

Exogenous Players in Mitochondria-Related CNS Disorders: Viral Pathogens and Unbalanced Microbiota in the Gut-Brain Axis
 , ,
, ,  ,
,  and
and 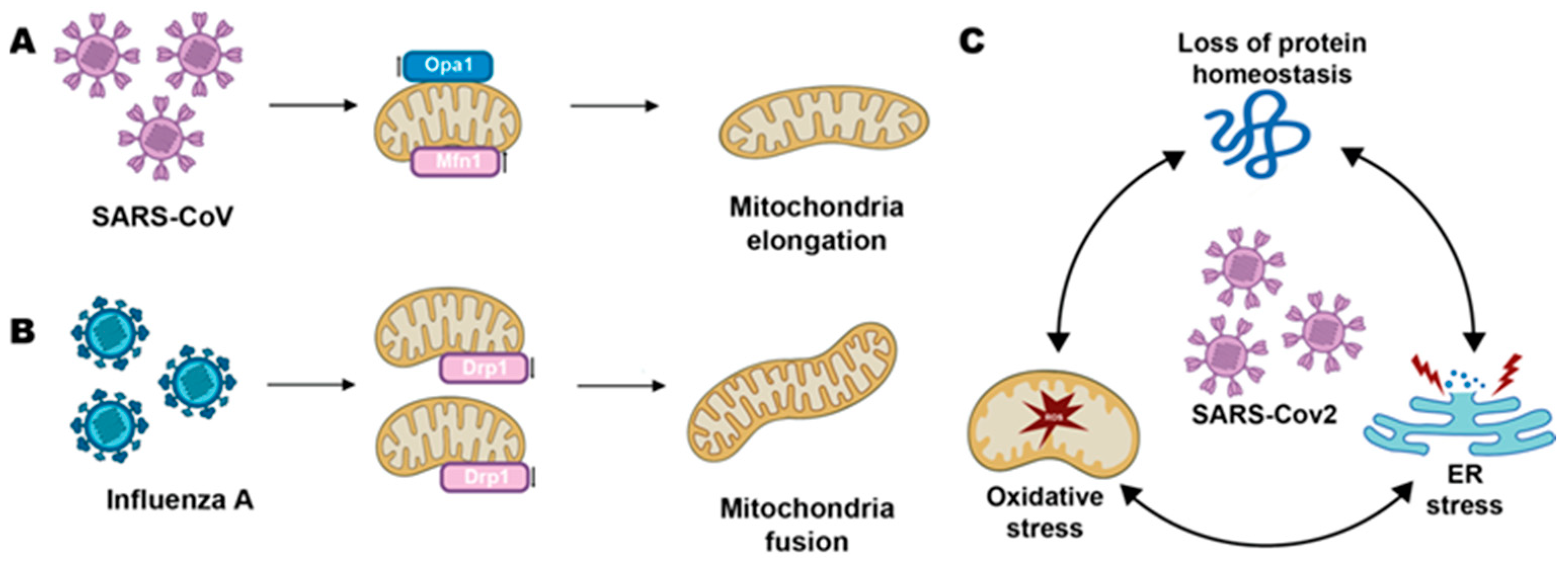{kind=link}
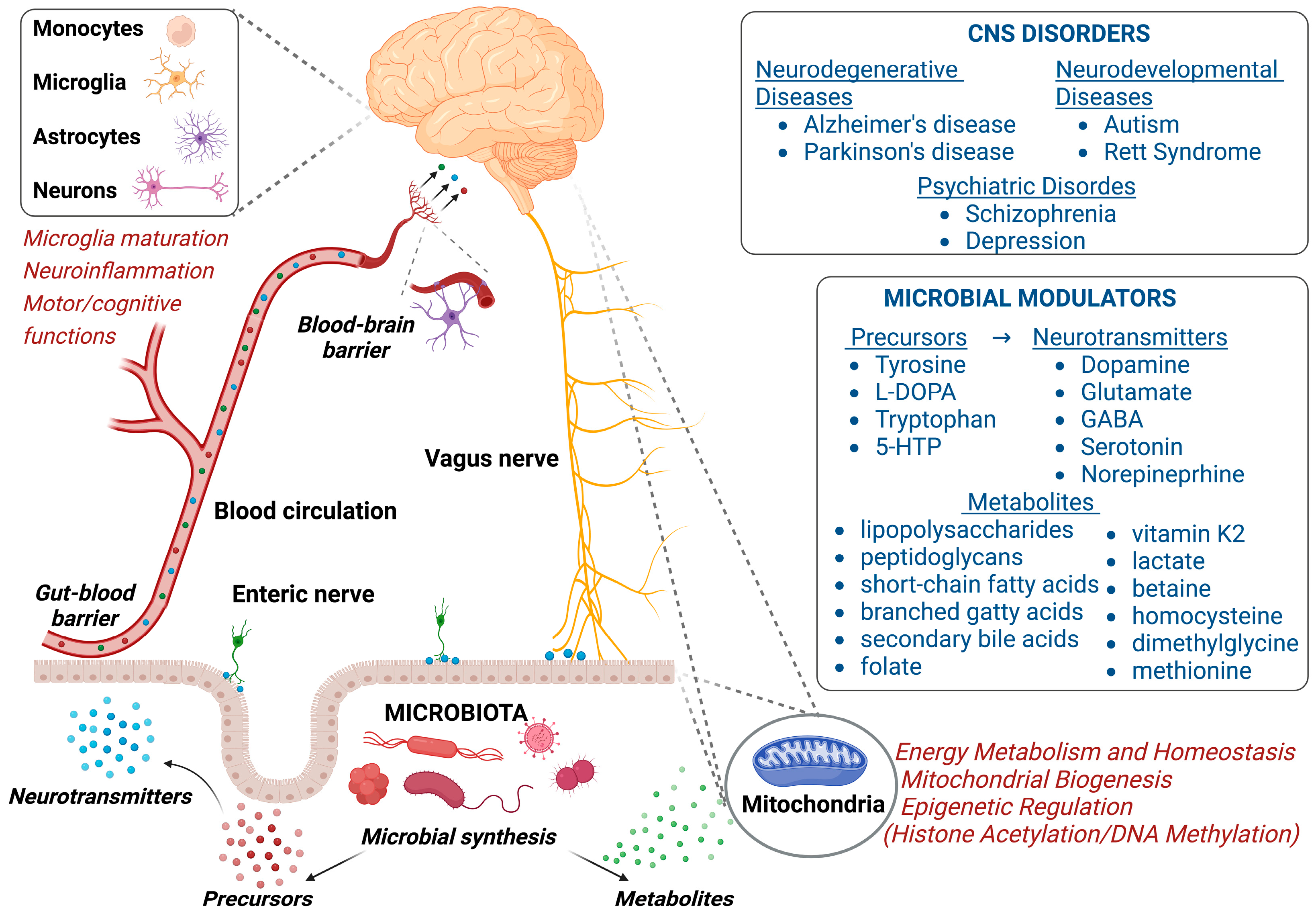{kind=link}
Abstract
:1. Introduction
2. Neurotropic Viruses
2.1. Poliovirus
2.2. Herpes Virus
2.3. Rabies Virus
2.4. West-Nile Virus
2.5. Zika Virus
3. SARS-CoV-2
4. Influenza A Virus
5. Unbalancing of the Gut-Brain Axis
5.1. Gut Bacterial Modulation of Mitochondrial Homeostatic Function
5.2. Microbiota and GBA Interplay in Mitochondria-Related CNS Disorders
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE-2 | angiotensin converting enzyme 2 |
| ARDS | acute respiratory distress syndrome |
| ASD | autism spectrum disorders |
| ATG5 | autophagy related 5 |
| BAD | Bcl-2 antagonist of cell death |
| BBB | brain blood barrier |
| BDNF | brain-derived neurotrophic factor |
| [Ca2+] | calcium concentration |
| CHPV | Chandipura Virus |
| CME | clathrin-mediated endocytosis |
| CNS | central nervous system |
| CoV | Coronavirus |
| COVID-19 | coronavirus disease 2019 |
| cyt | cytoplasmic |
| EC | endothelial cell |
| ER | endoplasmic reticulum |
| FTO | fat mass and obesity-associated gene |
| GBA | gut-brain axis |
| GBS | Guillain-Barre syndrome |
| GDFD | Growth retardation, developmental delay, coarse facies, and early death |
| GPCR | G-protein coupled receptor |
| HA | hemagglutinin |
| HAND | HIV-associated neurocognitive disorders |
| HCFC1 | Host Cell Factor C1 |
| HDAC | histone deacetylase |
| HIV | human immunodeficiency virus |
| HSE | HSV-1 encephalitis |
| HSV | Herpes simplex virus |
| HTLV-1 | Human T-cell leukemia virus type 1 |
| IFN-I | type I interferon |
| IP3R | inositol-1,4,5-triphosphate receptor |
| JEV | Japanese encephalitis virus |
| LDL | low-density lipoprotein |
| MAVS | mitochondria anti-viral signaling |
| MCU | mitochondrial calcium uniporter |
| MERS | Middle East Respiratory Syndrome |
| mt | mitochondrial |
| NA | neuraminidase |
| NDD | neurodevelopmental disorder |
| NMDAR | N-methyl-D-aspartate receptor |
| Nsp | non-structural protein |
| OMM | outer mitochondrial membrane |
| PI3K | phosphoinositide 3-kinase |
| PPA | propionate |
| PV | Poliovirus |
| RABV | Rabies virus |
| RIG-I | retinoic acid-inducible gene-I |
| ROS | reactive oxygen species |
| RSTS1 | Rubinstein-Taybi syndromes |
| RTT | Rett syndrome |
| RyR | Ryanodine receptor |
| SARS | Severe Acute Respiratory Syndrome |
| SCFA | short-chain fatty acid |
| SIRT1 | sirtuin 1 |
| Tat | trans-activator of transcription |
| TMPRSS2 | transmembrane serine protease 2 |
| VDAC | voltage-dependent anion channel |
| VGCC | voltage gated calcium channel |
| WNV | West Nile virus |
References
- Mattson, M.P.; Haughey, N.J.; Nath, A. Cell Death in HIV Dementia. Cell Death Differ. 2005, 12, 893–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasparotto, M.; Lee, Y.-S.; Palazzi, A.; Vacca, M.; Filippini, F. Nuclear and Cytoplasmatic Players in Mitochondria-Related CNS Disorders: Chromatin Modifications and Subcellular Trafficking. Biomolecules 2022, 12, 625. [Google Scholar] [CrossRef]
- Li, X.; Wu, K.; Zeng, S.; Zhao, F.; Fan, J.; Li, Z.; Yi, L.; Ding, H.; Zhao, M.; Fan, S.; et al. Viral Infection Modulates Mitochondrial Function. Int. J. Mol. Sci. 2021, 22, 4260. [Google Scholar] [CrossRef]
- Buckley, S.; Byrnes, S.; Cochrane, C.; Roche, M.; Estes, J.D.; Selemidis, S.; Angelovich, T.A.; Churchill, M.J. The Role of Oxidative Stress in HIV-Associated Neurocognitive Disorders. Brain Behav. Immun. Health 2021, 13, 100235. [Google Scholar] [CrossRef] [PubMed]
- Himanshu, D.; Tandon, R.; Kumar, S.; Sawlani, K.; Verma, S.; Misra, R.; Atam, V. Is International HIV Dementia Scale Good Enough to Diagnose HIV-Associated Neurocognitive Disorders? J. Family Med. Prim. Care 2022, 11, 5060. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, R.; Arora, H.; Seth, P. Mitochondrial Calcium Signaling in the Brain and Its Modulation by Neurotropic Viruses. Mitochondrion 2021, 59, 8–16. [Google Scholar] [CrossRef]
- Biasiotto, R.; Aguiari, P.; Rizzuto, R.; Pinton, P.; D’Agostino, D.M.; Ciminale, V. The P13 Protein of Human T Cell Leukemia Virus Type 1 (HTLV-1) Modulates Mitochondrial Membrane Potential and Calcium Uptake. Biochim. Biophys. Acta (BBA)—Bioenerg. 2010, 1797, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Leyrolle, Q.; Koistinen, V.; Kärkkäinen, O.; Layé, S.; Delzenne, N.; Hanhineva, K. Microbiota-Derived Metabolites as Drivers of Gut–Brain Communication. Gut. Microbes 2022, 14, 2102878. [Google Scholar] [CrossRef]
