
Anti-Huntington’s Effect of Rosiridin via Oxidative Stress/AchE Inhibition and Modulation of Succinate Dehydrogenase, Nitrite, and BDNF Levels against 3-Nitropropionic Acid in Rodents

 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Drugs and Chemicals
2.3. Acute Toxicity Studies
2.4. Experimental Design
2.5. Behavioral Estimation
2.5.1. Narrow Beam Walk Assessment
2.5.2. Hanging-Wire Test
2.5.3. Elevated Plus-Maze Test
2.6. Biochemical Estimation
2.6.1. Brain Homogenate
2.6.2. Brain Biochemical Parameters
2.6.3. Estimation of Brain-Derived Neurotrophic Factor (BDNF) Activity
2.6.4. Estimation of Succinate Dehydrogenase (SDH) Activity
2.6.5. Estimation of Glutamate
2.6.6. Nitrite Content Estimation
2.6.7. Evaluation of Acetylcholinesterase (AchE) Levels
2.7. Statistical Analysis
3. Results
3.1. Acute Toxicity Study
3.2. Behavioral Assessment
3.2.1. Effect of Rosiridin on the Hanging-Wire Test
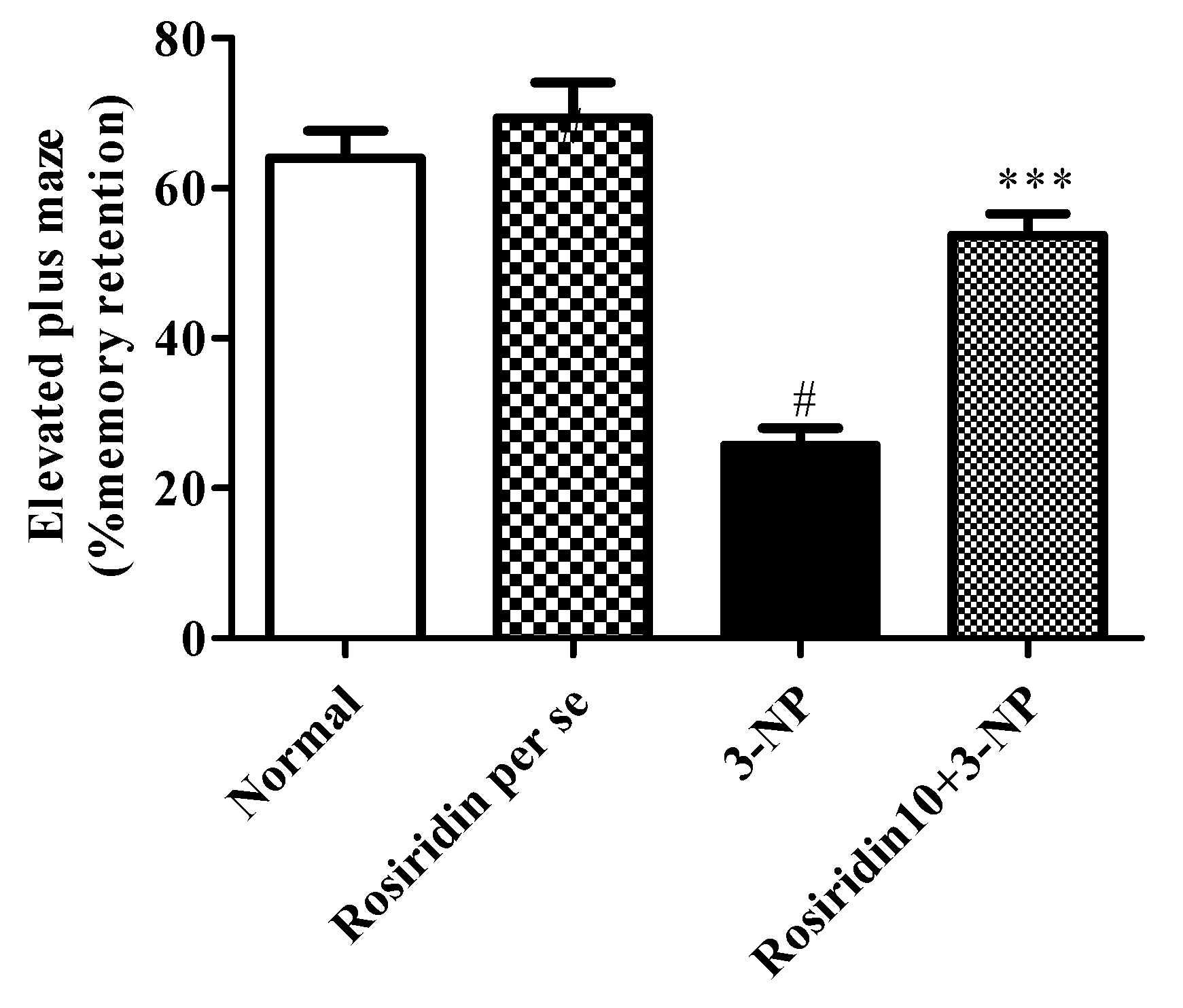
3.2.2. Effect of Rosiridin on the Elevated Plus-Maze Test
3.3. Biochemical Estimation
3.3.1. Effect of Rosiridin on Oxidative Stress Parameters
3.3.2. Effect of Rosiridin on Proinflammatory Biomarkers
3.3.3. Effect of Rosiridin on Brain-Derived Neurotrophic Factor (BDNF) Activity
3.3.4. Effect of Rosiridin on Succinate Dehydrogenase (SDH) Activity
3.3.5. Effect of Rosiridin on Glutamate Activity
3.3.6. Effect of Rosiridin on Nitrite Content
3.3.7. Effect of Rosiridin on Acetylcholinesterase (AchE) Levels
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roos, R.A.C. Huntington’s disease: A clinical review. Orphanet J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoker, T.B.; Mason, S.L.; Greenland, J.C.; Holden, S.T.; Santini, H.; Barker, R.A. Huntington’s disease: Diagnosis and management. Pract. Neurol. 2022, 22, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Caterino, M.; Squillaro, T.; Montesarchio, D.; Giordano, A.; Giancola, C.; Melone, M.A.B. Huntingtin protein: A new option for fixing the Huntington’s disease countdown clock. Neuropharmacology 2018, 135, 126–138. [Google Scholar] [CrossRef]
- Chao, T.-K.; Hu, J.; Pringsheim, T. Risk factors for the onset and progression of Huntington disease. Neurotoxicology 2017, 61, 79–99. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Finkbeiner, S. Huntington’s disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a007476. [Google Scholar] [CrossRef] [Green Version]
- Rawlins, M.D.; Wexler, N.S.; Wexler, A.R.; Tabrizi, S.J.; Douglas, I.; Evans, S.J.W.; Smeeth, L. The Prevalence of Huntington’s Disease. Neuroepidemiology 2016, 46, 144–153. [Google Scholar] [CrossRef]
- Feleus, S.; van Schaijk, M.; Roos, R.A.; de Bot, S.T. The Many Faces of Huntington’s Chorea Treatment: The Impact of Sudden Withdrawal of Tiapride after 40 Years of Use and a Systematic Review. J. Pers. Med. 2022, 12, 589. [Google Scholar] [CrossRef]
- Pan, L.; Feigin, A. Huntington’s disease: New frontiers in therapeutics. Curr. Neurol. Neurosci. Rep. 2021, 21, 10. [Google Scholar] [CrossRef]
- Gil-Mohapel, J.S.; Brocardo, P.R.; Christie, B. The role of oxidative stress in Huntington’s disease: Are antioxidants good therapeutic candidates? Curr. Drug Targets 2014, 15, 454–468. [Google Scholar] [CrossRef]
