

Novel Hominid-Specific IAPP Isoforms: Potential Biomarkers of Early Alzheimer’s Disease and Inhibitors of Amyloid Formation

 , ,
, ,  and
and 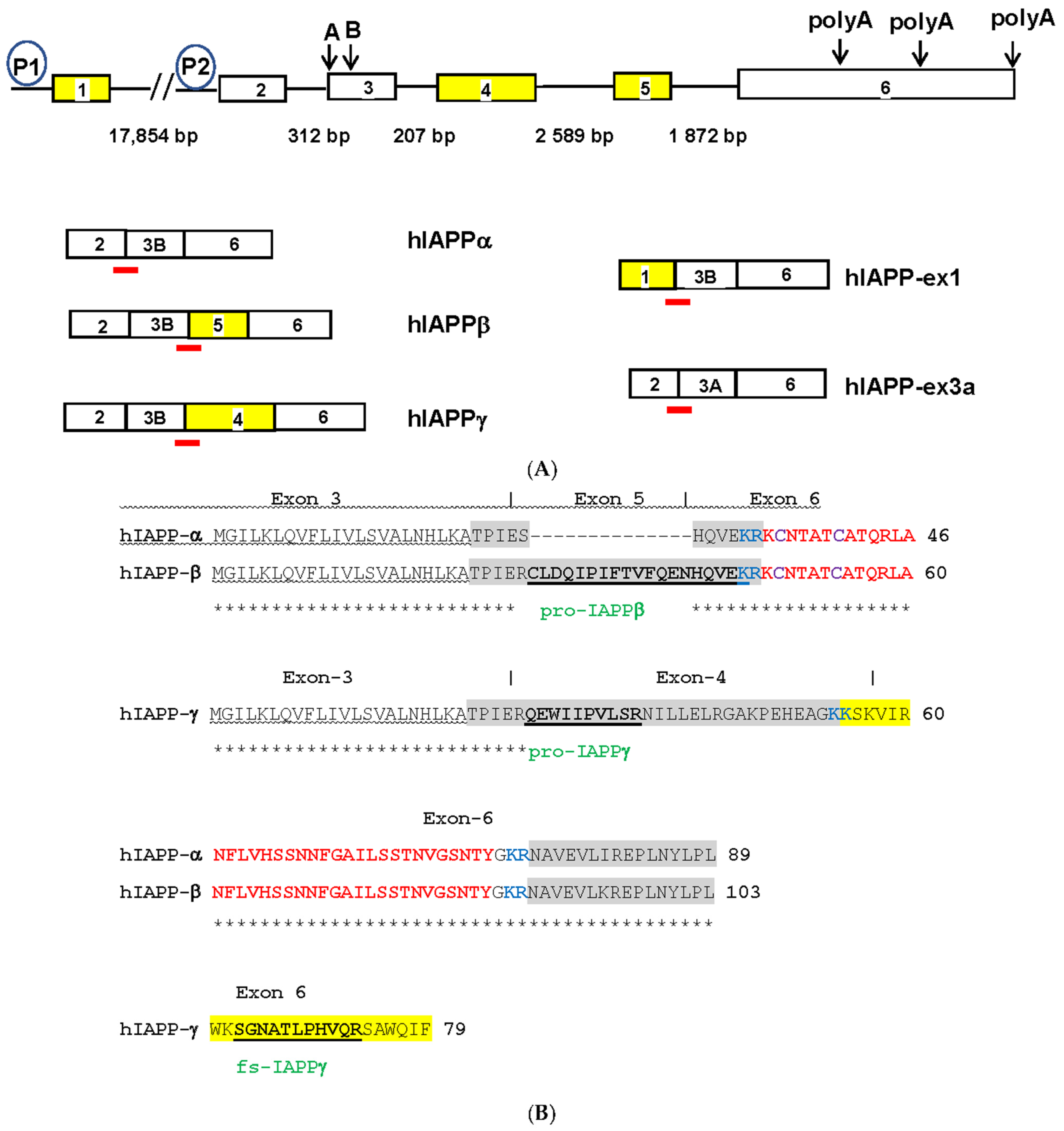{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Source of Tissue Samples
2.2. Participants
2.3. Bioinformatics and Sanger Sequencing
2.4. RNA Isolation, cDNA Synthesis, RT-qPCR, and ELISA
2.5. Selected Reaction Monitoring (SRM)-MS Assay
2.6. Thioflavin T In Vitro Assay for IAPP and Aβ Amyloid Formation
2.7. Statistical Data Analysis
3. Results
3.1. Novel Hominid-Specific Peptides Derived from hIAPP Isoforms as Potential Diagnostic and Therapeutic Targets for AD and T2DM
3.1.1. Identification of hIAPPβ and hIAPPγ Isoforms
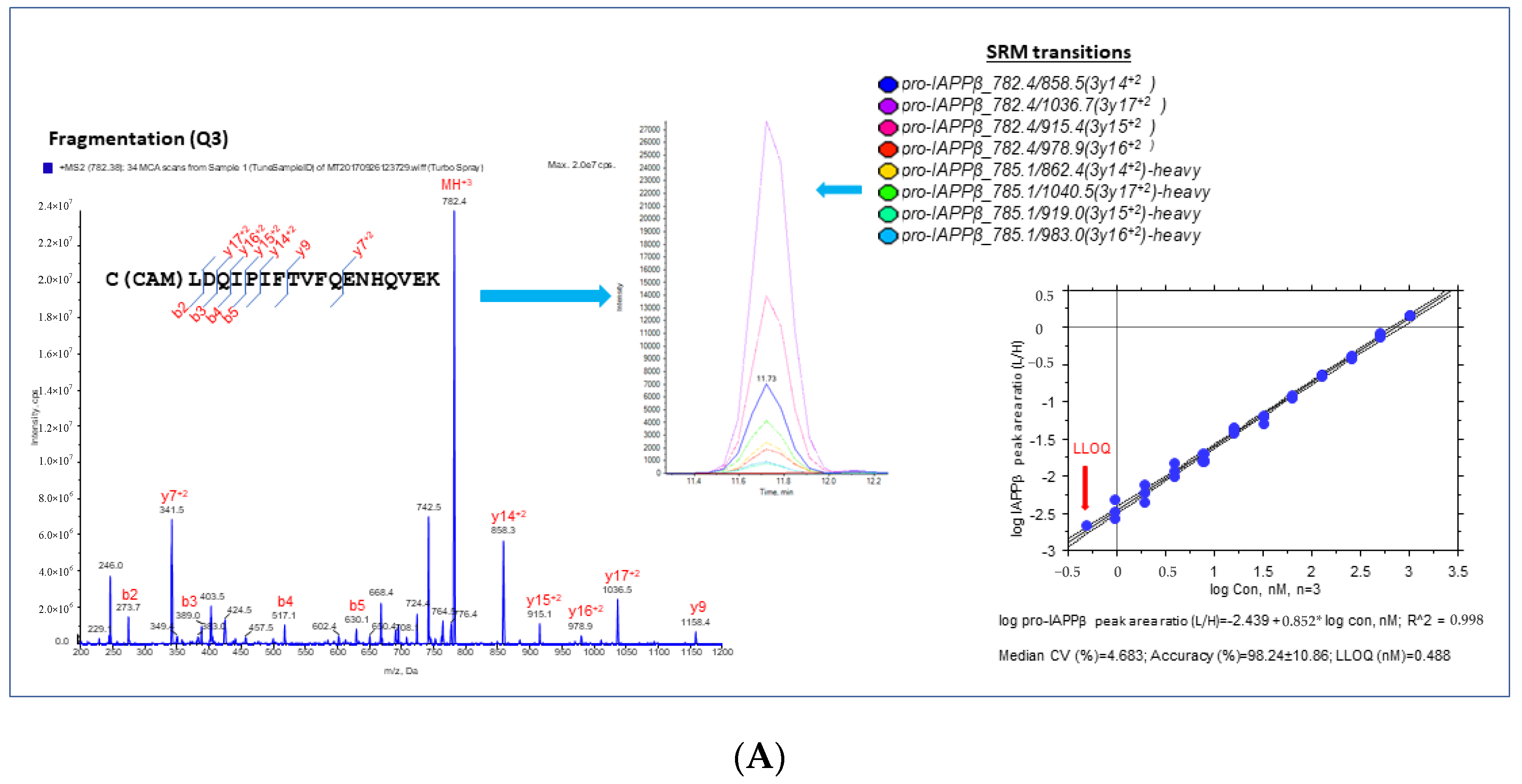
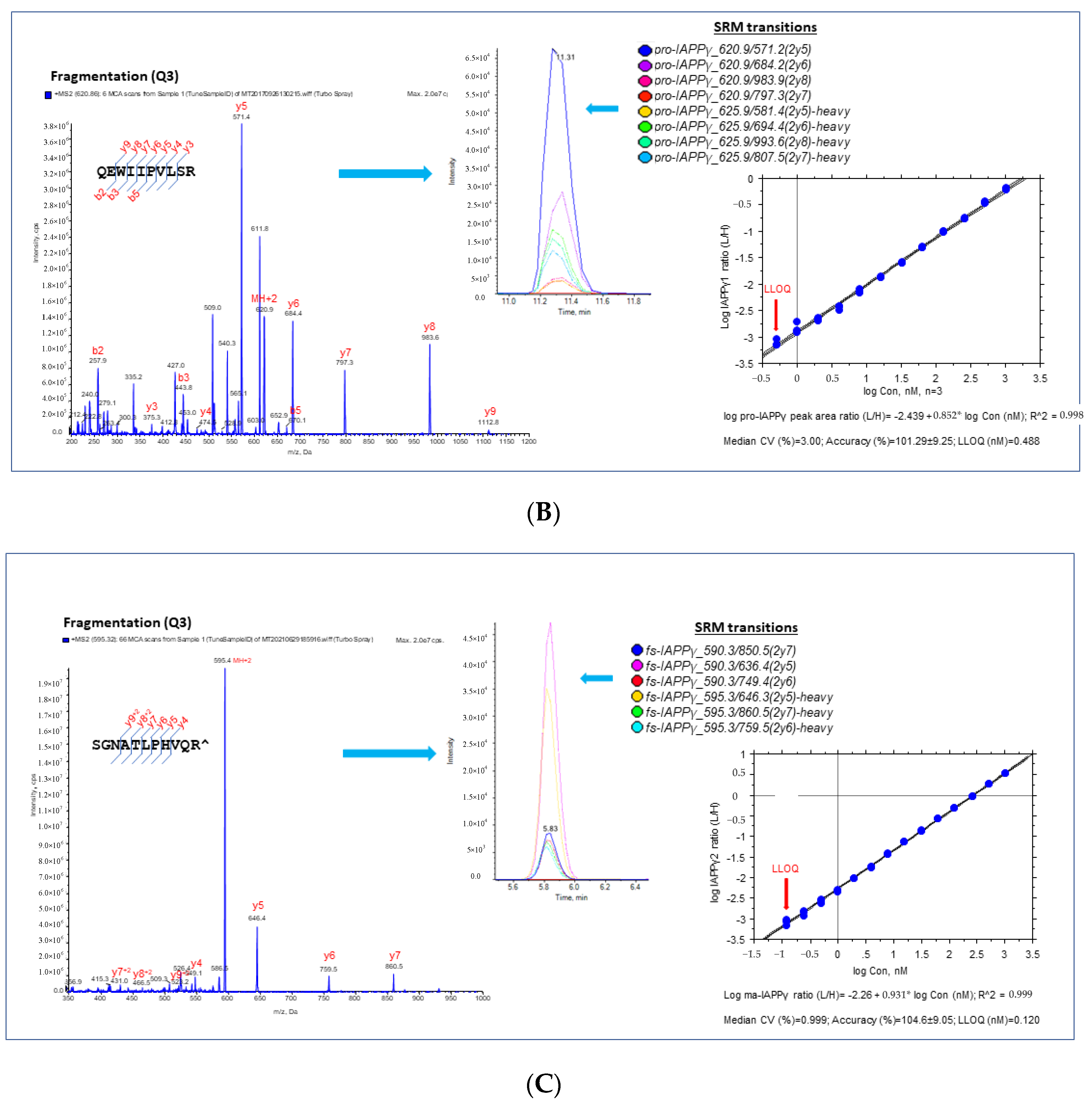
3.1.2. Validation and Quantification of hIAPPβ and hIAPPγ by MS-Based SRM Assay
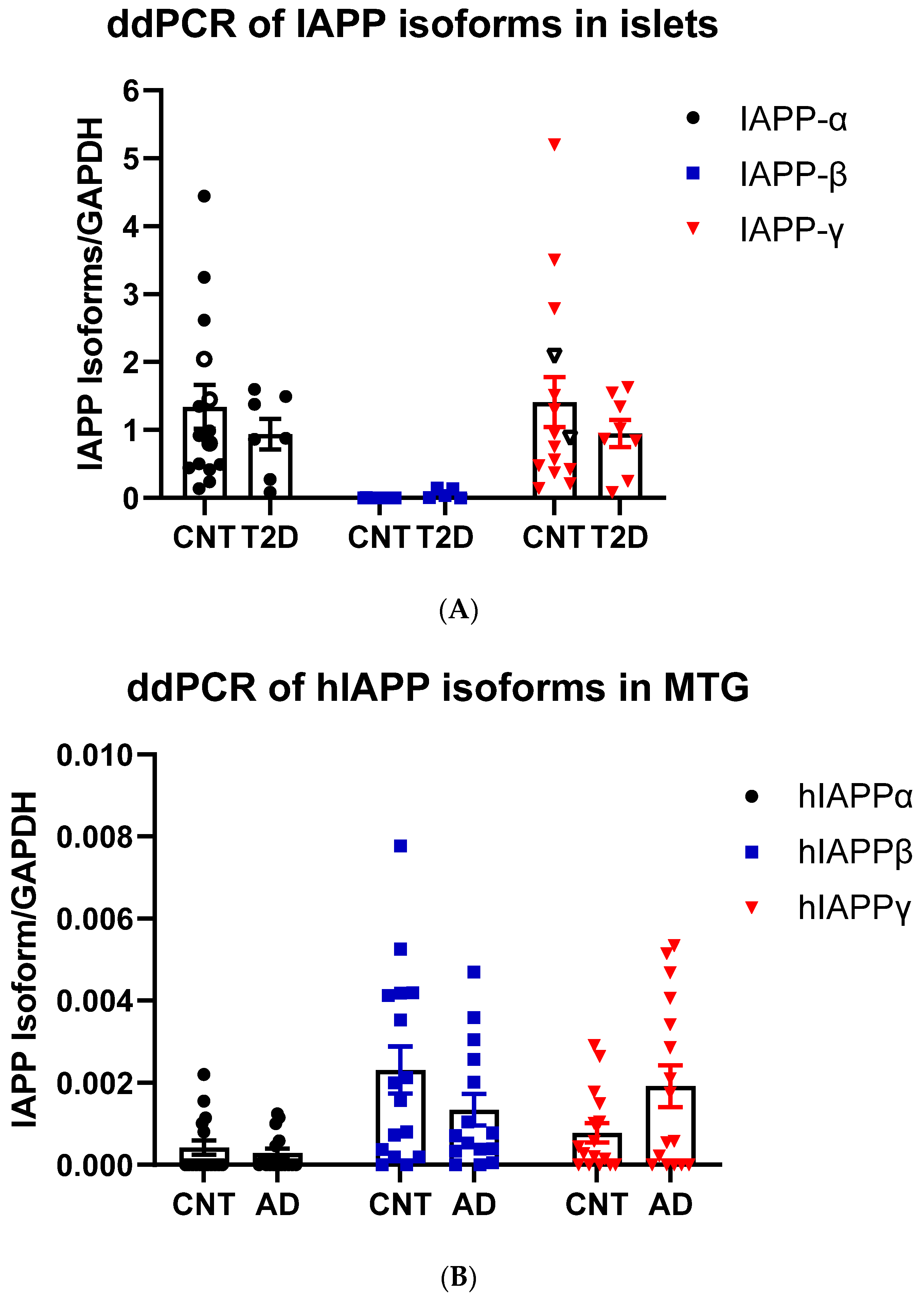
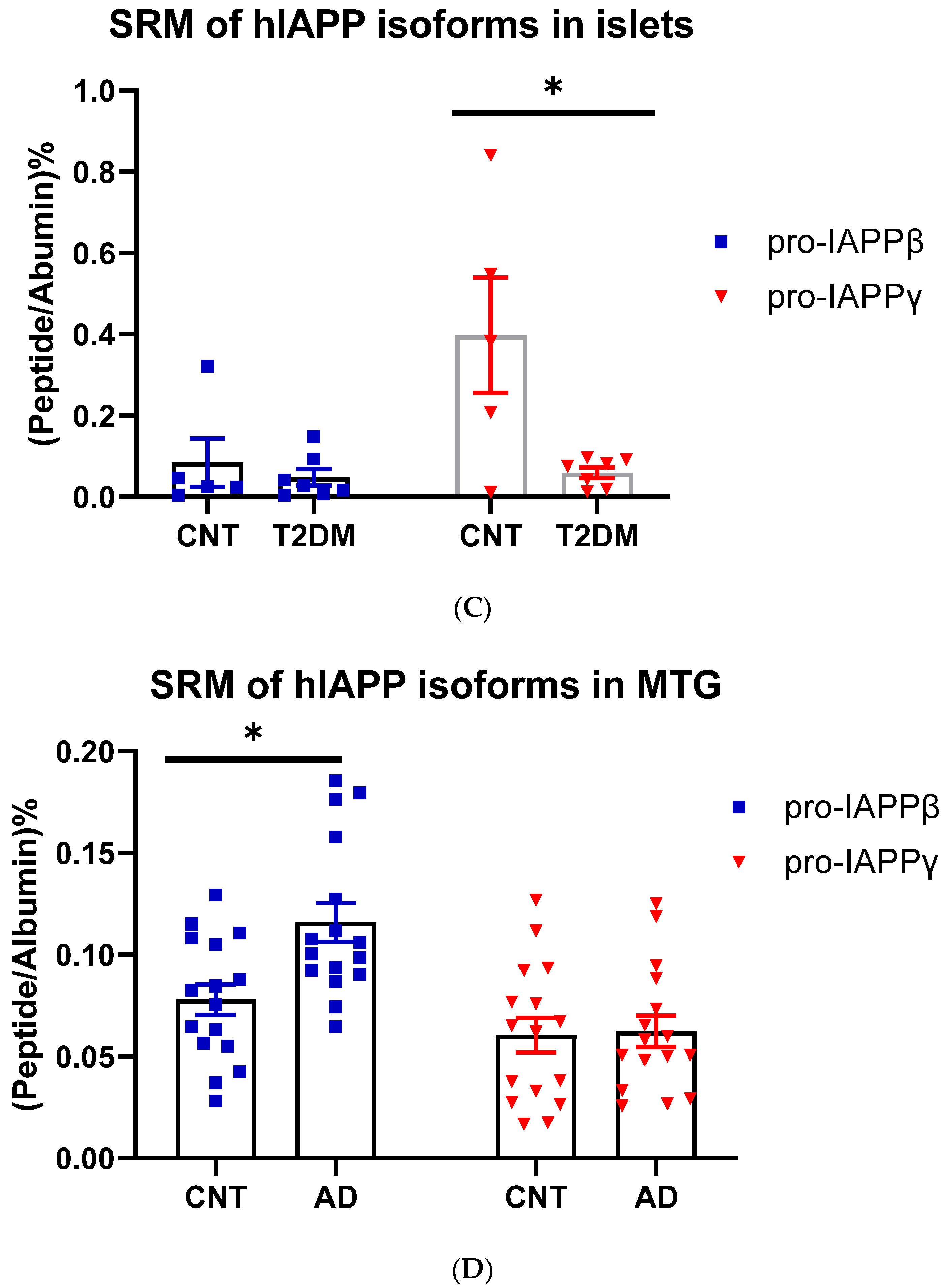
3.1.3. hIAPPβ and hIAPPγ in T2DM Islets and AD Cerebrum
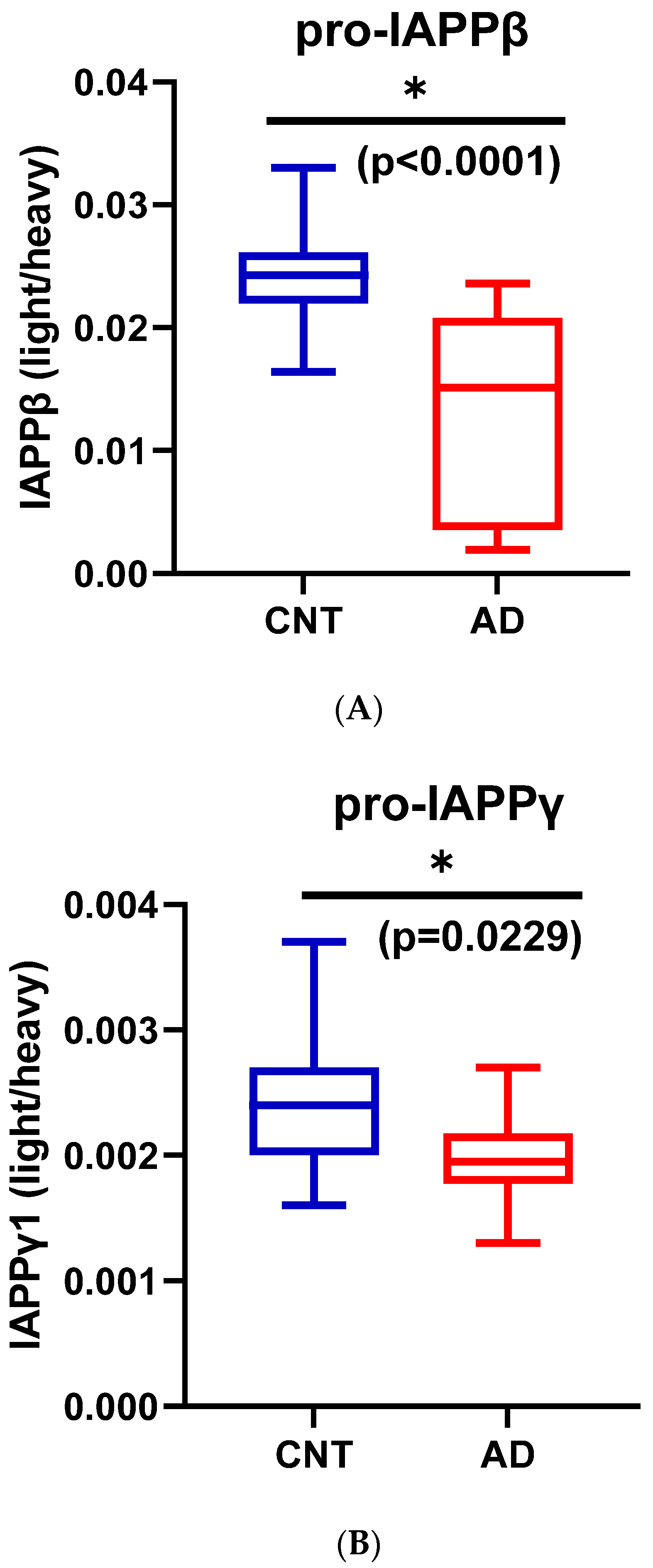
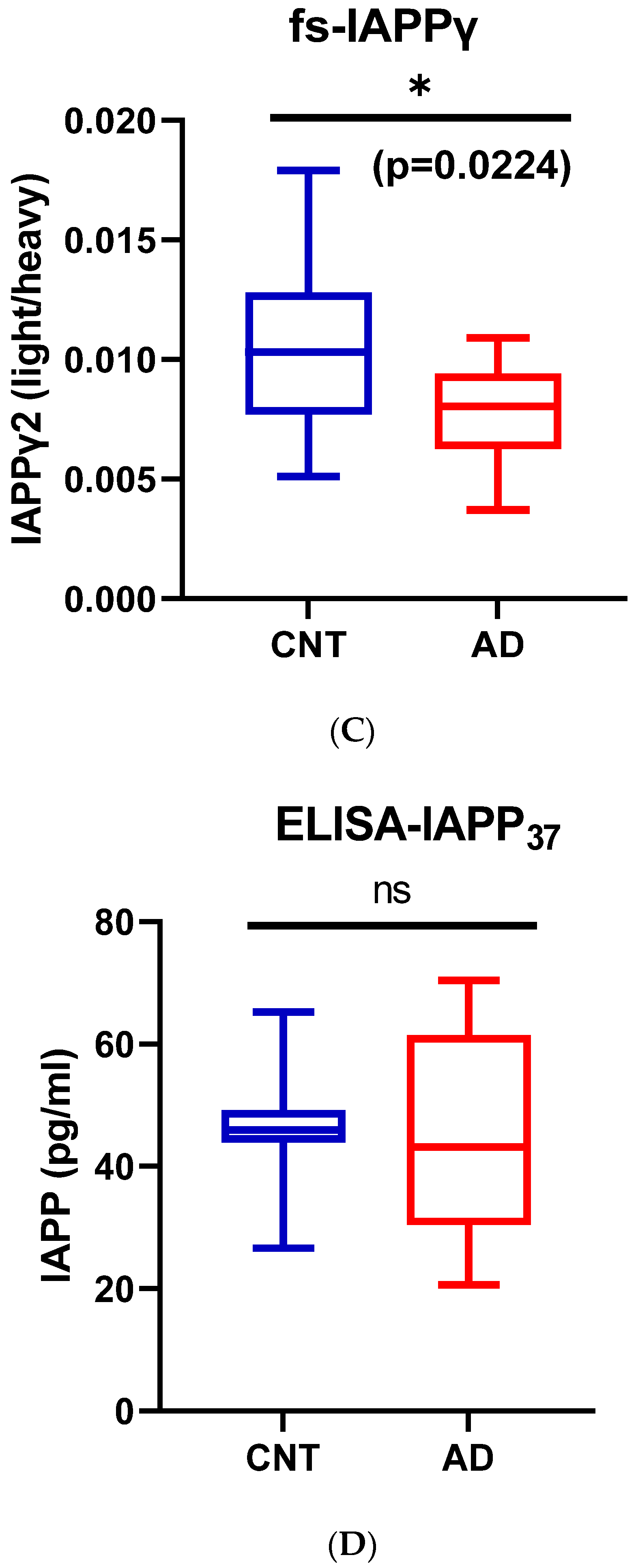
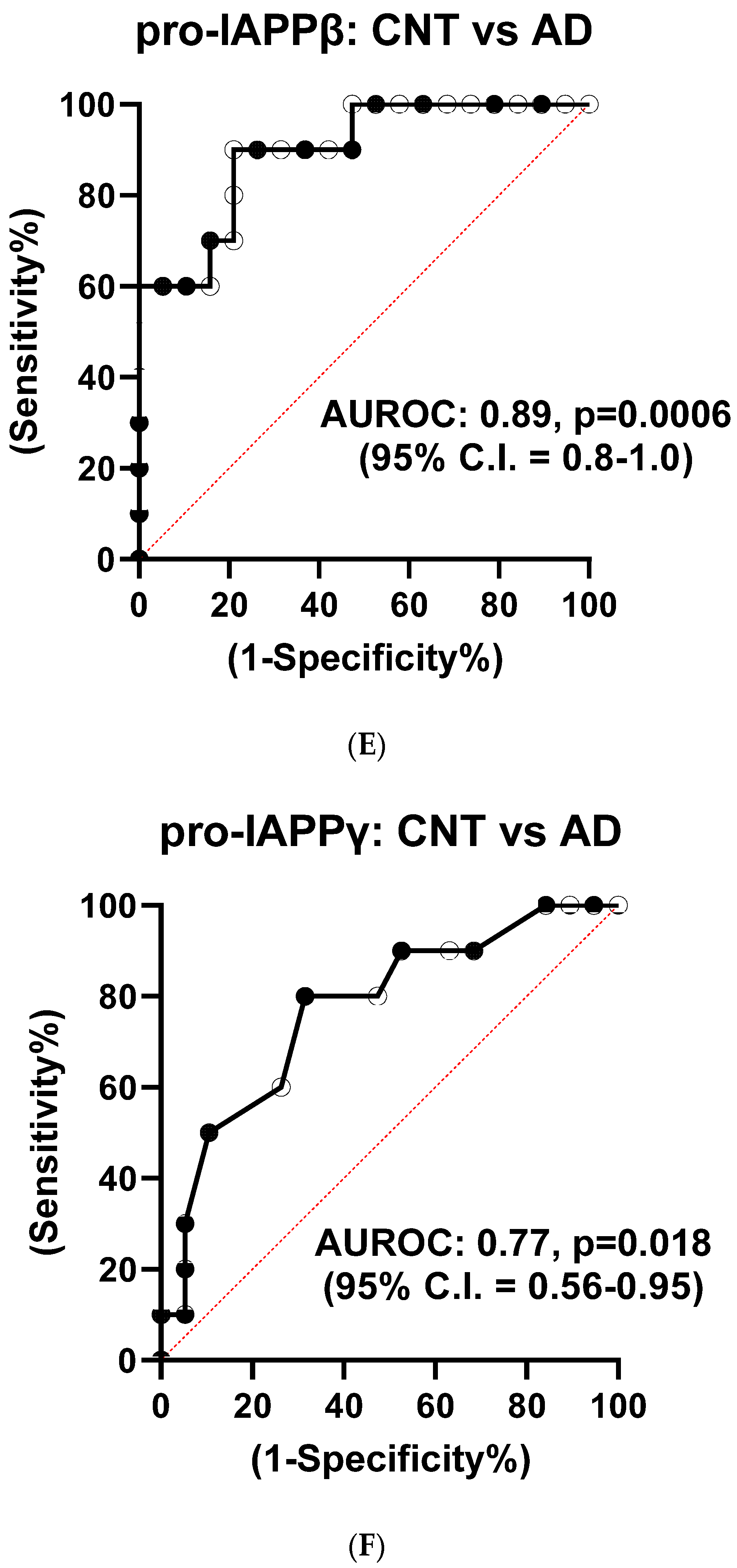
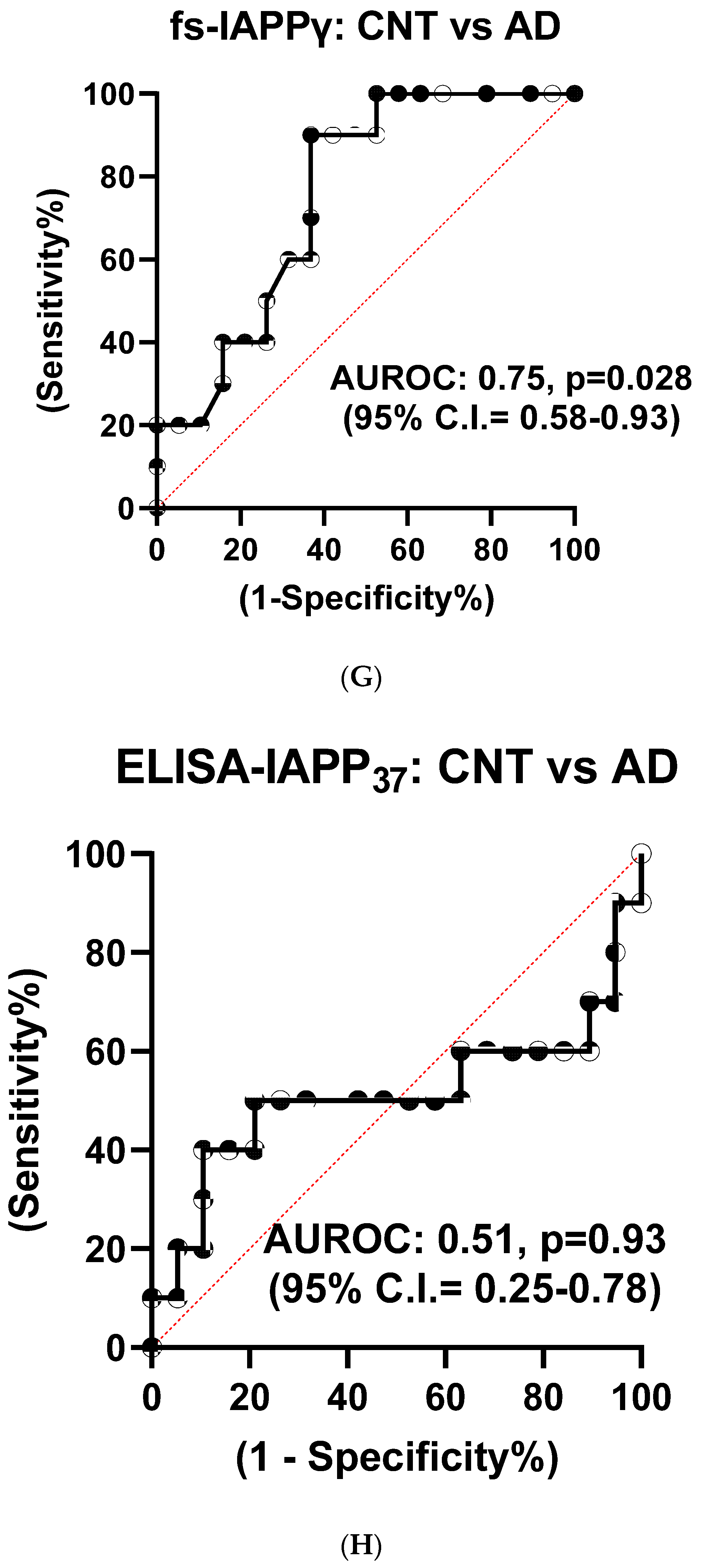
3.1.4. Reduction of hIAPPβ and hIAPPγ Peptides in AD Plasma Samples
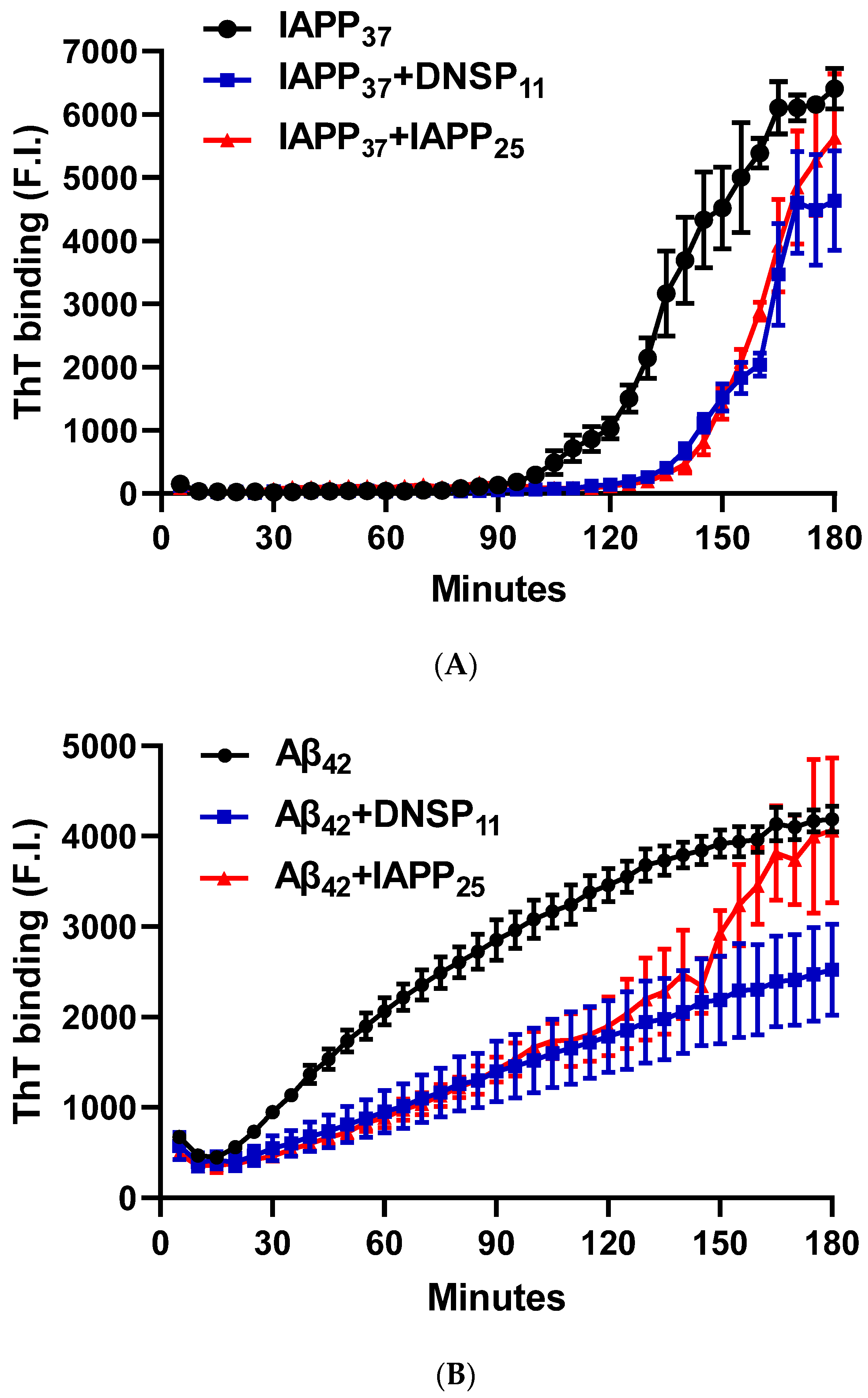
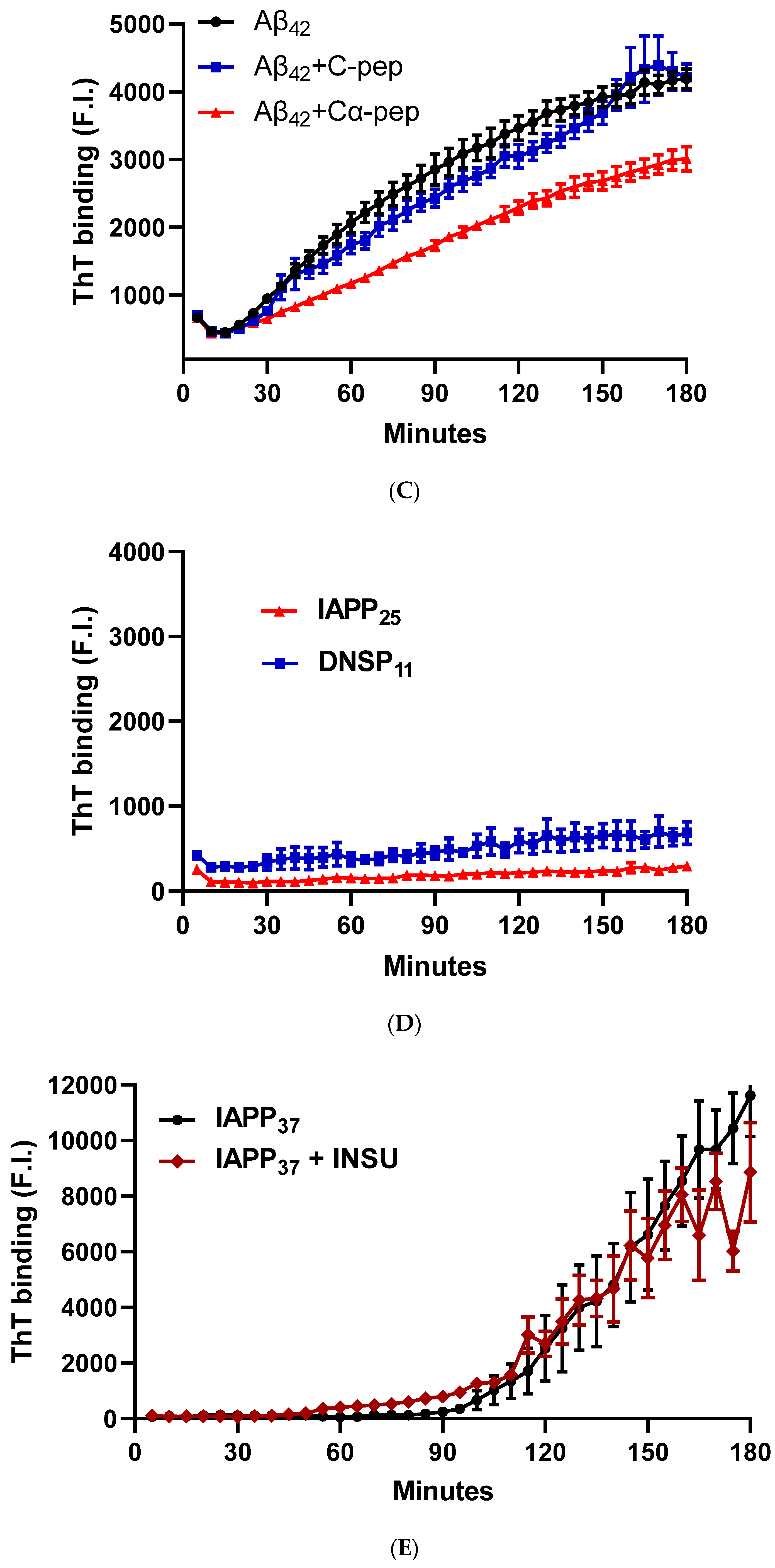
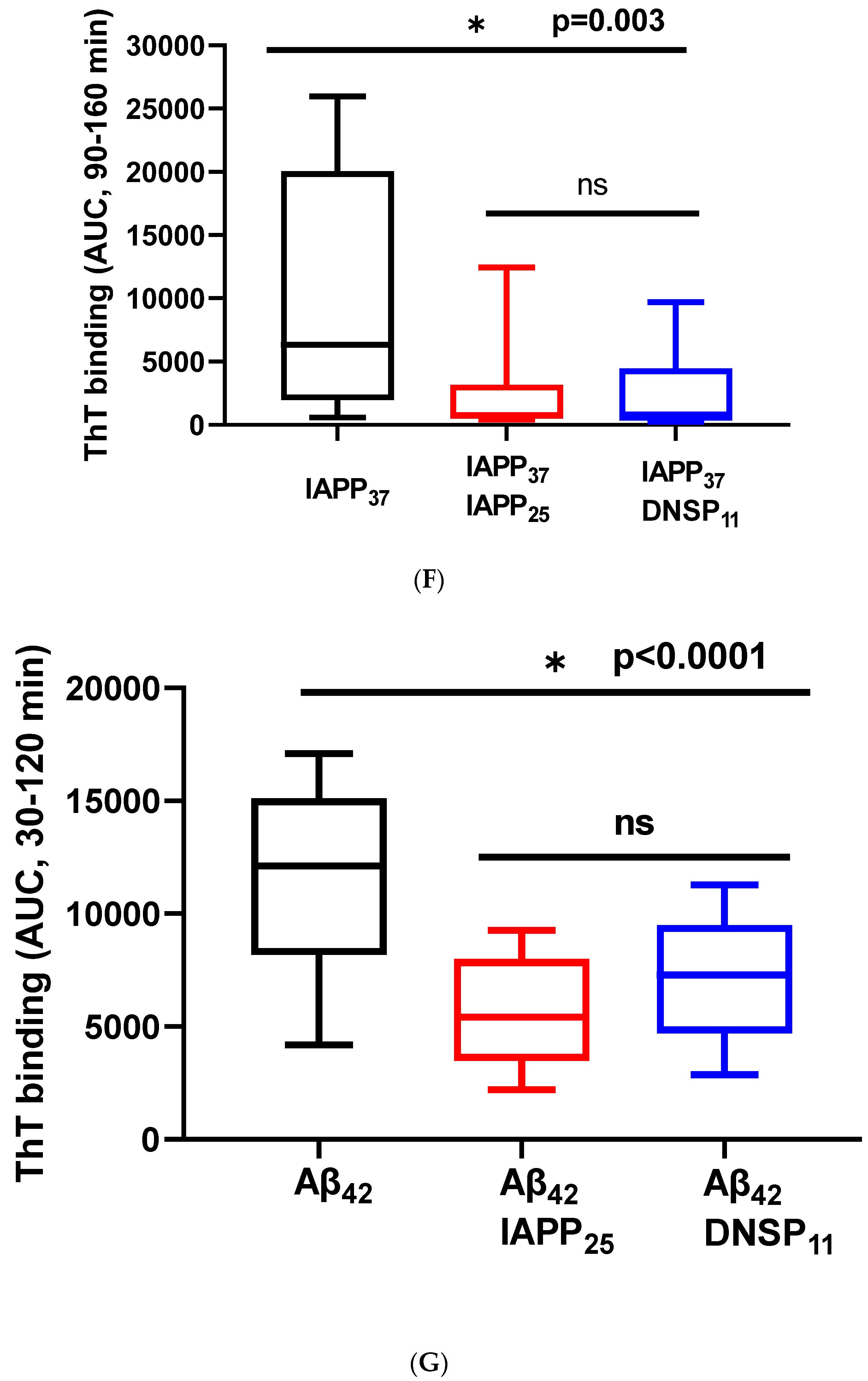
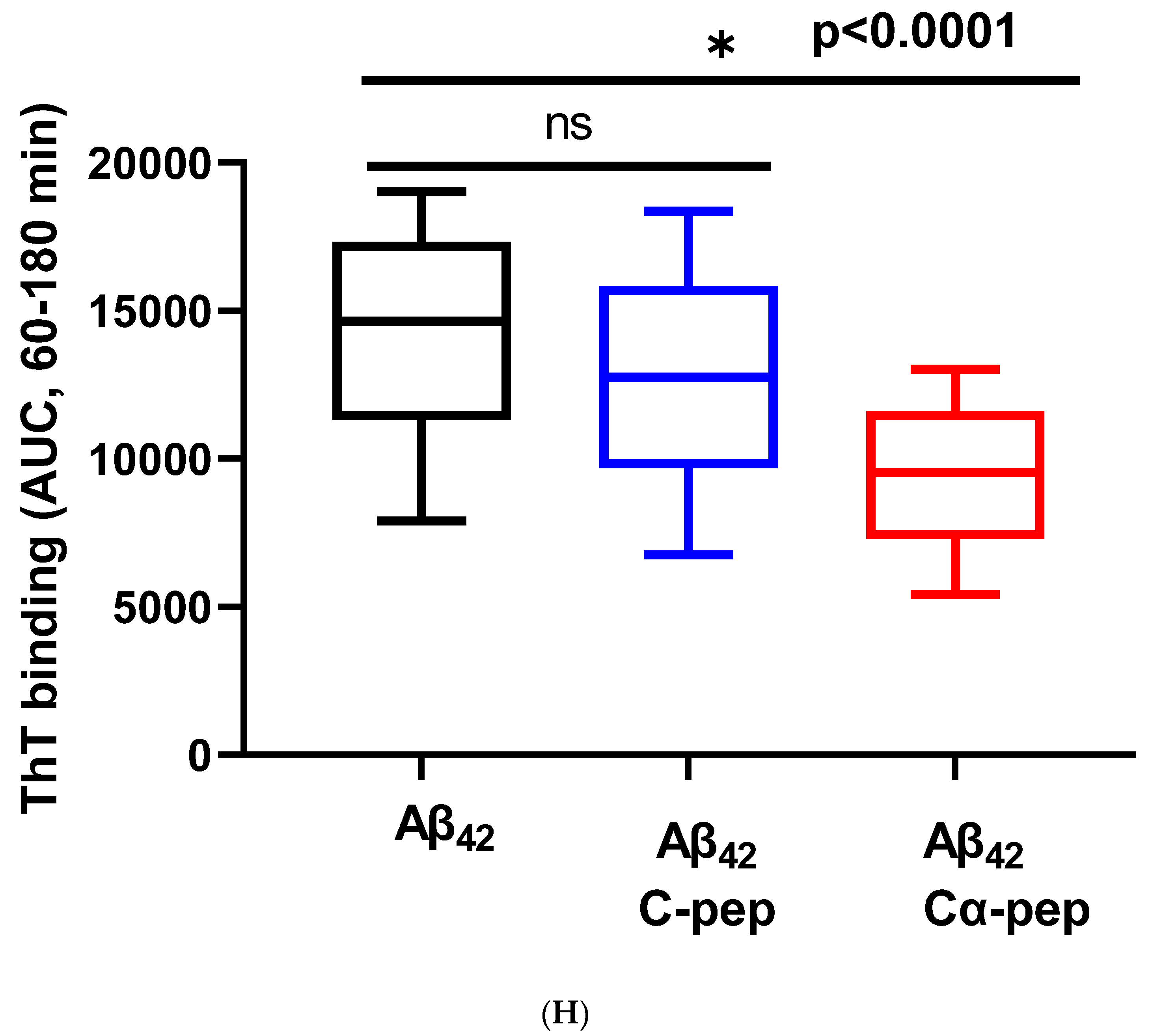
3.1.5. In Vitro Inhibition of IAPP37 and Ab42 Fibrillation by IAPP25 and DNSP11
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raleigh, D.; Zhang, X.; Hastoy, B.; Clark, A. The beta-cell assassin: IAPP cytotoxicity. J. Mol. Endocrinol. 2017, 59, R121–R140. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P. Amyloid in the islets of Langerhans: Thoughts and some historical aspects. Ups. J. Med. Sci. 2011, 116, 81–89. [Google Scholar] [CrossRef]
- Fortin, J.S.; Benoit-Biancamano, M.O. Wildlife sequences of islet amyloid polypeptide (IAPP) identify critical species variants for fibrillization. Amyloid 2015, 22, 194–202. [Google Scholar] [CrossRef]
- Bhowmick, D.C.; Singh, S.; Trikha, S.; Jeremic, A.M. The Molecular Physiopathogenesis of Islet Amyloidosis. Handb. Exp. Pharmacol. 2018, 245, 271–312. [Google Scholar] [PubMed] [Green Version]
- Milardi, D.; Gazit, E.; Radford, S.E.; Xu, Y.; Gallardo, R.U.; Caflisch, A.; Westermark, G.T.; Westermark, P.; Rosa, C.; Ramamoorthy, A. Proteostasis of Islet Amyloid Polypeptide: A Molecular Perspective of Risk Factors and Protective Strategies for Type II Diabetes. Chem. Rev. 2021, 121, 1845–1893. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.R.; Aseer, K.R.; Yao, Q.; Zhong, X.; Ghosh, P.; O’Connell, J.F.; Egan, J.M. Anti-Inflammatory and Pro-Autophagy Effects of the Cannabinoid Receptor CB2R: Possibility of Modulation in Type 1 Diabetes. Front. Pharmacol. 2021, 12, 809965. [Google Scholar] [CrossRef] [PubMed]