- Srikantha, P.; Mohajeri, M.H. The Possible Role of the Microbiota-Gut-Brain-Axis in Autism Spectrum Disorder. Int. J. Mol. Sci. 2019, 20, 2115. [Google Scholar] [CrossRef] [Green Version]
- Dash, S.; Syed, Y.A.; Khan, M.R. Understanding the Role of the Gut Microbiome in Brain Development and Its Association with Neurodevelopmental Psychiatric Disorders. Front. Cell Dev. Biol. 2022, 10, 808. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics Analysis Reveals Large Effects of Gut Microflora on Mammalian Blood Metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, Recognition and Regulation of Non-CpG Methylation in the Adult Mammalian Brain. Nat. Neurosci. 2014, 17, 215–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Frey, T.K.; Yang, J.J. Viral Calciomics: Interplays between Ca2+ and Virus. Cell Calcium. 2009, 46, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Mukherjee, S.; Sengupta, N.; Roy, A.; Dey, D.; Chakraborty, S.; Chattopadhyay, D.; Banerjee, A.; Basu, A. Network Analysis Reveals Common Host Protein/s Modulating Pathogenesis of Neurotropic Viruses. Sci. Rep. 2016, 6, 32593. [Google Scholar] [CrossRef] [Green Version]
- Brisac, C.; Téoulé, F.; Autret, A.; Pelletier, I.; Colbère-Garapin, F.; Brenner, C.; Lemaire, C.; Blondel, B. Calcium Flux between the Endoplasmic Reticulum and Mitochondrion Contributes to Poliovirus-Induced Apoptosis. J. Virol. 2010, 84, 12226–12235. [Google Scholar] [CrossRef] [Green Version]
- Campanella, M.; de Jong, A.S.; Lanke, K.W.H.; Melchers, W.J.G.; Willems, P.H.G.M.; Pinton, P.; Rizzuto, R.; van Kuppeveld, F.J.M. The Coxsackievirus 2B Protein Suppresses Apoptotic Host Cell Responses by Manipulating Intracellular Ca2+ Homeostasis. J. Biol. Chem. 2004, 279, 18440–18450. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Hsia, S.; Martin-Caraballo, M. Regulation of T-type Ca2+ Channel Expression by Interleukin-6 in Sensory-like ND7/23 Cells Post-herpes Simplex Virus (HSV-1) Infection. J. Neurochem. 2019, 151, 238–254. [Google Scholar] [CrossRef]
- Wnęk, M.; Ressel, L.; Ricci, E.; Rodriguez-Martinez, C.; Guerrero, J.C.V.; Ismail, Z.; Smith, C.; Kipar, A.; Sodeik, B.; Chinnery, P.F.; et al. Herpes Simplex Encephalitis Is Linked with Selective Mitochondrial Damage; a Post-Mortem and in Vitro Study. Acta Neuropathol. 2016, 132, 433–451. [Google Scholar] [CrossRef] [Green Version]
- Polansky, H.; Goral, B. How an Increase in the Copy Number of HSV-1 during Latency Can Cause Alzheimer’s Disease: The Viral and Cellular Dynamics According to the Microcompetition Model. J. Neurovirol. 2021, 27, 895–916. [Google Scholar] [CrossRef]
- Zan, J.; Liu, J.; Zhou, J.-W.; Wang, H.-L.; Mo, K.-K.; Yan, Y.; Xu, Y.-B.; Liao, M.; Su, S.; Hu, R.-L.; et al. Rabies Virus Matrix Protein Induces Apoptosis by Targeting Mitochondria. Exp. Cell Res. 2016, 347, 83–94. [Google Scholar] [CrossRef]
- Ubol, S.; Kasisith, J.; Pitidhammabhorn, D.; Tepsumethanol, V. Screening of Pro-Apoptotic Genes Upregulated in an Experimental Street Rabies Virus-Infected Neonatal Mouse Brain. Microbiol. Immunol. 2005, 49, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, B.H.; Wang, T. West Nile Virus Induced Cell Death in the Central Nervous System. Pathogens 2019, 8, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinschmidt, M.C.; Michaelis, M.; Ogbomo, H.; Doerr, H.-W.; Cinatl, J. Inhibition of Apoptosis Prevents West Nile Virus Induced Cell Death. BMC Microbiol. 2007, 7, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- del Carmen Parquet, M.; Kumatori, A.; Hasebe, F.; Morita, K.; Igarashi, A. West Nile Virus-Induced Bax-Dependent Apoptosis. FEBS Lett. 2001, 500, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrestha, B.; Gottlieb, D.; Diamond, M.S. Infection and Injury of Neurons by West NileEncephalitisVirus. J. Virol. 2003, 77, 13203–13213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, M.A.; Morrey, J.D.; Diamond, M.S. Caspase 3-Dependent Cell Death of Neurons Contributes to the Pathogenesis of West Nile Virus Encephalitis. J. Virol. 2007, 81, 2614–2623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmo, I.G.; Carvalho, T.G.; Costa, V.V.; Alves-Silva, J.; Ferrari, C.Z.; Izidoro-Toledo, T.C.; da Silva, J.F.; Teixeira, A.L.; Souza, D.G.; Marques, J.T.; et al. Zika Virus Promotes Neuronal Cell Death in a Non-Cell Autonomous Manner by Triggering the Release of Neurotoxic Factors. Front. Immunol. 2017, 8, 1016. [Google Scholar] [CrossRef] [Green Version]
