
Isolation, Characterization, and Autophagy Function of BECN1-Splicing Isoforms in Cancer Cells

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Cloning of BECN1 mRNA Isoforms
2.3. Transient Transfections and Treatments
2.4. Western Blotting Analysis
2.5. Immunofluorescence
2.6. Co-Immunoprecipitation
2.7. Antibodies
2.8. Statistical Analysis
3. Results
3.1. Cloning of BECN1 Transcript Variants from Ovarian Cancer Cells
3.2. Sequencing Analysis of BECN1 Transcript Variants
3.3. BECN1 Isoforms Show Different Alterations in Their Interactions with VPS34 and BCL2 While Maintaining Their Ability to Bind to ATG14
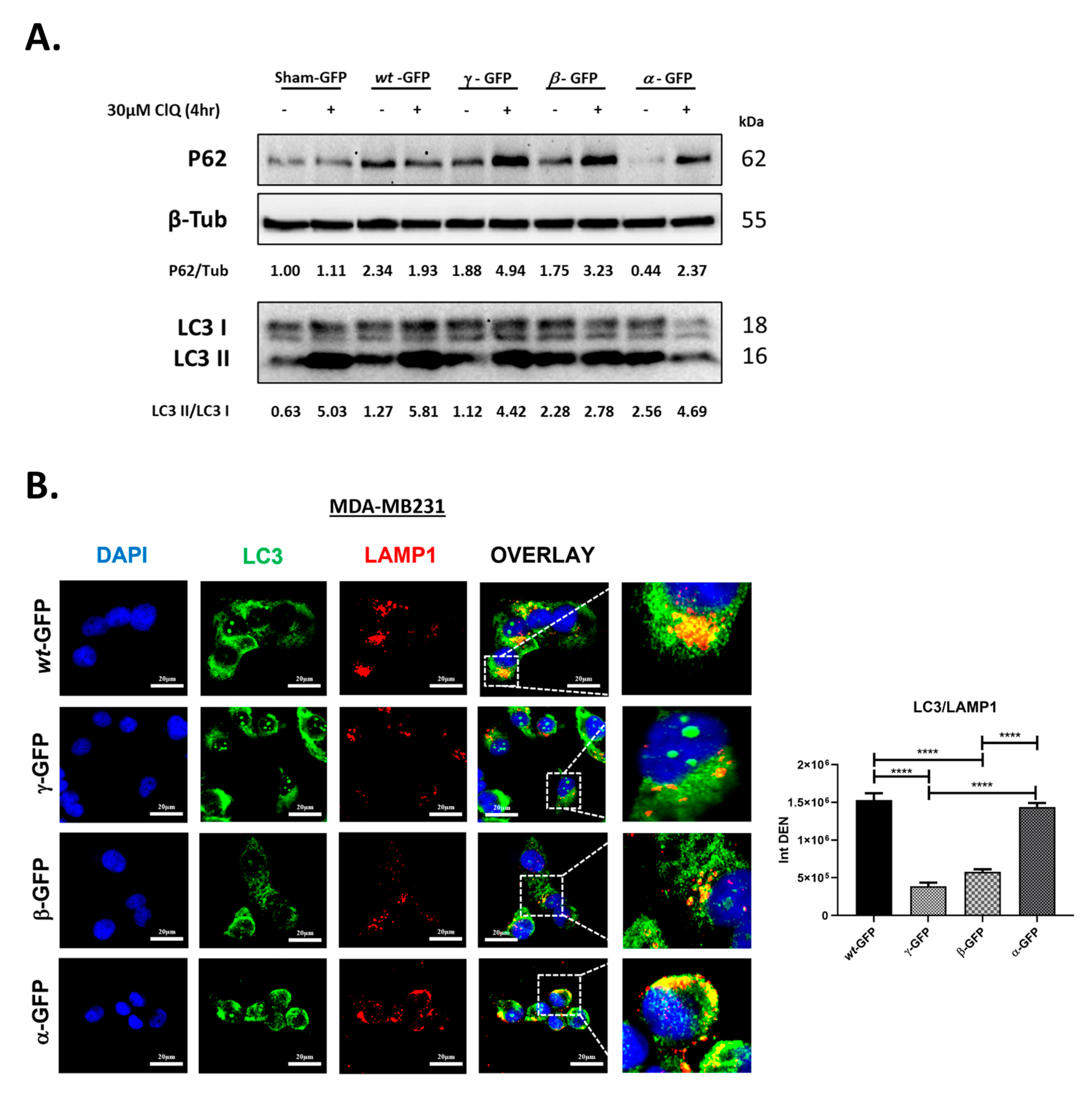
3.4. BECN1 Isoforms Have Idiosyncratic Effects on Autophagy
3.5. BECN1-α Interacts with PRKN Facilitating Mitophagy
4. Discussions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of Autophagy and Inhibition of Tumorigenesis by Beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Aita, V.M.; Liang, X.H.; Murty, V.V.; Pincus, D.L.; Yu, W.; Cayanis, E.; Kalachikov, S.; Gilliam, T.C.; Levine, B. Cloning and Genomic Organization of Beclin 1, a Candidate Tumor Suppressor Gene on Chromosome 17q21. Genomics 1999, 59, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of Tumorigenesis by Heterozygous Disruption of the Beclin 1 Autophagy Gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an Autophagy Gene Essential for Early Embryonic Development, Is a Haploinsufficient Tumor Suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicchini, M.; Chakrabarti, R.; Kongara, S.; Price, S.; Nahar, R.; Lozy, F.; Zhong, H.; Vazquez, A.; Kang, Y.; Karantza, V. Autophagy Regulator BECN1 Suppresses Mammary Tumorigenesis Driven by WNT1 Activation and Following Parity. Autophagy 2014, 10, 2036–2052. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Sebti, S.; Titone, R.; Zhou, Y.; Isidoro, C.; Ross, T.S.; Hibshoosh, H.; Xiao, G.; Packer, M.; Xie, Y.; et al. Decreased BECN1 MRNA Expression in Human Breast Cancer Is Associated with Estrogen Receptor-Negative Subtypes and Poor Prognosis. eBioMedicine 2015, 2, 255–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, G.; Morani, F.; Nicotra, G.; Fusco, N.; Peracchio, C.; Titone, R.; Alabiso, O.; Arisio, R.; Katsaros, D.; Benedetto, C.; et al. Expression and Clinical Significance of the Autophagy Proteins BECLIN 1 and LC3 in Ovarian Cancer. Biomed Res. Int. 2014, 2014, 462658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicotra, G.; Mercalli, F.; Peracchio, C.; Castino, R.; Follo, C.; Valente, G.; Isidoro, C. Autophagy-Active Beclin-1 Correlates with Favourable Clinical Outcome in Non-Hodgkin Lymphomas. Mod. Pathol. 2010, 23, 937–950. [Google Scholar] [CrossRef]