- Segerstolpe, A.; Palasantza, A.; Eliasson, P.; Andersson, E.M.; Andreasson, A.C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef] [Green Version]
- Kudva, Y.C.; Mueske, C.; Butler, P.C.; Eberhardt, N.L. A novel assay in vitro of human islet amyloid polypeptide amyloidogenesis and effects of insulin secretory vesicle peptides on amyloid formation. Biochem. J. 1998, 331 Pt 3, 809–813. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.R.; Zhu, M.; Zhang, P.; Mazucanti, C.H.; Huang, N.S.; Lang, D.L.; Chen, Q.; Auluck, P.; Marenco, S.; O’Connell, J.F.; et al. Novel Human Insulin Isoforms and Calpha-Peptide Product in Islets of Langerhans and Choroid Plexus. Diabetes 2021, 70, 2947–2956. [Google Scholar] [CrossRef]
- Chen, Y.C.; Taylor, A.J.; Verchere, C.B. Islet prohormone processing in health and disease. Diabetes Obes. Metab. 2018, 20 (Suppl. S2), 64–76. [Google Scholar] [CrossRef]
- Clark, A.; Nilsson, M.R. Islet amyloid: A complication of islet dysfunction or an aetiological factor in Type 2 diabetes? Diabetologia 2004, 47, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Asthana, S.; Mallick, B.; Alexandrescu, A.T.; Jha, S. IAPP in type II diabetes: Basic research on structure, molecular interactions, and disease mechanisms suggests potential intervention strategies. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1765–1782. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.A.; Bjorklund, A.; Gash, D.M.; Whone, A.; Van Laar, A.; Kordower, J.H.; Bankiewicz, K.; Kieburtz, K.; Saarma, M.; Booms, S.; et al. GDNF and Parkinson’s Disease: Where Next? A Summary from a Recent Workshop. J. Parkinsons. Dis. 2020, 10, 875–891. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Okada, Y.; Sato, M.; Sawai, H.; Funahashi, H.; Murase, T.; Hayakawa, T.; Manabe, T. Expression of glial cell line-derived neurotrophic factor family members and their receptors in pancreatic cancers. Surgery 2005, 138, 788–794. [Google Scholar] [CrossRef]
- Airavaara, M.; Pletnikova, O.; Doyle, M.E.; Zhang, Y.E.; Troncoso, J.C.; Liu, Q.R. Identification of novel GDNF isoforms and cis-antisense GDNFOS gene and their regulation in human middle temporal gyrus of Alzheimer disease. J. Biol. Chem. 2011, 286, 45093–45102. [Google Scholar] [CrossRef] [Green Version]