- Doñate-Macián, P.; Jungfleisch, J.; Pérez-Vilaró, G.; Rubio-Moscardo, F.; Perálvarez-Marín, A.; Diez, J.; Valverde, M.A. The TRPV4 Channel Links Calcium Influx to DDX3X Activity and Viral Infectivity. Nat. Commun. 2018, 9, 2307. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Wang, J.; Yang, Y.; Qu, S.; Wan, F.; Zhang, Z.; Wang, R.; Li, G.; Cong, H. Zika Virus Infection Induced Apoptosis by Modulating the Recruitment and Activation of Proapoptotic Protein Bax. J. Virol. 2021, 95, e01445-20. [Google Scholar] [CrossRef]
- Ledur, P.F.; Karmirian, K.; Pedrosa, C.d.S.G.; Souza, L.R.Q.; Assis-de-Lemos, G.; Martins, T.M.; Ferreira, J.d.C.C.G.; de Azevedo Reis, G.F.; Silva, E.S.; Silva, D.; et al. Zika Virus Infection Leads to Mitochondrial Failure, Oxidative Stress and DNA Damage in Human IPSC-Derived Astrocytes. Sci. Rep. 2020, 10, 1218. [Google Scholar] [CrossRef]
- Heidari, A.; Righetto, I.; Filippini, F. Electrostatic Variation of Haemagglutinin as a Hallmark of the Evolution of Avian Influenza Viruses. Sci. Rep. 2018, 8, 1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.; Ng, Y.; Tam, J.; Liu, D. Human Coronaviruses: A Review of Virus–Host Interactions. Diseases 2016, 4, 26. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and Evolution of Pathogenic Coronaviruses. Nat. Rev. Microbiol 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostafa, A.; Kandeil, A.; Shehata, M.; el Shesheny, R.; Samy, A.M.; Kayali, G.; Ali, M.A. Middle East Respiratory Syndrome Coronavirus (MERS-CoV): State of the Science. Microorganisms 2020, 8, 991. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Kopecky-Bromberg, S.A.; Martínez-Sobrido, L.; Frieman, M.; Baric, R.A.; Palese, P. Severe Acute Respiratory Syndrome Coronavirus Open Reading Frame (ORF) 3b, ORF 6, and Nucleocapsid Proteins Function as Interferon Antagonists. J. Virol. 2007, 81, 548–557. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.-S.; Qi, H.-Y.; Boularan, C.; Huang, N.-N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-9b Suppresses Innate Immunity by Targeting Mitochondria and the MAVS/TRAF3/TRAF6 Signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [Green Version]
- Kreimendahl, S.; Rassow, J. The Mitochondrial Outer Membrane Protein Tom70-Mediator in Protein Traffic, Membrane Contact Sites and Innate Immunity. Int. J. Mol. Sci. 2020, 21, 7262. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Fang, L.; Zhang, H.; Xia, S.; Xiao, S. Functions of Coronavirus Accessory Proteins: Overview of the State of the Art. Viruses 2021, 13, 1139. [Google Scholar] [CrossRef]
- Padhan, K.; Minakshi, R.; Towheed, M.A.B.; Jameel, S. Severe Acute Respiratory Syndrome Coronavirus 3a Protein Activates the Mitochondrial Death Pathway through P38 MAP Kinase Activation. J. Gen. Virol. 2008, 89, 1960–1969. [Google Scholar] [CrossRef]
- Davies, J.P.; Almasy, K.M.; McDonald, E.F.; Plate, L. Comparative Multiplexed Interactomics of SARS-CoV-2 and Homologous Coronavirus Nonstructural Proteins Identifies Unique and Shared Host-Cell Dependencies. ACS Infect. Dis. 2020, 6, 3174–3189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wei, L.; Jiang, D.; Wang, J.; Cong, X.; Fei, R. SARS-CoV Nucleocapsid Protein Induced Apoptosis of COS-1 Mediated by the Mitochondrial Pathway. Artif. Cells. Blood. Substit. Biotechnol. 2007, 35, 237–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.-M.; Ma, C.-W.; Chan, W.-Y.; Chan, H.Y.E. The SARS-Coronavirus Membrane Protein Induces Apoptosis through Modulating the Akt Survival Pathway. Arch. Biochem. Biophys. 2007, 459, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-Z.; Wang, S.-Y.; Zheng, Z.-Q.; Huang, Y.; Li, W.-W.; Xu, Z.-S.; Wang, Y.-Y. SARS-CoV-2 Membrane Glycoprotein M Antagonizes the MAVS-Mediated Innate Antiviral Response. Cell Mol. Immunol. 2021, 18, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Lui, R.N.; Sung, J.J. Covid-19 and the Digestive System. J. Gastroenterol. Hepatol. 2020, 35, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Saleh, J.; Peyssonnaux, C.; Singh, K.K.; Edeas, M. Mitochondria and Microbiota Dysfunction in COVID-19 Pathogenesis. Mitochondrion 2020, 54, 1–7. [Google Scholar] [CrossRef]