- Salwa, A.; Ferraresi, A.; Chinthakindi, M.; Vallino, L.; Vidoni, C.; Dhanasekaran, D.N.; Isidoro, C. BECN1 and BRCA1 Deficiency Sensitizes Ovarian Cancer to Platinum Therapy and Confers Better Prognosis. Biomedicines 2021, 9, 207. [Google Scholar] [CrossRef] [PubMed]
- Oberstein, A.; Jeffrey, P.D.; Shi, Y. Crystal Structure of the Bcl-XL-Beclin 1 Peptide Complex: Beclin 1 Is a Novel BH3-Only Protein. J. Biol. Chem. 2007, 282, 13123–13132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; He, L.; Che, K.H.; Funderburk, S.F.; Pan, L.; Pan, N.; Zhang, M.; Yue, Z.; Zhao, Y. Imperfect Interface of Beclin1 Coiled-Coil Domain Regulates Homodimer and Heterodimer Formation with Atg14L and UVRAG. Nat. Commun. 2012, 3, 662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, N.N.; Kobayashi, T.; Adachi, W.; Fujioka, Y.; Ohsumi, Y.; Inagaki, F. Structure of the Novel C-Terminal Domain of Vacuolar Protein Sorting 30/Autophagy-Related Protein 6 and Its Specific Role in Autophagy. J. Biol. Chem. 2012, 287, 16256–16266. [Google Scholar] [CrossRef] [Green Version]
- Furuya, N.; Yu, J.; Byfield, M.; Pattingre, S.; Levine, B. The Evolutionarily Conserved Domain of Beclin 1 Is Required for Vps34 Binding, Autophagy and Tumor Suppressor Function. Autophagy 2005, 1, 46–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, M.B.; Dhamija, S. Beclin 1 Phosphorylation-at the Center of Autophagy Regulation. Front. Cell Dev. Biol. 2018, 6, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Kihara, A.; Noda, T.; Ishihara, N.; Ohsumi, Y. Two Distinct Vps34 Phosphatidylinositol 3-Kinase Complexes Function in Autophagy and Carboxypeptidase Y Sorting in Saccharomyces Cerevisiae. J. Cell Biol. 2001, 152, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 Forms Two Distinct Phosphatidylinositol 3-Kinase Complexes with Mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Mizushima, N. Atg14 and UVRAG: Mutually Exclusive Subunits of Mammalian Beclin 1-PI3K Complexes. Autophagy 2009, 5, 534–536. [Google Scholar] [CrossRef] [Green Version]
- Michiorri, S.; Gelmetti, V.; Giarda, E.; Lombardi, F.; Romano, F.; Marongiu, R.; Nerini-Molteni, S.; Sale, P.; Vago, R.; Arena, G.; et al. The Parkinson-Associated Protein PINK1 Interacts with Beclin1 and Promotes Autophagy. Cell Death Differ. 2010, 17, 962–974. [Google Scholar] [CrossRef] [Green Version]
- Choubey, V.; Cagalinec, M.; Liiv, J.; Safiulina, D.; Hickey, M.A.; Kuum, M.; Liiv, M.; Anwar, T.; Eskelinen, E.-L.; Kaasik, A. BECN1 Is Involved in the Initiation of Mitophagy: It Facilitates PARK2 Translocation to Mitochondria. Autophagy 2014, 10, 1105–1119. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.W.; Jeon, J.; Go, G.; Lee, J.H.; Lee, S.H. The Dual Role of Autophagy in Cancer Development and a Therapeutic Strategy for Cancer by Targeting Autophagy. Int. J. Mol. Sci. 2021, 22, 179. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sánchez-Álvarez, M.; Lolo, F.-N.; Trionfetti, F.; Strippoli, R.; Cordani, M. Autophagy and the Lysosomal System in Cancer. Cells 2021, 10, 2752. [Google Scholar] [CrossRef] [PubMed]
