
Cancer-Associated Fibroblast Heterogeneity, Activation and Function: Implications for Prostate Cancer



Abstract
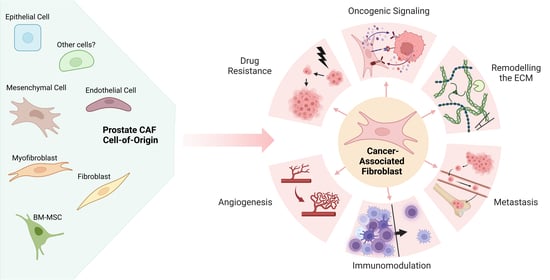
:
1. Introduction
1.1. Fibroblasts Function to Maintain Normal Adult Prostate Tissue Homeostasis
1.1.1. Fibroblasts Cross-Talk with Prostate Epithelial Cells
1.1.2. The Role of Fibroblasts during Ageing
1.1.3. Normal Fibroblasts Facilitate Wound Repair and Inflammation
1.2. Identification of Fibroblasts in Normal Adult Prostate Tissue

{kind=link}
{kind=link}
{kind=link}
| Cell Type (Species) | Alternative Nomenclature | Protein and mRNA Expression Markers 1 | Functions | Cell-of-Origin | Reference 2 |
|---|---|---|---|---|---|
| Fibroblasts (Human and Mouse) | Resting fibroblasts or Quiescent adult Fibroblasts | VIM, PDGFR-α, PDPN, αSMA, FSP1, NG2, Col1a1, DCN, STC1, NKTR, CAV1, CFH, Fn, PARVA, SPARC, Col1a2, Col5a1, Fbln1/2, Cd34, FGF2, FGF7, SNAPC2, AR, S100A16, FAP, ANXA2P3, SPRED2, PTGDS, RSPO3 | Maintain normal tissue homeostasis ECM deposition | Mesenchymal cells | [23,24,25,26] Breast and pancreas: [60,61] Muscular organs: [18] Cervix [62] |
| Periacinar Fibroblasts (Human) | Periacinar myofibroblasts | VIM, αSMA, COL1A1, COL3A1, FN, P4HB, COL4, Laminin, NMII | Maintain normal tissue homeostasis ECM deposition | Mesenchymal cells | [25] Pancreas: [63] |
| Fibrocytes (Human) | Bone marrow-derived cells, fibroblasts or myofibroblasts | CD45, COL1A1 | Collage type-I production Inflammatory secretome production during fibrosis/scarring | Bone-marrow-derived mesenchymal stem cells | Liver: [13,64] |
| Myofibroblasts (Human and Mouse) | Activated fibroblasts or Normal activated fibroblasts | VIM, αSMA, FAP, PDPN, AR, TGFβR1/2, FSP1, COL1A1, COL3A1, CFH, PARVA, SPARC, pSMAD2/3, FN, TNC | Wound healing | Resident fibroblasts (in response to TGF-β) | [25,65,66,67] (reviewed by [11,22,68]) |
| Smooth muscle cells (Human) | αSMA, CNN1 | Maintain normal tissue homeostasis (AR signaling) | Mesenchymal cells | [23,25] | |
| Fibromuscular stromal cells (Human) | CD49a, CD49e, CD51/61, CD30, CD29, CD55, CD56, CD59, CD79, CD81, CD90, CD99, CD131 | Maintain normal tissue homeostasis | Mesenchymal cells | [69] | |
| Endothelial cells (Human and Mouse) | VIM, CD31, CD34, CD105, VEGFR, CD200 | Forms a layer that lines blood vessels Modulates exchanges (e.g., signal molecules, EVs, gases) between the bloodstream and surrounding tissues. | Mesenchymal cells | [23] (reviewed by [70]) Pancreas: [61] |
2. Prostate Cancer-Associated Fibroblast Origin, Activation and Recruitment
2.1. The Heterogeneous Origin of CAFs in Prostate Cancer
2.1.1. Resident Fibroblasts within the TME Differentiate into CAFs
2.1.2. Local Mesenchymal Cell Derived CAFs
2.1.3. Bone-Marrow Derived CAFs
2.1.4. Endothelial Cell Derived CAFs
2.1.5. Epithelial-to-Mesenchymal Transition and CAF Generation
2.2. CAF Activation
2.2.1. TGFβ-Signaling Causes CAF Activation
2.2.2. The PI3K/AKT/PTEN Signaling Pathway Mediates CAF Activity
2.2.3. Notch Signaling Mediated CAF Activation