- Swain, O.; Romano, S.K.; Miryala, R.; Tsai, J.; Parikh, V.; Umanah, G.K.E. SARS-CoV-2 Neuronal Invasion and Complications: Potential Mechanisms and Therapeutic Approaches. J. Neurosci. 2021, 41, 5338–5349. [Google Scholar] [CrossRef]
- Kumari, P.; Rothan, H.A.; Natekar, J.P.; Stone, S.; Pathak, H.; Strate, P.G.; Arora, K.; Brinton, M.A.; Kumar, M. Neuroinvasion and Encephalitis Following Intranasal Inoculation of SARS-CoV-2 in K18-HACE2 Mice. Viruses 2021, 13, 132. [Google Scholar] [CrossRef]
- Baig, A.M.; Khaleeq, A.; Ali, U.; Syeda, H. Evidence of the COVID-19 Virus Targeting the CNS: Tissue Distribution, Host–Virus Interaction, and Proposed Neurotropic Mechanisms. ACS Chem. Neurosci. 2020, 11, 995–998. [Google Scholar] [CrossRef] [Green Version]
- Rappoport, J.Z.; Benmerah, A.; Simon, S.M. Analysis of the AP-2 Adaptor Complex and Cargo during Clathrin-Mediated Endocytosis. Traffic 2005, 6, 539–547. [Google Scholar] [CrossRef]
- Al-Thomali, A.W.; Al-kuraishy, H.M.; Al-Gareeb, A.I.; K. Al-buhadiliy, A.; de Waard, M.; Sabatier, J.-M.; Khan Khalil, A.A.; Saad, H.M.; Batiha, G.E.-S. Role of Neuropilin 1 in COVID-19 Patients with Acute Ischemic Stroke. Biomedicines 2022, 10, 2032. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, B.; Daly, J.L.; Simón-Gracia, L.; Klein, K.; Weeratunga, S.; Antón-Plágaro, C.; Tobi, A.; Hodgson, L.; Lewis, P.A.; Heesom, K.J.; et al. ESCPE-1 Mediates Retrograde Endosomal Sorting of the SARS-CoV-2 Host Factor Neuropilin-1. Proc. Natl. Acad. Sci. USA 2022, 119, e2201980119. [Google Scholar] [CrossRef] [PubMed]
- Baindara, P.; Roy, D.; Mandal, S.M.; Schrum, A.G. Conservation and Enhanced Binding of SARS-CoV-2 Omicron Spike Protein to Coreceptor Neuropilin-1 Predicted by Docking Analysis. Infect. Dis. Rep. 2022, 14, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cao, Y.; Yamada, S.; Thirunavukkarasu, M.; Nin, V.; Joshi, M.; Rishi, M.T.; Bhattacharya, S.; Camacho-Pereira, J.; Sharma, A.K.; et al. Cardiomyopathy and Worsened Ischemic Heart Failure in SM22-α Cre-Mediated Neuropilin-1 Null Mice. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1401–1412. [Google Scholar] [CrossRef] [Green Version]
- Saleki, K.; Banazadeh, M.; Miri, N.S.; Azadmehr, A. Triangle of Cytokine Storm, Central Nervous System Involvement, and Viral Infection in COVID-19: The Role of SFasL and Neuropilin-1. Rev. Neurosci. 2022, 33, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Issitt, T.; Bosseboeuf, E.; de Winter, N.; Dufton, N.; Gestri, G.; Senatore, V.; Chikh, A.; Randi, A.M.; Raimondi, C. Neuropilin-1 Controls Endothelial Homeostasis by Regulating Mitochondrial Function and Iron-Dependent Oxidative Stress. iScience 2019, 11, 205–223. [Google Scholar] [CrossRef] [Green Version]
- Krasemann, S.; Haferkamp, U.; Pfefferle, S.; Woo, M.S.; Heinrich, F.; Schweizer, M.; Appelt-Menzel, A.; Cubukova, A.; Barenberg, J.; Leu, J.; et al. The Blood-Brain Barrier Is Dysregulated in COVID-19 and Serves as a CNS Entry Route for SARS-CoV-2. Stem. Cell. Rep. 2022, 17, 307–320. [Google Scholar] [CrossRef]
- Stefano, G.B.; Büttiker, P.; Weissenberger, S.; Martin, A.; Ptacek, R.; Kream, R.M. Editorial: The Pathogenesis of Long-Term Neuropsychiatric COVID-19 and the Role of Microglia, Mitochondria, and Persistent Neuroinflammation: A Hypothesis. Med. Sci. Monit. 2021, 27, e933015-1. [Google Scholar] [CrossRef]
- Griffin, D.E. Emergence and Re-Emergence of Viral Diseases of the Central Nervous System. Prog. Neurobiol. 2010, 91, 95–101. [Google Scholar] [CrossRef]
- Shao, W.; Li, X.; Goraya, M.; Wang, S.; Chen, J.-L. Evolution of Influenza A Virus by Mutation and Re-Assortment. Int. J. Mol. Sci. 2017, 18, 1650. [Google Scholar] [CrossRef]
- Righetto, I.; Milani, A.; Cattoli, G.; Filippini, F. Comparative Structural Analysis of Haemagglutinin Proteins from Type A Influenza Viruses: Conserved and Variable Features. BMC Bioinform. 2014, 15, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bruin, A.C.M.; Funk, M.; Spronken, M.I.; Gultyaev, A.P.; Fouchier, R.A.M.; Richard, M. Hemagglutinin Subtype Specificity and Mechanisms of Highly Pathogenic Avian Influenza Virus Genesis. Viruses 2022, 14, 1566. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Wang, R.; Zhang, J.; Sun, X.; Zou, Z.; Wang, S.; Jin, M. Insights into Human Astrocyte Response to H5N1 Infection by Microarray Analysis. Viruses 2015, 7, 2618–2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studahl, M. Influenza Virus and CNS Manifestations. J. Clin. Virol 2003, 28, 225–232. [Google Scholar] [CrossRef]