- Mwangi, S.; Anitha, M.; Mallikarjun, C.; Ding, X.; Hara, M.; Parsadanian, A.; Larsen, C.P.; Thule, P.; Sitaraman, S.V.; Anania, F.; et al. Glial cell line-derived neurotrophic factor increases beta-cell mass and improves glucose tolerance. Gastroenterology 2008, 134, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Mwangi, S.M.; Usta, Y.; Raja, S.M.; Anitha, M.; Chandrasekharan, B.; Parsadanian, A.; Sitaraman, S.V.; Srinivasan, S. Glial cell line-derived neurotrophic factor enhances neurogenin3 gene expression and beta-cell proliferation in the developing mouse pancreas. Am. J. Physiol. Gastrointest Liver Physiol. 2010, 299, G283–G292. [Google Scholar] [CrossRef] [Green Version]
- Lucini, C.; Maruccio, L.; Facello, B.; Cocchia, N.; Tortora, G.; Castaldo, L. Cellular localization of GDNF and its GFRalpha1/RET receptor complex in the developing pancreas of cat. J. Anat. 2008, 213, 565–572. [Google Scholar] [CrossRef]
- Bradley, L.H.; Fuqua, J.; Richardson, A.; Turchan-Cholewo, J.; Ai, Y.; Kelps, K.A.; Glass, J.D.; He, X.; Zhang, Z.; Grondin, R.; et al. Dopamine neuron stimulating actions of a GDNF propeptide. PLoS ONE 2010, 5, e9752. [Google Scholar] [CrossRef]
- Immonen, T.; Alakuijala, A.; Hytonen, M.; Sainio, K.; Poteryaev, D.; Saarma, M.; Pasternack, M.; Sariola, H. A proGDNF-related peptide BEP increases synaptic excitation in rat hippocampus. Exp. Neurol. 2008, 210, 793–796. [Google Scholar] [CrossRef]
- Stenslik, M.J.; Potts, L.F.; Sonne, J.W.; Cass, W.A.; Turchan-Cholewo, J.; Pomerleau, F.; Huettl, P.; Ai, Y.; Gash, D.M.; Gerhardt, G.A.; et al. Methodology and effects of repeated intranasal delivery of DNSP-11 in a rat model of Parkinson’s disease. J. Neurosci. Methods 2015, 251, 120–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.W.; DeCarli, C.; Despa, F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 2013, 74, 517–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, R.; Zhang, M.; Chen, H.; Jiang, B.; Zheng, J. Cross-Seeding Interaction between beta-Amyloid and Human Islet Amyloid Polypeptide. ACS Chem. Neurosci. 2015, 6, 1759–1768. [Google Scholar] [CrossRef]
- Moreno-Gonzalez, I.; Edwards Iii, G.; Salvadores, N.; Shahnawaz, M.; Diaz-Espinoza, R.; Soto, C. Molecular interaction between type 2 diabetes and Alzheimer’s disease through cross-seeding of protein misfolding. Mol. Psychiatry 2017, 22, 1327–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, B.L.; Yarchoan, M.; Hwang, H.M.; Louneva, N.; Blair, J.A.; Palm, R.; Smith, M.A.; Lee, H.G.; Arnold, S.E.; Casadesus, G. Neuroprotective effects of the amylin analogue pramlintide on Alzheimer’s disease pathogenesis and cognition. Neurobiol. Aging 2014, 35, 793–801. [Google Scholar] [CrossRef]