- Kumar, A.; Ratan, R.R. Oxidative Stress and Huntington’s Disease: The Good, The Bad, and The Ugly. J. Huntingt. Dis. 2016, 5, 217–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabrizi, S.J.; Ghosh, R.; Leavitt, B.R. Huntingtin lowering strategies for disease modification in Huntington’s disease. Neuron 2019, 101, 801–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, B.; Jung, R.; Oh, H.; Owens, G.E.; Lee, H.; Kwak, S.; Lee, R.; Cotman, S.L.; Lee, J.-M.; MacDonald, M.E.; et al. Novel DNA Aptamers that Bind to Mutant Huntingtin and Modify Its Activity. Molecular therapy. Nucleic Acids. 2018, 11, 416–428. [Google Scholar] [CrossRef] [Green Version]
- Gipson, T.A.; Neueder, A.; Wexler, N.S.; Bates, G.P.; Housman, D. Aberrantly spliced HTT, a new player in Huntington’s disease pathogenesis. RNA Biol. 2013, 10, 1647–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stack, E.C.; Matson, W.R.; Ferrante, R.J. Evidence of oxidant damage in Huntington’s disease: Translational strategies using antioxidants. Ann. N. Y. Acad. Sci. 2008, 1147, 79–92. [Google Scholar] [CrossRef]
- Kumar, P.; Kalonia, H.; Kumar, A. Possible GABAergic mechanism in the neuroprotective effect of gabapentin and lamotrigine against 3-nitropropionic acid induced neurotoxicity. Eur. J. Pharmacol. 2012, 674, 265–274. [Google Scholar] [CrossRef]
- Dhir, A.; Akula, K.K.; Kulkarni, S.K. Tiagabine, a GABA uptake inhibitor, attenuates 3-nitropropionic acid-induced alterations in various behavioral and biochemical parameters in rats. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 835–843. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington disease: New insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef]
- Weir, D.W.; Sturrock, A.; Leavitt, B.R. Development of biomarkers for Huntington’s disease. Lancet Neurol. 2011, 10, 573–590. [Google Scholar] [CrossRef]
- Panossian, A.; Wikman, G. Effects of adaptogens on the central nervous system and the molecular mechanisms associated with their stress—protective activity. Pharmaceuticals 2010, 3, 188–224. [Google Scholar] [CrossRef]
- Panossian, A.; Wikman, G.; Wagner, H. Plant adaptogens III. Earlier and more recent aspects and concepts on their mode of action. Phytomedicine 1999, 6, 287–300. [Google Scholar] [CrossRef]
- Panossian, A.; Wikman, G. Pharmacology of Schisandra chinensis Bail.: An overview of Russian research and uses in medicine. J. Ethnopharmacol. 2008, 118, 183–212. [Google Scholar] [CrossRef] [PubMed]
- Panossian, A.; Wikman, G. Effect of adaptogens on the central nervous system. Arq. Bras. Fitomed. Cient. 2005, 3, 29–51. [Google Scholar]
- Schriner, S.E.; Avanesian, A.; Liu, Y.; Luesch, H.; Jafari, M. Protection of human cultured cells against oxidative stress by Rhodiola rosea without activation of antioxidant defenses. Free Radic. Biol. Med. 2009, 47, 577–584. [Google Scholar] [CrossRef]
- Lee, M.-W.; Lee, Y.-A.; Park, H.-M.; Toh, S.-H.; Lee, E.-J.; Jang, H.-D.; Kim, Y.-H. Antioxidative phenolic compounds from the roots of Rhodiola sachalinensis A. Bor. Arch. Pharm. Res. 2000, 23, 455–458. [Google Scholar] [CrossRef]