- Siegers, J.Y.; van de Bildt, M.W.G.; Lin, Z.; Leijten, L.M.; Lavrijssen, R.A.M.; Bestebroer, T.; Spronken, M.I.J.; de Zeeuw, C.I.; Gao, Z.; Schrauwen, E.J.A.; et al. Viral Factors Important for Efficient Replication of Influenza A Viruses in Cells of the Central Nervous System. J. Virol. 2019, 93, e02273-18. [Google Scholar] [CrossRef] [Green Version]
- Michalicová, A.; Bhide, K.; Bhide, M.; Kováč, A. How Viruses Infiltrate the Central Nervous System. Acta. Virol. 2017, 61, 393–400. [Google Scholar] [CrossRef]
- Davis, C.O.; Kim, K.-Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular Degradation of Axonal Mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of Mitochondria from Astrocytes to Neurons after Stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Ward, M.C. Neurovirulence of Influenza A Virus. J. Neurovirol. 1996, 2, 139–151. [Google Scholar] [CrossRef]
- Mori, I.; Yokochi, T.; Kimura, Y. Role of Influenza A Virus Hemagglutinin in Neurovirulence for Mammalians. Med. Microbiol. Immunol. 2002, 191, 1–4. [Google Scholar] [CrossRef]
- Hatta, M.; Gao, P.; Halfmann, P.; Kawaoka, Y. Molecular Basis for High Virulence of Hong Kong H5N1 Influenza A Viruses. Science 2001, 293, 1840–1842. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Pu, J.; Sun, Y.; Bi, Y.; Jiang, Z.; Xu, G.; Zhang, H.; Cao, J.; Chang, K.-C.; Liu, J.; et al. Neurovirulence of Avian Influenza Virus Is Dependent on the Interaction of Viral NP Protein with FMRP in the Murine Brain. J. Virol. 2021, 95, e01272-20. [Google Scholar] [CrossRef]
- Righetto, I.; Filippini, F. Normal Modes Analysis and Surface Electrostatics of Haemagglutinin Proteins as Fingerprints for High Pathogenic Type A Influenza Viruses. BMC Bioinform. 2020, 21, 354. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, G.; Wang, C.; Jiang, M.; Gao, W.; Wang, M.; Sun, H.; Sun, Y.; Chang, K.-C.; Liu, J.; et al. Enhanced Pathogenicity and Neurotropism of Mouse-Adapted H10N7 Influenza Virus Are Mediated by Novel PB2 and NA Mutations. J. Gen. Virol. 2017, 98, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhu, Y.; Lin, X.; Ren, C.; Zhao, J.; Wang, F.; Gao, X.; Xiao, R.; Zhao, L.; Chen, H.; et al. Influenza M2 Protein Regulates MAVS-Mediated Signaling Pathway through Interacting with MAVS and Increasing ROS Production. Autophagy 2019, 15, 1163–1181. [Google Scholar] [CrossRef] [PubMed]
- Pila-Castellanos, I.; Molino, D.; McKellar, J.; Lines, L.; da Graca, J.; Tauziet, M.; Chanteloup, L.; Mikaelian, I.; Meyniel-Schicklin, L.; Codogno, P.; et al. Mitochondrial Morphodynamics Alteration Induced by Influenza Virus Infection as a New Antiviral Strategy. PLoS Pathog. 2021, 17, e1009340. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Xu, S.; Wei, Y.; Zhang, X.; Wang, Q.; Jia, Y.; Wang, W.; Han, L.; Chen, Z.; Wang, Z.; et al. The PB1 Protein of Influenza A Virus Inhibits the Innate Immune Response by Targeting MAVS for NBR1-Mediated Selective Autophagic Degradation. PLoS Pathog. 2021, 17, e1009300. [Google Scholar] [CrossRef] [PubMed]
- Long, J.C.D.; Fodor, E. The PB2 Subunit of the Influenza A Virus RNA Polymerase Is Imported into the Mitochondrial Matrix. J. Virol. 2016, 90, 8729–8738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Ji, Q.; Cheng, F.; Chen, D.; Geng, T.; Huang, Y.; Zhang, J.; He, Y.; Song, T. The LncRNAs Involved in Regulating the RIG-I Signaling Pathway. Front. Cell Infect. Microbiol. 2022, 12, 1664. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Platt, M.P.; Gilley, R.P.; Brown, D.; Dube, P.H.; Yu, Y.; Gonzalez-Juarbe, N. Influenza Causes MLKL-Driven Cardiac Proteome Remodeling during Convalescence. Circ. Res. 2021, 128, 570–584. [Google Scholar] [CrossRef]
- Tran, A.T.; Cortens, J.P.; Du, Q.; Wilkins, J.A.; Coombs, K.M. Influenza Virus Induces Apoptosis via BAD-Mediated Mitochondrial Dysregulation. J. Virol. 2013, 87, 1049–1060. [Google Scholar] [CrossRef] [Green Version]
- Bian, Q.; Lu, J.; Zhang, L.; Chi, Y.; Li, Y.; Guo, H. Highly Pathogenic Avian Influenza A Virus H5N1 Non-structural Protein 1 Is Associated with Apoptotic Activation of the Intrinsic Mitochondrial Pathway. Exp. Ther. Med. 2017, 14, 4041–4046. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, Y.; Zhang, Q.; Song, Y.; Wang, L.; Zhu, Z. Interactions Between Intestinal Microbiota and Neural Mitochondria: A New Perspective on Communicating Pathway from Gut to Brain. Front. Microbiol. 2022, 13, 493. [Google Scholar] [CrossRef]