- Castino, R.; Bellio, N.; Nicotra, G.; Follo, C.; Trincheri, N.F.; Isidoro, C. Cathepsin D-Bax Death Pathway in Oxidative Stressed Neuroblastoma Cells. Free Radic. Biol. Med. 2007, 42, 1305–1316. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Gómez-Sintes, R.; Boya, P. Lysosomal Membrane Permeabilization and Cell Death. Traffic 2018, 19, 918–931. [Google Scholar] [CrossRef]
- Shin, H.R.; Zoncu, R. The Lysosome at the Intersection of Cellular Growth and Destruction. Dev. Cell 2020, 54, 226–238. [Google Scholar] [CrossRef]
- Rubinstein, A.D.; Kimchi, A. Life in the Balance-a Mechanistic View of the Crosstalk between Autophagy and Apoptosis. J. Cell Sci. 2012, 125, 5259–5268. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Gao, P.; Zhang, J. Crosstalk between Autophagy and Apoptosis: Potential and Emerging Therapeutic Targets for Cardiac Diseases. Int. J. Mol. Sci. 2016, 17, 332. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep Surveying of Alternative Splicing Complexity in the Human Transcriptome by High-Throughput Sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Matlin, A.J.; Clark, F.; Smith, C.W.J. Understanding Alternative Splicing: Towards a Cellular Code. Nat. Rev. Mol. Cell Biol. 2005, 6, 386–398. [Google Scholar] [CrossRef]
- Li, D.; McIntosh, C.S.; Mastaglia, F.L.; Wilton, S.D.; Aung-Htut, M.T. Neurodegenerative Diseases: A Hotbed for Splicing Defects and the Potential Therapies. Transl. Neurodegener. 2021, 10, 16. [Google Scholar] [CrossRef]
- Pajares, M.J.; Ezponda, T.; Catena, R.; Calvo, A.; Pio, R.; Montuenga, L.M. Alternative Splicing: An Emerging Topic in Molecular and Clinical Oncology. Lancet Oncol. 2007, 8, 349–357. [Google Scholar] [CrossRef]
- Li, Y.; Sahni, N.; Pancsa, R.; McGrail, D.J.; Xu, J.; Hua, X.; Coulombe-Huntington, J.; Ryan, M.; Tychhon, B.; Sudhakar, D.; et al. Revealing the Determinants of Widespread Alternative Splicing Perturbation in Cancer. Cell Rep. 2017, 21, 798–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahles, A.; Lehmann, K.-V.; Toussaint, N.C.; Hüser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Cancer Genome Atlas Research Network; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8705 Patients. Cancer Cell 2018, 34, 211–224.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Rodríguez, P.; Klionsky, D.J.; Joseph, B. Autophagy Regulation by RNA Alternative Splicing and Implications in Human Diseases. Nat. Commun. 2022, 13, 2735. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.-N.; Liu, Q.-Q.; Zhang, S.-P.; Yuan, N.; Cao, Y.; Cai, J.-Y.; Lin, W.-W.; Xu, F.; Wang, Z.-J.; Chen, B.; et al. Alternative Messenger RNA Splicing of Autophagic Gene Beclin 1 in Human B-Cell Acute Lymphoblastic Leukemia Cells. Asian Pac. J. Cancer Prev. 2014, 15, 2153–2158. [Google Scholar] [CrossRef] [Green Version]