2.2.4. Extracellular Vesicles Regulate CAF Activation
2.2.5. Inflammation-Mediated CAF Activation
2.2.6. Senescence and CAF Activation
3. Identification of Prostate CAFs
| CAF Subtype | Alternative Nomenclature | Protein and mRNA Expression Markers 1 | Functions | Cell-of-Origin | Reference 2 |
|---|---|---|---|---|---|
| Identified in prostate cancer | |||||
| CAFs (generic) (Human and mouse) | Activated fibroblasts or Activated Myofibroblasts or Tumor-associated fibroblasts (TAFs) | Vimentin, αSMA, TGFβ, AR, TGFβR1/2, FAP, CD90, CD105, COL1A1/2, FSP1, PDPN, CD26, PDGFR-α/β, TNC, ASPN, POSTN, EGF, FGF7/2/10, IGF1, HGF, VEGF, OGN, Fibronectin, FBLN1, CTSK, PARVA, ZEB1, SPARC | ECM remodeling Immune Modulation Angiogenesis Paracrine signaling to prostate cancer cells to promote growth, proliferation and survival. | Resident fibroblasts Endothelial cells Vascular mural cells Epithelial cells Bone marrow-derived cells Mesenchymal stem cells | [11,25,102,103,153] |
| CD90+ (Human) | Reactive stroma fibroblasts or Tumorigenic fibroblasts or Tumor adjacent fibroblasts | CD90, ASPN, VEGF, FGF2, PATCH, TGFβ, IL6 | Tumorigenic | Unknown | [77,103] |
| CCL2+ (Human) | CAF-0 | Vimentin, CD90, αSMA, PDPN, LRP1Low, GLRX, PKMLow, CD63Low, TGFβ, CCL2 | CCL2 release to attract TAMs | Unknown (resident fibroblasts predicted) | [57] |
| CXCL12+ (Human) | CAF-1 or SDF1+ CAFs | Vimentin, CD90Low, αSMA, PDPN, LRP1 GLRXLow, PKMLow, CD63Low, TGFβ, CXCL12 | CXCL12/SDF1α release to attract immune cells and activate AKT signaling via CXCR4 to promote cancer cell growth and survival | Unknown, likely resident fibroblasts | [57,154] |
| CD105+ (Human and Mouse) | Fibroblasts promoting neuroendocrine differentiation or endoglin+ CAFs | CD105, αSMA, TNC, SFRP1 | Promotes neuroendocrine differentiation of prostate adenocarcinoma | Unknown | [6,155] |
| TGFβR2+ (Mouse) | TGFβR2, FGF2, Acta2, Tgfβ, Vimentin | Angiogenesis Prostate cancer cell proliferation | Unknown (myofibroblasts predicted) | [104] | |
| TGFβR2- (Human and Mouse) | TGFβR2 negative, αSMA, AR, Wnt3a, CXCL16, CXCL1 | Promotes prostate cancer cell adhesion to bone collagen-I fibers to facilitate skeletal metastasis via CXCL1/CXCL16 secretion | Unknown | [133,156] | |
| FGF2+ (Human) | Prostate cancer SC-9 cells | FGF2/7, TGFβhigh, VEGFhigh, COL1A1, TNC, ACTA2, EGF, IGF1 | Predicted functions: ECM remodeling; collagen Deposition; paracrine TGFβ signaling to prostate cancer cells; angiogenesis | Unknown | [102] |
| HGF+ (Human) | Prostate cancer SC-8 cells | HGF, TGFβ, VEGF, TNC, ACTA2, EGF, FGF7, IGF1, | Predicted functions: ECM remodeling and collagen deposition; paracrine TGFβ signaling to prostate cancer cells; angiogenesis. | Unknown | [102] |
| FGF10+ (Mouse) | FGF10 | Paracrine FGF10 signaling to prostate cancer cells causes increased AR expression and activated AKT. | Mesenchymal cells | [100] | |
| PDGFR+ (Human) | PDGFR | Paracrine PDGFR signaling to prostate cancer cells increased cell motility and invasion. | Resident fibroblasts | [95] | |