- Zhu, H.; Tao, Q.; Ang, T.F.A.; Massaro, J.; Gan, Q.; Salim, S.; Zhu, R.Y.; Kolachalama, V.B.; Zhang, X.; Devine, S.; et al. Association of Plasma Amylin Concentration With Alzheimer Disease and Brain Structure in Older Adults. JAMA Netw. Open 2019, 2, e199826. [Google Scholar] [CrossRef] [Green Version]
- Makimattila, S.; Fineman, M.S.; Yki-Jarvinen, H. Deficiency of total and nonglycosylated amylin in plasma characterizes subjects with impaired glucose tolerance and type 2 diabetes. J. Clin. Endocrinol. Metab. 2000, 85, 2822–2827. [Google Scholar] [CrossRef]
- Zhu, H.; Stern, R.A.; Tao, Q.; Bourlas, A.; Essis, M.D.; Chivukula, M.; Rosenzweig, J.; Steenkamp, D.; Xia, W.; Mercier, G.A.; et al. An amylin analog used as a challenge test for Alzheimer’s disease. Alzheimers Dement. 2017, 3, 33–43. [Google Scholar] [CrossRef]
- Zhu, H.; Xue, X.; Wang, E.; Wallack, M.; Na, H.; Hooker, J.M.; Kowall, N.; Tao, Q.; Stein, T.D.; Wolozin, B.; et al. Amylin receptor ligands reduce the pathological cascade of Alzheimer’s disease. Neuropharmacology 2017, 119, 170–181. [Google Scholar] [CrossRef]
- Garcia-Vinuales, S.; Ilie, I.M.; Santoro, A.M.; Romanucci, V.; Zarrelli, A.; Di Fabio, G.; Caflisch, A.; Milardi, D. Silybins inhibit human IAPP amyloid growth and toxicity through stereospecific interactions. Biochim. Biophys. Acta Proteins Proteom. 2022, 1870, 140772. [Google Scholar] [CrossRef]
- Sciacca, M.F.M.; Romanucci, V.; Zarrelli, A.; Monaco, I.; Lolicato, F.; Spinella, N.; Galati, C.; Grasso, G.; D’Urso, L.; Romeo, M.; et al. Inhibition of Abeta Amyloid Growth and Toxicity by Silybins: The Crucial Role of Stereochemistry. ACS Chem. Neurosci. 2017, 8, 1767–1778. [Google Scholar] [CrossRef]
- Leibson, C.L.; Rocca, W.A.; Hanson, V.A.; Cha, R.; Kokmen, E.; O’Brien, P.C.; Palumbo, P.J. Risk of dementia among persons with diabetes mellitus: A population-based cohort study. Am. J. Epidemiol. 1997, 145, 301–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, A.; Stolk, R.P.; Hofman, A.; van Harskamp, F.; Grobbee, D.E.; Breteler, M.M. Association of diabetes mellitus and dementia: The Rotterdam Study. Diabetologia 1996, 39, 1392–1397. [Google Scholar] [CrossRef] [Green Version]
- Mullins, R.J.; Mustapic, M.; Chia, C.W.; Carlson, O.; Gulyani, S.; Tran, J.; Li, Y.; Mattson, M.P.; Resnick, S.; Egan, J.M.; et al. A Pilot Study of Exenatide Actions in Alzheimer’s Disease. Curr. Alzheimer. Res. 2019, 16, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, L.M.; Vanderstichele, H.; Knapik-Czajka, M.; Clark, C.M.; Aisen, P.S.; Petersen, R.C.; Blennow, K.; Soares, H.; Simon, A.; Lewczuk, P.; et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Annals. Neurol. 2009, 65, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar]