- Cheng, B.; Xu, A.; Qiao, M.; Wu, Q.; Wang, W.; Mei, Y.; Wu, M. BECN1s, a Short Splice Variant of BECN1, Functions in Mitophagy. Autophagy 2015, 11, 2048–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Marín, A. Characterization and Prediction of Alternative Splice Sites. Gene 2006, 366, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Thanaraj, T.A.; Clark, F. Human GC-AG Alternative Intron Isoforms with Weak Donor Sites Show Enhanced Consensus at Acceptor Exon Positions. Nucleic Acids Res. 2001, 29, 2581–2593. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Levine, B. The Autophagy Effector Beclin 1: A Novel BH3-Only Protein. Oncogene 2008, 27 (Suppl. 1), S137–S148. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.F.; Perugini, M.A.; Pettikiriarachchi, A.; Evangelista, M.; Keizer, D.W.; Yao, S.; Fairlie, W.D. The BECN1 N-Terminal Domain Is Intrinsically Disordered. Autophagy 2016, 12, 460–471. [Google Scholar] [CrossRef] [Green Version]
- Strappazzon, F.; Vietri-Rudan, M.; Campello, S.; Nazio, F.; Florenzano, F.; Fimia, G.M.; Piacentini, M.; Levine, B.; Cecconi, F. Mitochondrial BCL-2 Inhibits AMBRA1-Induced Autophagy. EMBO J. 2011, 30, 1195–1208. [Google Scholar] [CrossRef] [PubMed]
- Lindmo, K.; Brech, A.; Finley, K.D.; Gaumer, S.; Contamine, D.; Rusten, T.E.; Stenmark, H. The PI 3-Kinase Regulator Vps15 Is Required for Autophagic Clearance of Protein Aggregates. Autophagy 2008, 4, 500–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, S.M.; Wrobel, L.; Rubinsztein, D.C. Post-Translational Modifications of Beclin 1 Provide Multiple Strategies for Autophagy Regulation. Cell Death Differ. 2019, 26, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T. How to Interpret LC3 Immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th Edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Kumar, A.; Shaha, C. SESN2 Facilitates Mitophagy by Helping Parkin Translocation through ULK1 Mediated Beclin1 Phosphorylation. Sci. Rep. 2018, 8, 615. [Google Scholar] [CrossRef]
- Zhang, L.; Zheng, X.-C.; Huang, Y.-Y.; Ge, Y.-P.; Sun, M.; Chen, W.-L.; Liu, W.-B.; Li, X.-F. Carbonyl Cyanide 3-Chlorophenylhydrazone Induced the Imbalance of Mitochondrial Homeostasis in the Liver of Megalobrama Amblycephala: A Dynamic Study. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2021, 244, 109003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in Cell Death, Autophagy, and Mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [Green Version]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, Å.B. Microtubule-Associated Protein 1 Light Chain 3 (LC3) Interacts with Bnip3 Protein to Selectively Remove Endoplasmic Reticulum and Mitochondria via Autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.-X.; Yin, X.-M. Mitophagy: Mechanisms, Pathophysiological Roles, and Analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [Green Version]
- Van Humbeeck, C.; Cornelissen, T.; Hofkens, H.; Mandemakers, W.; Gevaert, K.; de Strooper, B.; Vandenberghe, W. Parkin Interacts with Ambra1 to Induce Mitophagy. J. Neurosci. 2011, 31, 10249–10261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, Á.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.-C.; et al. Disruption of the Beclin 1-BCL2 Autophagy Regulatory Complex Promotes Longevity in Mice. Nature 2018, 558, 136–140. [Google Scholar] [CrossRef]
- Tripathi, R.; Ash, D.; Shaha, C. Beclin-1-P53 Interaction Is Crucial for Cell Fate Determination in Embryonal Carcinoma Cells. J. Cell. Mol. Med. 2014, 18, 2275–2286. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-A.; Huang, D.-Y.; Lin, W.-W. Beclin-1-Independent Autophagy Positively Regulates Internal Ribosomal Entry Site-Dependent Translation of Hypoxia-Inducible Factor 1α under Nutrient Deprivation. Oncotarget 2014, 5, 7525–7539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.-Y.; Du, Z.-X.; Meng, X.; Zong, Z.-H.; Wang, H.-Q. Beclin 1 Enhances Proteasome Inhibition-Mediated Cytotoxicity of Thyroid Cancer Cells in Macroautophagy-Independent Manner. J. Clin. Endocrinol. Metab. 2013, 98, E217–E226. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maheshwari, C.; Vidoni, C.; Titone, R.; Castiglioni, A.; Lora, C.; Follo, C.; Isidoro, C. Isolation, Characterization, and Autophagy Function of BECN1-Splicing Isoforms in Cancer Cells. Biomolecules 2022, 12, 1069. https://doi.org/10.3390/biom12081069
Maheshwari C, Vidoni C, Titone R, Castiglioni A, Lora C, Follo C, Isidoro C. Isolation, Characterization, and Autophagy Function of BECN1-Splicing Isoforms in Cancer Cells. Biomolecules. 2022; 12(8):1069. https://doi.org/10.3390/biom12081069
Chicago/Turabian StyleMaheshwari, Chinmay, Chiara Vidoni, Rossella Titone, Andrea Castiglioni, Claudia Lora, Carlo Follo, and Ciro Isidoro. 2022. "Isolation, Characterization, and Autophagy Function of BECN1-Splicing Isoforms in Cancer Cells" Biomolecules 12, no. 8: 1069. https://doi.org/10.3390/biom12081069
APA StyleMaheshwari, C., Vidoni, C., Titone, R., Castiglioni, A., Lora, C., Follo, C., & Isidoro, C. (2022). Isolation, Characterization, and Autophagy Function of BECN1-Splicing Isoforms in Cancer Cells. Biomolecules, 12(8), 1069. https://doi.org/10.3390/biom12081069