- Blaser, M.J. The Microbiome Revolution. J. Clin. Invest. 2014, 124, 4162–4165. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-Gut Microbiota Metabolic Interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the Microbiota, Immune and Nervous Systems in Health and Disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Midtvedt, A.-C.; Midtvedt, T. Production of Short Chain Fatty Acids by the Intestinal Microflora during the First 2 Years of Human Life. J. Pediatr. Gastroenterol. Nutr. 1992, 15, 395–403. [Google Scholar] [CrossRef]
- Pistollato, F.; Sumalla Cano, S.; Elio, I.; Masias Vergara, M.; Giampieri, F.; Battino, M. Role of Gut Microbiota and Nutrients in Amyloid Formation and Pathogenesis of Alzheimer Disease. Nutr. Rev. 2016, 74, 624–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulak, A. Brain-Gut-Microbiota Axis in Parkinson’s Disease. World J. Gastroenterol. 2015, 21, 10609. [Google Scholar] [CrossRef] [PubMed]
- Cristiano, C.; Lama, A.; Lembo, F.; Mollica, M.P.; Calignano, A.; Mattace Raso, G. Interplay Between Peripheral and Central Inflammation in Autism Spectrum Disorders: Possible Nutritional and Therapeutic Strategies. Front. Physiol. 2018, 9, 184. [Google Scholar] [CrossRef] [PubMed]
- Neier, K.; Grant, T.E.; Palmer, R.L.; Chappell, D.; Hakam, S.M.; Yasui, K.M.; Rolston, M.; Settles, M.L.; Hunter, S.S.; Madany, A.; et al. Sex Disparate Gut Microbiome and Metabolome Perturbations Precede Disease Progression in a Mouse Model of Rett Syndrome. Commun. Biol. 2021, 4, 1408. [Google Scholar] [CrossRef]
- Arnold, J.W.; Roach, J.; Azcarate-Peril, M.A. Emerging Technologies for Gut Microbiome Research. Trends Microbiol. 2016, 24, 887–901. [Google Scholar] [CrossRef] [Green Version]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The Role of Short-Chain Fatty Acids in the Interplay between Diet, Gut Microbiota, and Host Energy Metabolism. J. Lipid. Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, H.; Singh, Y.; Singh, S.; Singh, R.B. Gut Microbiome-Mediated Epigenetic Regulation of Brain Disorder and Application of Machine Learning for Multi-Omics Data Analysis. Genome 2021, 64, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Ren, S.; Gil, G.; Dent, P. Bile Acids as Regulatory Molecules. J. Lipid. Res. 2009, 50, 1509–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joyce, S.A.; Gahan, C.G.M. Bile Acid Modifications at the Microbe-Host Interface: Potential for Nutraceutical and Pharmaceutical Interventions in Host Health. Annu. Rev. Food Sci. Technol. 2016, 7, 313–333. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, H.; Odamaki, T.; Hashikura, N.; Abe, F.; Xiao, J. Differences in Folate Production by Bifidobacteria of Different Origins. Biosci. Microbiota Food Health 2015, 34, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Marley, M.G.; Meganathan, R.; Bentley, R. Menaquinone (Vitamin K2) Biosynthesis in Escherichia Coli: Synthesis of o-Succinylbenzoate Does Not Require the Decarboxylase Activity of the Ketoglutarate Dehydrogenase Complex. Biochemistry 1986, 25, 1304–1307. [Google Scholar] [CrossRef]
- Mao, J.-H.; Kim, Y.-M.; Zhou, Y.-X.; Hu, D.; Zhong, C.; Chang, H.; Brislawn, C.J.; Fansler, S.; Langley, S.; Wang, Y.; et al. Genetic and Metabolic Links between the Murine Microbiome and Memory. Microbiome 2020, 8, 53. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Knauf, C. How Gut Microbes Talk to Organs: The Role of Endocrine and Nervous Routes. Mol. Metab. 2016, 5, 743–752. [Google Scholar] [CrossRef]
- Luu, M.; Visekruna, A. Short-chain Fatty Acids: Bacterial Messengers Modulating the Immunometabolism of T Cells. Eur. J. Immunol. 2019, 49, 842–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melbye, P.; Olsson, A.; Hansen, T.H.; Søndergaard, H.B.; Bang Oturai, A. Short-Chain Fatty Acids and Gut Microbiota in Multiple Sclerosis. Acta Neurol. Scand. 2019, 139, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Vitetta, L. Butyrate in Inflammatory Bowel Disease Therapy. Gastroenterology 2020, 158, 1511. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Niyazov, D.M.; Rossignol, D.A.; Goldenthal, M.; Kahler, S.G.; Frye, R.E. Clinical and Molecular Characteristics of Mitochondrial Dysfunction in Autism Spectrum Disorder. Mol. Diagn. Ther. 2018, 22, 571–593. [Google Scholar] [CrossRef] [Green Version]