- Mazucanti, C.H.; Liu, Q.R.; Lang, D.; Huang, N.; O’Connell, J.F.; Camandola, S.; Egan, J.M. Insulin is produced in choroid plexus and its release is regulated by serotonergic signaling. JCI Insight 2019, 4, e131682. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.R.; Rubio, F.J.; Bossert, J.M.; Marchant, N.J.; Fanous, S.; Hou, X.; Shaham, Y.; Hope, B.T. Detection of molecular alterations in methamphetamine-activated Fos-expressing neurons from a single rat dorsal striatum using fluorescence-activated cell sorting (FACS). J. Neurochem. 2014, 128, 173–185. [Google Scholar] [CrossRef] [Green Version]
- Majbour, N.; Aasly, J.; Abdi, I.; Ghanem, S.; Erskine, D.; van de Berg, W.; El-Agnaf, O. Disease-Associated alpha-Synuclein Aggregates as Biomarkers of Parkinson Disease Clinical Stage. Neurology 2022, 99, e2417–e2427. [Google Scholar]
- Chia, C.W.; Odetunde, J.O.; Kim, W.; Carlson, O.D.; Ferrucci, L.; Egan, J.M. GIP contributes to islet trihormonal abnormalities in type 2 diabetes. J. Clin. Endocrinol. Metab. 2014, 99, 2477–2485. [Google Scholar] [CrossRef] [Green Version]
- Boguski, M.S.; Lowe, T.M.; Tolstoshev, C.M. dbEST-database for “expressed sequence tags”. Nat. Genet. 1993, 4, 332–333. [Google Scholar] [CrossRef] [PubMed]
- Christmanson, L.; Rorsman, F.; Stenman, G.; Westermark, P.; Betsholtz, C. The human islet amyloid polypeptide (IAPP) gene. Organization, chromosomal localization and functional identification of a promoter region. FEBS Lett. 1990, 267, 160–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prufer, K.; Racimo, F.; Patterson, N.; Jay, F.; Sankararaman, S.; Sawyer, S.; Heinze, A.; Renaud, G.; Sudmant, P.H.; de Filippo, C.; et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 2014, 505, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artenstein, A.W.; Opal, S.M. Proprotein convertases in health and disease. N. Engl. J. Med. 2011, 365, 2507–2518. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Dore, V.; Bourgeat, P.; Burnham, S.C.; Laws, S.; Salvado, O.; Masters, C.L.; Rowe, C.C. Abeta-amyloid and Tau Imaging in Dementia. Semin. Nucl. Med. 2017, 47, 75–88. [Google Scholar] [CrossRef]
