, J.; Liu, L.; Zhang, X.; Chen, P.; Liu, B. MMP-2 and 9 in Chronic Kidney Disease. Int. J. Mol. Sci. 2017, 18, 776. [Google Scholar] [CrossRef] [Green Version]
- Ślusarz, A.; Nichols, L.A.; Grunz-Borgmann, E.A.; Chen, G.; Akintola, A.D.; Catania, J.M.; Burghardt, R.C.; Trzeciakowski, J.P.; Parrish, A.R. Overexpression of MMP-7 increases collagen 1A2 in the aging kidney. Physiol. Rep. 2013, 1, e0009(1-20). [Google Scholar] [CrossRef]
- Kim, C.S.; Kim, I.J.; Choi, J.S.; Bae, E.H.; Ma, S.K.; Kim, S.W. Tamoxifen ameliorates obstructive nephropathy through Src and the PI3K/Akt/mTOR pathway. Biol. Cell 2019, 111, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.X.; Zhang, S.; Cui, X.; Zhang, J.; Yu, H.; Khalaf, F.K.; Malhotra, D.; Kennedy, D.J.; Shapiro, J.I.; Tian, J.; et al. Na/K-ATPase/src complex mediates regulation of CD40 in renal parenchyma. Nephrol. Dial. Transplant. 2018, 33, 1138–1149. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Song, Y.; Wang, Y. pNaKtide ameliorates renal interstitial fibrosis through inhibition of sodium-potassium adenosine triphosphatase-mediated signaling pathways in unilateral ureteral obstruction mice. Nephrol. Dial. Transplant. 2019, 34, 242–252. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Z.; Xie, J.X.; Li, X.; Tian, J.; Cai, T.; Cui, H.; Ding, H.; Shapiro, J.I.; Xie, Z. Na/K-ATPase Mimetic pNaKtide Peptide Inhibits the Growth of Human Cancer Cells. J. Biol. Chem. 2011, 286, 32394–32403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.; Li, L.; Menon, M.C.; Zhang, W.; Fu, J.; Kidd, B.; Keung, K.L.; Woytovich, C.; Greene, I.; Xiao, W.; et al. Genomic Analysis of Kidney Allograft Injury Identifies Hematopoietic Cell Kinase as a Key Driver of Renal Fibrosis. J. Am. Soc. Nephrol. 2017, 28, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Dorotea, D.; Lee, S.; Lee, S.J.; Lee, G.; Son, J.B.; Choi, H.G.; Ahn, S.-M.; Ha, H. KF-1607, a Novel Pan Src Kinase Inhibitor, Attenuates Obstruction-Induced Tubulointerstitial Fibrosis in Mice. Biomol. Ther. 2021, 29, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Pabla, N.; Dong, G.; Jiang, M.; Huang, S.; Kumar, M.V.; Messing, R.O.; Dong, Z. Inhibition of PKCδ reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J. Clin. Investig. 2011, 121, 2709–2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arany, I.; Megyesi, J.K.; Kaneto, H.; Price, P.M.; Safirstein, R.L. Cisplatin-induced cell death is EGFR/src/ERK signaling dependent in mouse proximal tubule cells. Am. J. Physiol. Renal. Physiol. 2004, 287, F543–F549. [Google Scholar] [CrossRef] [Green Version]
- Leclercq, G.; Haegel, H.; Schneider, A.; Giusti, A.M.; Marrer-Berger, E.; Boetsch, C.; Walz, A.-C.; Pulko, V.; Sam, J.; Challier, J.; et al. Src/lck inhibitor dasatinib reversibly switches off cytokine release and T cell cytotoxicity following stimulation with T cell bispecific antibodies. J. Immunother. Cancer 2021, 9, e002582. [Google Scholar] [CrossRef]
- Liu, F.; Wang, L.; Qi, H.; Wang, J.; Wang, Y.; Jiang, W.; Xu, L.; Liu, N.; Zhuang, S. Nintedanib, a triple tyrosine kinase inhibitor, attenuates renal fibrosis in chronic kidney disease. Clin. Sci. 2017, 131, 2125–2143. [Google Scholar] [CrossRef]
- Feng, L.; Li, W.; Chao, Y.; Huan, Q.; Lu, F.; Yi, W.; Jun, W.; Binbin, C.; Na, L.; Shougang, Z. Synergistic Inhibition of Renal Fibrosis by Nintedanib and Gefitinib in a Murine Model of Obstructive Nephropathy. Kidney Dis. 2021, 7, 34–49. [Google Scholar] [CrossRef]
- Amoui, M.; Dráber, P.; Dráberová, L. Src family-selective tyrosine kinase inhibitor, PP1, inhibits both FcɛRI- and Thy-1-mediated activation of rat basophilic leukemia cells. Eur. J. Immunol. 1997, 27, 1881–1886. [Google Scholar] [CrossRef]
- Montero, J.C.; Seoane, S.; Ocaña, A.; Pandiella, A. Inhibition of SRC family kinases and receptor tyrosine kinases by dasatinib: Possible combinations in solid tumors. Clin Cancer Res. 2011, 17, 5546–5552. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Member of SFKs | Organs/Cells | Models | Mechanisms | References |