- Linh, P.T.; Kim, Y.H.; Hong, S.P.; Jian, J.J.; Kang, J.S. Quantitative determination of salidroside and tyrosol from the underground part ofRhodiola rosea by high performance liquid chromatography. Arch. Pharm. Res. 2000, 23, 349–352. [Google Scholar] [CrossRef]
- Bol’shakova, I.V.; Lozovskaia, E.L.; Sapezhinskiĭ, I. Antioxidant properties of a series of extracts from medicinal plants. Biofizika 1997, 42, 480–483. [Google Scholar]
- Lazarova, M.B.; Petkov, V.D.; Markovska, V.L.; Petkov, V.V.; Mosharrof, A. Effects of meclofenoxate and Extr. Rhodiolae roseae L. on electroconvulsive shock-impaired learning and memory in rats. Methods Find. Exp. Clin. Pharmacol. 1986, 8, 547–552. [Google Scholar]
- Bucci, L.R. Selected herbals and human exercise performance. Am. J. Clin. Nutr. 2000, 72, 624S–636S. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, M.; Nakamura, S.; Li, X.; Matsuda, H. Reinvestigation of absolute stereostructure of (−)-rosiridol: Structures of monoterpene glycosides, rosiridin, rosiridosides A, B, and C, from Rhodiola sachalinensis. Chem. Pharm. Bull. 2008, 56, 695–700. [Google Scholar] [CrossRef] [Green Version]
- Alshehri, S.; Imam, S.S. Rosinidin Attenuates Lipopolysaccharide-Induced Memory Impairment in Rats: Possible Mechanisms of Action Include Antioxidant and Anti-Inflammatory Effects. Biomolecules 2021, 11, 1747. [Google Scholar] [CrossRef] [PubMed]
- Dhadde, S.B.; Nagakannan, P.; Roopesh, M.; Anand Kumar, S.R.; Thippeswamy, B.S.; Veerapur, V.P.; Badami, S. Effect of embelin against 3-nitropropionic acid-induced Huntington’s disease in rats. Biomed. Pharmacother. 2016, 77, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Alshehri, S.; Al-Abbasi, F.A.; Ghoneim, M.M.; Imam, S.S.; Afzal, M.; Alharbi, K.S.; Nadeem, M.S.; Sayyed, N.; Kazmi, I. Anti-Huntington’s Effect of Butin in 3-Nitropropionic Acid-Treated Rats: Possible Mechanism of Action. Neurotox. Res. 2022, 40, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Mehan, S.; Monga, V.; Rani, M.; Dudi, R.; Ghimire, K. Neuroprotective effect of solanesol against 3-nitropropionic acid-induced Huntington’s disease-like behavioral, biochemical, and cellular alterations: Restoration of coenzyme-Q10-mediated mitochondrial dysfunction. Indian J. Pharmacol. 2018, 50, 309–319. [Google Scholar] [CrossRef]
- Sandhir, R.; Mehrotra, A.; Kamboj, S.S. Lycopene prevents 3-nitropropionic acid-induced mitochondrial oxidative stress and dysfunctions in nervous system. Neurochem. Int. 2010, 57, 579–587. [Google Scholar] [CrossRef]
- Wollenman, L.C.; Ploeg, M.R.V.; Miller, M.L.; Zhang, Y.; Bazil, J.N. The effect of respiration buffer composition on mitochondrial metabolism and function. PLoS ONE. 2017, 12, e0187523. [Google Scholar] [CrossRef] [Green Version]
- George, E. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar]
- Aebi, H.; Wyss, S.R.; Scherz, B.; Skvaril, F. Heterogeneity of erythrocyte catalase II. Isolation and characterization of normal and variant erythrocyte catalase and their subunits. Eur. J. Biochem. 1974, 48, 137–145. [Google Scholar] [CrossRef]