- Fuqua, J.L.; Littrell, O.M.; Lundblad, M.; Turchan-Cholewo, J.; Abdelmoti, L.G.; Galperin, E.; Bradley, L.H.; Cass, W.A.; Gash, D.M.; Gerhardt, G.A. Dynamic changes in dopamine neuron function after DNSP-11 treatment: Effects in vivo and increased ERK 1/2 phosphorylation in vitro. Peptides 2014, 54, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.R.; Pan, C.H.; Hishimoto, A.; Li, C.Y.; Xi, Z.X.; Llorente-Berzal, A.; Viveros, M.P.; Ishiguro, H.; Arinami, T.; Onaivi, E.S.; et al. Species differences in cannabinoid receptor 2 (CNR2 gene): Identification of novel human and rodent CB2 isoforms, differential tissue expression and regulation by cannabinoid receptor ligands. Genes Brain Behav. 2009, 8, 519–530. [Google Scholar] [CrossRef]
- Li, Y.; Li, C.; Li, S.; Peng, Q.; An, N.A.; He, A.; Li, C.Y. Human exonization through differential nucleosome occupancy. Proc. Natl. Acad. Sci. USA 2018, 115, 8817–8822. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Westermark, G.; Chan, S.J.; Steiner, D.F. Altered gene structure and tissue expression of islet amyloid polypeptide in the chicken. Mol. Endocrinol. 1994, 8, 713–721. [Google Scholar]
- Toshimori, H.; Narita, R.; Nakazato, M.; Asai, J.; Mitsukawa, T.; Kangawa, K.; Matsuo, H.; Matsukura, S. Islet amyloid polypeptide (IAPP) in the gastrointestinal tract and pancreas of man and rat. Cell Tissue Res. 1990, 262, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Dupin, M.; Fortin, T.; Larue-Triolet, A.; Surault, I.; Beaulieu, C.; Gouel-Cheron, A.; Allaouchiche, B.; Asehnoune, K.; Roquilly, A.; Venet, F.; et al. Impact of Serum and Plasma Matrices on the Titration of Human Inflammatory Biomarkers Using Analytically Validated SRM Assays. J. Proteome Res. 2016, 15, 2366–2378. [Google Scholar] [CrossRef] [Green Version]
- Guzel, C.; Govorukhina, N.I.; Stingl, C.; Dekker, L.J.M.; Boichenko, A.; van der Zee, A.G.J.; Bischoff, R.P.H.; Luider, T.M. Comparison of Targeted Mass Spectrometry Techniques with an Immunoassay: A Case Study for HSP90alpha. Proteom. Clin. Appl. 2018, 12, 1700107. [Google Scholar] [CrossRef]
- El Haj, M.; Antoine, P.; Amouyel, P.; Lambert, J.C.; Pasquier, F.; Kapogiannis, D. Apolipoprotein E (APOE) epsilon4 and episodic memory decline in Alzheimer’s disease: A review. Ageing Res. Rev. 2016, 27, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanlon, C.S.; Rubinsztein, D.C. Arginine residues at codons 112 and 158 in the apolipoprotein E gene correspond to the ancestral state in humans. Atherosclerosis 1995, 112, 85–90. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, A.M.; Bennett, C.; Dickson, D.; Anestis, S.F.; Watts, D.P.; Webster, T.H.; Fontenot, M.B.; Bradley, B.J. The apolipoprotein E (APOE) gene appears functionally monomorphic in chimpanzees (Pan troglodytes). PLoS ONE 2012, 7, e47760. [Google Scholar] [CrossRef] [Green Version]