- Uittenbogaard, M.; Brantner, C.A.; Chiaramello, A. Epigenetic Modifiers Promote Mitochondrial Biogenesis and Oxidative Metabolism Leading to Enhanced Differentiation of Neuroprogenitor Cells. Cell Death Dis. 2018, 9, 360. [Google Scholar] [CrossRef] [Green Version]
- Blevins, H.M.; Xu, Y.; Biby, S.; Zhang, S. The NLRP3 Inflammasome Pathway: A Review of Mechanisms and Inhibitors for the Treatment of Inflammatory Diseases. Front Aging Neurosci. 2022, 14, 879021. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, H.; Rao, X.; Liu, L.; Zheng, P.; Li, W.; Zhou, W.; Chai, T.; Ji, P.; Song, J.; et al. Proteomic Profiling of Lysine Acetylation Indicates Mitochondrial Dysfunction in the Hippocampus of Gut Microbiota-Absent Mice. Front Mol. Neurosci. 2021, 14, 594332. [Google Scholar] [CrossRef]
- el Hayek, L.; Khalifeh, M.; Zibara, V.; Abi Assaad, R.; Emmanuel, N.; Karnib, N.; El-Ghandour, R.; Nasrallah, P.; Bilen, M.; Ibrahim, P.; et al. Lactate Mediates the Effects of Exercise on Learning and Memory through SIRT1-Dependent Activation of Hippocampal Brain-Derived Neurotrophic Factor (BDNF). J. Neurosci. 2019, 39, 2369–2382. [Google Scholar] [CrossRef] [Green Version]
- Cohen, H.Y.; Miller, C.; Bitterman, K.J.; Wall, N.R.; Hekking, B.; Kessler, B.; Howitz, K.T.; Gorospe, M.; de Cabo, R.; Sinclair, D.A. Calorie Restriction Promotes Mammalian Cell Survival by Inducing the SIRT1 Deacetylase. Science 2004, 305, 390–392. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.-H.; Sommer, F.; Falk-Paulsen, M.; Ulas, T.; Best, P.; Fazio, A.; Kachroo, P.; Luzius, A.; Jentzsch, M.; Rehman, A.; et al. Exposure to the Gut Microbiota Drives Distinct Methylome and Transcriptome Changes in Intestinal Epithelial Cells during Postnatal Development. Genome. Med. 2018, 10, 27. [Google Scholar] [CrossRef]
- Vaquero, A.; Scher, M.; Lee, D.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Human SirT1 Interacts with Histone H1 and Promotes Formation of Facultative Heterochromatin. Mol. Cell 2004, 16, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.B.; Johansen, L.J.; Powell, L.D.; Quig, D.; Rubin, R.A. Gastrointestinal Flora and Gastrointestinal Status in Children with Autism—Comparisons to Typical Children and Correlation with Autism Severity. BMC Gastroenterol. 2011, 11, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motil, K.J.; Caeg, E.; Barrish, J.O.; Geerts, S.; Lane, J.B.; Percy, A.K.; Annese, F.; McNair, L.; Skinner, S.A.; Lee, H.-S.; et al. Gastrointestinal and Nutritional Problems Occur Frequently throughout Life in Girls and Women with Rett Syndrome. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 292–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milani, D.; Manzoni, F.M.P.; Pezzani, L.; Ajmone, P.; Gervasini, C.; Menni, F.; Esposito, S. Rubinstein-Taybi Syndrome: Clinical Features, Genetic Basis, Diagnosis, and Management. Ital. J. Pediatr. 2015, 41, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossignol, D.A.; Frye, R.E. Mitochondrial Dysfunction in Autism Spectrum Disorders: A Systematic Review and Meta-Analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef] [Green Version]
- MacFabe, D.F. Short-Chain Fatty Acid Fermentation Products of the Gut Microbiome: Implications in Autism Spectrum Disorders. Microb. Ecol. Health Dis. 2012, 23, 19260. [Google Scholar] [CrossRef]
- Jones, L.L.; McDonald, D.A.; Borum, P.R. Acylcarnitines: Role in Brain. Prog. Lipid. Res. 2010, 49, 61–75. [Google Scholar] [CrossRef]
- Justice, M.J.; Buchovecky, C.M.; Kyle, S.M.; Djukic, A. A Role for Metabolism in Rett Syndrome Pathogenesis. Rare Dis. 2013, 1, e27265. [Google Scholar] [CrossRef] [Green Version]
- Derecki, N.C.; Cronk, J.C.; Kipnis, J. Microglia Involvement in Rett Syndrome. In Neuroinflammation; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 221–233. [Google Scholar]
- Borghi, E.; Borgo, F.; Severgnini, M.; Savini, M.; Casiraghi, M.; Vignoli, A. Rett Syndrome: A Focus on Gut Microbiota. Int. J. Mol. Sci. 2017, 18, 344. [Google Scholar] [CrossRef] [Green Version]
- Strati, F.; Cavalieri, D.; Albanese, D.; de Felice, C.; Donati, C.; Hayek, J.; Jousson, O.; Leoncini, S.; Pindo, M.; Renzi, D.; et al. Altered Gut Microbiota in Rett Syndrome. Microbiome 2016, 4, 41. [Google Scholar] [CrossRef]