|---|---|---|---|---|
| Scr | Kidney | renal ischemia/reperfusion model | Reduces renal injury by activating STAT3, ERK1/2, and NF-κB signaling pathway | [12] |
| Kidney | unilateral ureteral obstruction | Mediates the activation of TGF-β1 signal, NF-κB, Smad-3 epidermal growth factor receptor and STAT3, and EGFR transactivation | [51,52,53] | |
| Kidney | unilateral ureteral obstruction | Regulates phosphorylation and localization of YAP | [54] | |
| Kidney | STZ-induced type 1 diabetes | Mediates phosphorylation of EGFR and MAPK | [55] | |
| Kidney | diabetic db/db | Induces activation of p38 MAPK activation | [56] | |
| Kidney | LPS-induced septic AKI | Mediates NF-κB and MAPK signaling pathways | [57] | |
| podocytes | HIV-associated nephropathy (HIVAN) | Activates of STAT3 and MAPK1, 2 Mediates cell proliferation and dedifferentiation of podocytes | [58] | |
| HK-2 | hypoxia | Decreases MMP-2 activity and aggravates renal interstitial fibrosis | [37] | |
| HK-2 | ER stress | Activates mTOR pathway | [59] | |
| Fyn | Kidney | STZ-induced type 1 diabetes | Suppresses Nrf2 expression | [49] |
| Kidney | type 2 diabetes-induced nephropathy | Promotes the output of Nrf2 from nucleus | [60] | |
| Kidney | obstructive fibrosis | Mediates STAT3 activation | [61] | |
| Kidney | lupus nephritis | Mediates ITAM phosphorylation to promote inflammation | [62] | |
| Podocytes | apoptosis | Activates of Fyn-induced TRPC6 phosphorylation | [63] | |
| NRK-52E | oxidative stress | Mediates degradation of Nrf2 | [64,65] | |
| Lyn | Kidney | lupus nephritis | Mediates ITAMi phosphorylation to homeostasis | [62] |
| Compounds | Targeted SFKs | Effects | Reference |
|---|---|---|---|
| PP2 | Src/Fyn | Improves mitochondrial dysfunction and renal injury induced by LPS, | [13] |
| Src | Reduces collagen deposition and improves fibrosis in kidney | [137] | |
| KF-1607 | Src | Inhibits renal inflammation and oxidative stress, prevents tubulointerstitial fibrosis | [143] |
| PP1 | Src | Relieves renal injury in mouse model of renal ischemia/reperfusion (I/R) | [12] |
| Src | Reduces the expression of VCAM-1 in human mesangial cells (HRMC) treated with LPS and alleviates monocyte adhesion and inflammatory reaction | [85] | |
| Src | Reduces the damage and death of renal cells induced by cisplatin | [144] | |
| Src | Inhibits apoptosis after cisplatin treatment by Src/ERK signaling pathway | [145] | |
| Src | Inhibits the activation and proliferation of renal interstitial fibroblasts, regulates the expression of cyclin, and improves fibrosis | [51] | |
| dasatinib | Src/lck/Hck/c-Abl | Decreases inflammatory macrophage infiltration and renal oxidative stress, reduces renal expression of α-SMA and fibronectin, and improves fibrosis | [53,142,146] |
| nintedanib | Src/Lck/Lyn | Inhibits inflammation and renal fibrosis | [147,148] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, N.; Lin, G.; Zhang, H.; Sun, J.; Gui, M.; Liu, Y.; Li, W.; Liu, J.; Tang, J. Src Family Kinases: A Potential Therapeutic Target for Acute Kidney Injury. Biomolecules 2022, 12, 984. https://doi.org/10.3390/biom12070984
Li N, Lin G, Zhang H, Sun J, Gui M, Liu Y, Li W, Liu J, Tang J. Src Family Kinases: A Potential Therapeutic Target for Acute Kidney Injury. Biomolecules. 2022; 12(7):984. https://doi.org/10.3390/biom12070984
Chicago/Turabian StyleLi, Nannan, Guoxin Lin, Hao Zhang, Jian Sun, Ming Gui, Yan Liu, Wei Li, Jishi Liu, and Juan Tang. 2022. "Src Family Kinases: A Potential Therapeutic Target for Acute Kidney Injury" Biomolecules 12, no. 7: 984. https://doi.org/10.3390/biom12070984
APA StyleLi, N., Lin, G., Zhang, H., Sun, J., Gui, M., Liu, Y., Li, W., Liu, J., & Tang, J. (2022). Src Family Kinases: A Potential Therapeutic Target for Acute Kidney Injury. Biomolecules, 12(7), 984. https://doi.org/10.3390/biom12070984