- Okaichi, Y.; Ishikura, Y.; Akimoto, K.; Kawashima, H.; Toyoda-Ono, Y.; Kiso, Y.; Okaichi, H. Arachidonic acid improves aged rats’ spatial cognition. Physiology & behavior 2005, 84, 617–623. [Google Scholar]
- Farias, J.G.; Puebla, M.; Acevedo, A.; Tapia, P.J.; Gutierrez, E.; Zepeda, A.; Calaf, G.; Juantok, C.; Reyes, J.G. Oxidative stress in rat testis and epididymis under intermittent hypobaric hypoxia: Protective role of ascorbate supplementation. J. Androl. 2010, 31, 314–321. [Google Scholar] [CrossRef]
- Etemad, A.; Sheikhzadeh, F.; Ahmadiasl, N. Evaluation of brain-derived neurotrophic factor in diabetic rats. Neurol. Res. 2015, 37, 217–222. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.; Ohnishi, T.; Winter, D.B.; Wu, J.T. Biochemical and EPR probes for structure-function studies of iron sulfur centers of succinate dehydrogenase. Adv. Exp. Med. Biol. 1976, 74, 182–227. [Google Scholar] [CrossRef] [PubMed]
- Pagel, P.; Blome, J.; Wolf, H.U. High-performance liquid chromatographic separation and measurement of various biogenic compounds possibly involved in the pathomechanism of Parkinson’s disease. J. Chromatogr. B Biomed. Sci. Appl. 2000, 746, 297–304. [Google Scholar] [CrossRef]
- Heinrikson, R.L.; Meredith, S.C. Amino acid analysis by reverse-phase high-performance liquid chromatography: Precolumn derivatization with phenylisothiocyanate. Anal. Biochem. 1984, 136, 65–74. [Google Scholar] [CrossRef]
- Sastry, K.V.H.; Moudgal, R.P.; Mohan, J.; Tyagi, J.S.; Rao, G.S. Spectrophotometric determination of serum nitrite and nitrate by copper-cadmium alloy. Anal. Biochem. 2002, 306, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Johri, A.; Beal, M.F. Antioxidants in Huntington’s disease. Biochim. Biophys. Acta 2012, 1822, 664–674. [Google Scholar] [CrossRef] [Green Version]
- Túnez, I.; Tasset, I.; Pérez-De La Cruz, V.; Santamaría, A. 3-Nitropropionic acid as a tool to study the mechanisms involved in Huntington’s disease: Past, present and future. Molecules 2010, 15, 878–916. [Google Scholar] [CrossRef] [Green Version]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease BT—Polyglutamine Disorders; Nóbrega, C., Pereira de Almeida, L., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 59–83. [Google Scholar] [CrossRef]
- Ross, C.A.; Reilmann, R. E1 Diagnostic criteria for huntington’s disease based on natural history. J. Neurol. Neurosurg. Psychiatry 2016, 87, A45. [Google Scholar] [CrossRef]
- Kaur, N.; Jamwal, S.; Deshmukh, R.; Gauttam, V.; Kumar, P. Beneficial effect of rice bran extract against 3-nitropropionic acid induced experimental Huntington’s disease in rats. Toxicol. Rep. 2015, 2, 1222–1232. [Google Scholar] [CrossRef] [Green Version]
- Brouillet, E.; Jacquard, C.; Bizat, N.; Blum, D. 3-Nitropropionic acid: A mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J. Neurochem. 2005, 95, 1521–1540. [Google Scholar] [CrossRef] [PubMed]