- di Fede, E.; Ottaviano, E.; Grazioli, P.; Ceccarani, C.; Galeone, A.; Parodi, C.; Colombo, E.A.; Bassanini, G.; Fazio, G.; Severgnini, M.; et al. Insights into the Role of the Microbiota and of Short-Chain Fatty Acids in Rubinstein–Taybi Syndrome. Int. J. Mol. Sci. 2021, 22, 3621. [Google Scholar] [CrossRef]
- Daoud, H.; Zhang, D.; McMurray, F.; Yu, A.; Luco, S.M.; Vanstone, J.; Jarinova, O.; Carson, N.; Wickens, J.; Shishodia, S.; et al. Identification of a Pathogenic FTO Mutation by next-Generation Sequencing in a Newborn with Growth Retardation and Developmental Delay. J. Med. Genet. 2016, 53, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.v.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.-J.; Chen, Q.; et al. Reversible Methylation of M6Am in the 5′ Cap Controls MRNA Stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Ma, L.; Zhang, H.; Cao, Y.; Wang, C.; Hou, N.; Huang, N.; von Deneen, K.M.; Zhao, C.; Shi, Y.; et al. Fto Deficiency Reduces Anxiety- and Depression-Like Behaviors in Mice via Alterations in Gut Microbiota. Theranostics 2019, 9, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-C.; Sloan, J.L.; Scharer, G.; Brebner, A.; Quintana, A.M.; Achilly, N.P.; Manoli, I.; Coughlin, C.R.; Geiger, E.A.; Schneck, U.; et al. An X-Linked Cobalamin Disorder Caused by Mutations in Transcriptional Coregulator HCFC1. Am. J. Hum. Genet. 2013, 93, 506–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esser, A.J.; Mukherjee, S.; Dereven’kov, I.A.; Makarov, S.v.; Jacobsen, D.W.; Spiekerkoetter, U.; Hannibal, L. Versatile Enzymology and Heterogeneous Phenotypes in Cobalamin Complementation Type C Disease. iScience 2022, 25, 104981. [Google Scholar] [CrossRef]
- Elesela, S.; Lukacs, N.W. Role of Mitochondria in Viral Infections. Life 2021, 11, 232. [Google Scholar] [CrossRef]
- Obrenovich, M. Leaky Gut, Leaky Brain? Microorganisms 2018, 6, 107. [Google Scholar] [CrossRef] [Green Version]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive Impact of Non-Antibiotic Drugs on Human Gut Bacteria. Nature 2018, 555, 623–628. [Google Scholar] [CrossRef]
- Holmes, M.; Flaminio, Z.; Vardhan, M.; Xu, F.; Li, X.; Devinsky, O.; Saxena, D. Cross Talk between Drug-resistant Epilepsy and the Gut Microbiome. Epilepsia 2020, 61, 2619–2628. [Google Scholar] [CrossRef]
- Kitamura, S.; Sugihara, K.; Kuwasako, M.; Tatsumi, K. The Role of Mammalian Intestinal Bacteria in the Reductive Metabolism of Zonisamide. J. Pharm. Pharmacol. 2011, 49, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yuan, J.; Wei, L.; Zhou, L.; Mei, K.; Yue, J.; Gao, H.; Zhang, M.; Jia, L.; Kang, Q.; et al. SATB2 Is a Sensitive Marker for Lower Gastrointestinal Well-Differentiated Neuroendocrine Tumors. Int. J. Clin. Exp. Pathol. 2015, 8, 7072–7082. [Google Scholar] [PubMed]
- Ni, H.; Chen, Y.; Xia, W.; Wang, C.; Hu, C.; Sun, L.; Tang, W.; Cui, H.; Shen, T.; Liu, Y.; et al. SATB2 Defect Promotes Colitis and Colitis-Associated Colorectal Cancer by Impairing Cl−/HCO3− Exchange and Homeostasis of Gut Microbiota. J. Crohns. Colitis 2021, 15, 2088–2102. [Google Scholar] [CrossRef]
- Gu, W.; Wang, H.; Huang, X.; Kraiczy, J.; Singh, P.N.P.; Ng, C.; Dagdeviren, S.; Houghton, S.; Pellon-Cardenas, O.; Lan, Y.; et al. SATB2 Preserves Colon Stem Cell Identity and Mediates Ileum-Colon Conversion via Enhancer Remodeling. Cell. Stem. Cell 2022, 29, 101–115.e10. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Righetto, I.; Gasparotto, M.; Casalino, L.; Vacca, M.; Filippini, F. Exogenous Players in Mitochondria-Related CNS Disorders: Viral Pathogens and Unbalanced Microbiota in the Gut-Brain Axis. Biomolecules 2023, 13, 169. https://doi.org/10.3390/biom13010169
Righetto I, Gasparotto M, Casalino L, Vacca M, Filippini F. Exogenous Players in Mitochondria-Related CNS Disorders: Viral Pathogens and Unbalanced Microbiota in the Gut-Brain Axis. Biomolecules. 2023; 13(1):169. https://doi.org/10.3390/biom13010169
Chicago/Turabian StyleRighetto, Irene, Matteo Gasparotto, Laura Casalino, Marcella Vacca, and Francesco Filippini. 2023. "Exogenous Players in Mitochondria-Related CNS Disorders: Viral Pathogens and Unbalanced Microbiota in the Gut-Brain Axis" Biomolecules 13, no. 1: 169. https://doi.org/10.3390/biom13010169
APA StyleRighetto, I., Gasparotto, M., Casalino, L., Vacca, M., & Filippini, F. (2023). Exogenous Players in Mitochondria-Related CNS Disorders: Viral Pathogens and Unbalanced Microbiota in the Gut-Brain Axis. Biomolecules, 13(1), 169. https://doi.org/10.3390/biom13010169