- De Franco, E.; Owens, N.D.L.; Montaser, H.; Wakeling, M.N.; SaarimäkiVire, J.; Ibrahim, H.; Triantou, A.; Balboa, D.; Caswell, R.C.; Johnson, M.B.; et al. Primate-Specific ZNF808 Is Essential for Pancreatic Development in Humans. 2021. Available online: https://www.medrxiv.org/content/10.1101/2021.08.23.21262262v1 (accessed on 10 November 2022).
- Ding, Y.; Luan, W.; Shen, X.; Wang, Z.; Cao, Y. LncRNA BDNF-AS as ceRNA regulates the miR-9-5p/BACE1 pathway affecting neurotoxicity in Alzheimer’s disease. Arch. Gerontol. Geriatr. 2022, 99, 104614. [Google Scholar] [CrossRef]
- Wang, S.; Fan, Y.; Xu, Y.; Zhang, L.; Cai, L.; Lv, B. GDNFOS1 knockdown decreases the invasion and viability of glioblastoma cells. Exp. Ther. Med. 2019, 18, 1315–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.Y.; Zhang, Y.; Wang, Z.; Zhang, Y.; Cao, C.; Zhang, P.W.; Lu, S.J.; Li, X.M.; Yu, Q.; Zheng, X.; et al. A human-specific de novo protein-coding gene associated with human brain functions. PLoS Comput. Biol. 2010, 6, e1000734. [Google Scholar] [CrossRef]
- Yang, Y.; Xie, B.; Ju, C.; Jin, H.; Ye, X.; Yao, L.; Jia, M.; Sun, Z.; Yuan, Y. The Association of Decreased Serum Gdnf Level with Hyperglycemia and Depression in Type 2 Diabetes Mellitus. Endocr. Pract. 2019, 25, 951–965. [Google Scholar] [CrossRef]
- Saini, R.K.; Goyal, D.; Goyal, B. Targeting Human Islet Amyloid Polypeptide Aggregation and Toxicity in Type 2 Diabetes: An Overview of Peptide-Based Inhibitors. Chem. Res. Toxicol. 2020, 33, 2719–2738. [Google Scholar] [CrossRef] [PubMed]
- Hishimoto, A.; Pletnikova, O.; Lang, D.L.; Troncoso, J.C.; Egan, J.M.; Liu, Q.R. Neurexin 3 transmembrane and soluble isoform expression and splicing haplotype are associated with neuron inflammasome and Alzheimer’s disease. Alzheimers Res. Ther. 2019, 11, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Meyer, S.; Schaeverbeke, J.M.; Verberk, I.M.W.; Gille, B.; De Schaepdryver, M.; Luckett, E.S.; Gabel, S.; Bruffaerts, R.; Mauroo, K.; Thijssen, E.H.; et al. Comparison of ELISA- and SIMOA-based quantification of plasma Abeta ratios for early detection of cerebral amyloidosis. Alzheimers Res. Ther. 2020, 12, 162. [Google Scholar] [CrossRef]
- Meng, X.; Li, T.; Wang, X.; Lv, X.; Sun, Z.; Zhang, J.; Su, F.; Kang, S.; Kim, S.; An, S.S.A.; et al. Association between increased levels of amyloid-beta oligomers in plasma and episodic memory loss in Alzheimer’s disease. Alzheimers Res. Ther. 2019, 11, 89. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, E.H.; La Joie, R.; Strom, A.; Fonseca, C.; Iaccarino, L.; Wolf, A.; Spina, S.; Allen, I.E.; Cobigo, Y.; Heuer, H.; et al. Plasma phosphorylated tau 217 and phosphorylated tau 181 as biomarkers in Alzheimer’s disease and frontotemporal lobar degeneration: A retrospective diagnostic performance study. Lancet Neurol. 