- Lum, P.T.; Sekar, M.; Gan, S.H.; Bonam, S.R.; Shaikh, M.F. Protective effect of natural products against Huntington’s disease: An overview of scientific evidence and understanding their mechanism of action. ACS Chem. Neurosci. 2021, 12, 391–418. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, H.N.; El-Tanbouly, D.M.; Zaki, H.F. Topiramate mitigates 3-nitropropionic acid-induced striatal neurotoxicity via modulation of AMPA receptors. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2018, 118, 227–234. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog. Neurobiol. 2007, 81, 294–330. [Google Scholar] [CrossRef]
- Jamwal, S.; Kumar, P. Spermidine ameliorates 3-nitropropionic acid (3-NP)-induced striatal toxicity: Possible role of oxidative stress, neuroinflammation, and neurotransmitters. Physiol. Behav. 2016, 155, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, J.; Yang, L.; Zhou, S.-M.; Guan, S.-Y.; Yang, L.-K.; Shi, Q.-X.; Zhao, M.-G.; Yang, Q. Effect of Praeruptorin C on 3-nitropropionic acid induced Huntington’s disease-like symptoms in mice. Biomed. Pharmacother. 2017, 86, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Aliaghaei, A.; Boroujeni, M.E.; Ahmadi, H.; Bayat, A.-H.; Tavirani, M.R.; Abdollahifar, M.A.; Pooyafar, M.H.; Mansouri, V. Dental pulp stem cell transplantation ameliorates motor function and prevents cerebellar atrophy in rat model of cerebellar ataxia. Cell Tissue Res. 2019, 376, 179–187. [Google Scholar] [CrossRef]
- Khan, A.; Jamwal, S.; Bijjem, K.R.V.; Prakash, A.; Kumar, P. Neuroprotective effect of hemeoxygenase-1/glycogen synthase kinase-3β modulators in 3-nitropropionic acid-induced neurotoxicity in rats. Neuroscience 2015, 287, 66–77. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afzal, M.; Sayyed, N.; Alharbi, K.S.; Alzarea, S.I.; Alshammari, M.S.; Alomar, F.A.; Alenezi, S.K.; Quazi, A.M.; Alzarea, A.I.; Kazmi, I. Anti-Huntington’s Effect of Rosiridin via Oxidative Stress/AchE Inhibition and Modulation of Succinate Dehydrogenase, Nitrite, and BDNF Levels against 3-Nitropropionic Acid in Rodents. Biomolecules 2022, 12, 1023. https://doi.org/10.3390/biom12081023
Afzal M, Sayyed N, Alharbi KS, Alzarea SI, Alshammari MS, Alomar FA, Alenezi SK, Quazi AM, Alzarea AI, Kazmi I. Anti-Huntington’s Effect of Rosiridin via Oxidative Stress/AchE Inhibition and Modulation of Succinate Dehydrogenase, Nitrite, and BDNF Levels against 3-Nitropropionic Acid in Rodents. Biomolecules. 2022; 12(8):1023. https://doi.org/10.3390/biom12081023
Chicago/Turabian StyleAfzal, Muhammad, Nadeem Sayyed, Khalid Saad Alharbi, Sami I. Alzarea, Mohammed Salem Alshammari, Fadhel A. Alomar, Sattam Khulaif Alenezi, Anwarulabedin Mohsin Quazi, Abdulaziz I. Alzarea, and Imran Kazmi. 2022. "Anti-Huntington’s Effect of Rosiridin via Oxidative Stress/AchE Inhibition and Modulation of Succinate Dehydrogenase, Nitrite, and BDNF Levels against 3-Nitropropionic Acid in Rodents" Biomolecules 12, no. 8: 1023. https://doi.org/10.3390/biom12081023
APA StyleAfzal, M., Sayyed, N., Alharbi, K. S., Alzarea, S. I., Alshammari, M. S., Alomar, F. A., Alenezi, S. K., Quazi, A. M., Alzarea, A. I., & Kazmi, I. (2022). Anti-Huntington’s Effect of Rosiridin via Oxidative Stress/AchE Inhibition and Modulation of Succinate Dehydrogenase, Nitrite, and BDNF Levels against 3-Nitropropionic Acid in Rodents. Biomolecules, 12(8), 1023. https://doi.org/10.3390/biom12081023