2021, 20, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Mengel, D.; Keshavan, A.; Rissman, R.A.; Billinton, A.; Perkinton, M.; Percival-Alwyn, J.; Schultz, A.; Properzi, M.; Johnson, K.; et al. Learnings about the complexity of extracellular tau aid development of a blood-based screen for Alzheimer’s disease. Alzheimers Dement. 2019, 15, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Andreasson, U.; Zetterberg, H.; Blennow, K.; Alzheimer’s Disease Neuroimaging, I. Association of Plasma Neurofilament Light With Neurodegeneration in Patients With Alzheimer Disease. JAMA Neurol. 2017, 74, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Kapogiannis, D.; Mustapic, M.; Shardell, M.D.; Berkowitz, S.T.; Diehl, T.C.; Spangler, R.D.; Tran, J.; Lazaropoulos, M.P.; Chawla, S.; Gulyani, S.; et al. Association of Extracellular Vesicle Biomarkers with Alzheimer Disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol. 2019, 76, 1340–1351. [Google Scholar] [CrossRef]
- Ashton, N.J.; Leuzy, A.; Karikari, T.K.; Mattsson-Carlgren, N.; Dodich, A.; Boccardi, M.; Corre, J.; Drzezga, A.; Nordberg, A.; Ossenkoppele, R.; et al. The validation status of blood biomarkers of amyloid and phospho-tau assessed with the 5-phase development framework for AD biomarkers. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2140–2156. [Google Scholar] [CrossRef]














Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.-R.; Zhu, M.; Chen, Q.; Mustapic, M.; Kapogiannis, D.; Egan, J.M. Novel Hominid-Specific IAPP Isoforms: Potential Biomarkers of Early Alzheimer’s Disease and Inhibitors of Amyloid Formation. Biomolecules 2023, 13, 167. https://doi.org/10.3390/biom13010167
Liu Q-R, Zhu M, Chen Q, Mustapic M, Kapogiannis D, Egan JM. Novel Hominid-Specific IAPP Isoforms: Potential Biomarkers of Early Alzheimer’s Disease and Inhibitors of Amyloid Formation. Biomolecules. 2023; 13(1):167. https://doi.org/10.3390/biom13010167
Chicago/Turabian StyleLiu, Qing-Rong, Min Zhu, Qinghua Chen, Maja Mustapic, Dimitrios Kapogiannis, and Josephine M. Egan. 2023. "Novel Hominid-Specific IAPP Isoforms: Potential Biomarkers of Early Alzheimer’s Disease and Inhibitors of Amyloid Formation" Biomolecules 13, no. 1: 167. https://doi.org/10.3390/biom13010167
APA StyleLiu, Q. -R., Zhu, M., Chen, Q., Mustapic, M., Kapogiannis, D., & Egan, J. M. (2023). Novel Hominid-Specific IAPP Isoforms: Potential Biomarkers of Early Alzheimer’s Disease and Inhibitors of Amyloid Formation. Biomolecules, 13(1), 167. https://doi.org/10.3390/biom13010167