| PDGFRβ+ (Human) | CAF-S1 | PDGFRβ, VIM, αSMA, CAV1, SPARC, ETS1 | Cell adhesion and angiogenesis | Unknown (mesenchymal cells predicted) | [36] |
| PDGFRα+ (Human) | CAF-S2 | PDGFRα, VIM, αSMA, CREB3L1, PLAGL1 | ECM production and angiogenesis | Unknown (mesenchymal cells predicted) | [36] |
| Vimhi/αSMAhi (Human) | CAF-S3 | VIM, FAP, αSMA, TNC, CAV1, MAFB, HOXB2 | Fiber contraction to increase ECM stiffness and angiogenesis | Unknown (myeloid cells predicted) | [25,36,94] |
| Bone marrow derived (Human and Mouse) | Prostate cancer bone metastatic stromal cells | COl1A1, αSMA, VIM, EPHA3, PTN, FSCN1, FN1, TGFβ1, TGFβR1/2, FGF2, CD109, PDGFRβ | Pro-tumorigenic ECM remodeling Skeletal system development Cell adhesion Angiogenesis Wound healing EMT Wnt signaling | Bone marrow resident fibroblasts, bone-marrow-derived mesenchymal stem cells or hematopoietic stem cells | [21] Breast cancer: [56] Gastric cancer: [83] |
| Identified in other solid cancers | |||||
| MHCII+ (Human and Mouse) | Antigen presenting fibroblasts | MHCII, PDPN, CD74, COL1A1/2, PDPN, H2-AB1, FAP, VIM | Antigen-specific CD4+ T cell activation | Resident fibroblasts | Pancreatic cancer: [157] |
| LY6C+ (Human and Mouse) | Inflammatory fibroblasts | LY6C, PDPN, IL-1R1, IL-6, COL1A41, HAS1, CXCL12, FAP, VIM | Activates NF-kB and JAK/STAT signaling to promote cancer cell proliferation. | Resident fibroblasts | Pancreatic cancer: [157,158,159] |
| Endo180R+ (Human and Mouse) | Matrix remodeling CAFs or uPARP+ CAFs | Endo180R, PDGFRα, Fibulin-1, ACTA2, FAP, Vim, Sparc, PDGFRβ PDPN | ECM remodeling Collagen internalization and degradation Angiogenesis | Resident fibroblasts | Breast cancer: [110,160,161] |
| CD31+ (Human and Mouse) | Vascular CAFs | Nidogen-2, CD31, ACTA2, FSP1, PDGFRβ | Angiogenesis | Pericytes or endothelial cells | Breast cancer: [110] Pancreatic cancer: [88] |
| Active CD31+ (Human and Mouse) | Actively cycling vascular CAFs | Nidogen-2, Ki-67, CD31, ACTA2, PDGFRβ | Proliferating CD31-CAFs. | Perivascular cells | Breast cancer: [110] |
| Developmental (Human and Mouse) | SCRG1, Sparc, Mia, TRL | Cell differentiation Tissue development and morphogenesis | Predicted: mesenchymal stem cells, or malignant epithelial cells | Breast cancer: [110] | |
| FSP1+ (Mouse) | Fibroblast-like CAFs | FSP-1, VEGFA, TNC | Blood vessel remodeling Apoptosis evasion | Resident fibroblasts | Breast cancer: [93] |
| NG2+ (Human and Mouse) | NG2, αSMA, PDGFRβ | Predicted function: Integrin-dependent PI3K/AKT signaling and chemoresistance | Unknown | Breast and pancreatic cancer: [61,162] | |
| CD10+ (Human) | CD10, GPR77 | Stimulates cancer stem cell activity via activation of NF-κB signaling | Unknown | Breast cancer: [163] | |
3.1. RNA-Based Identification of CAF Subpopulations
3.2. Protein-Based Assays to Identify CAFs
4. Prostate Cancer-Associated Fibroblast Functions
4.1. CAFs Remodel the Extracellular Matrix to Facilitate Tumor Growth and Progression
4.1.1. CAF-Mediated Collagen Deposition
4.1.2. Tenascin C (TNC)
4.1.3. Hyaluronan
4.1.4. Fibronectin
4.1.5. Matrix Metalloproteinases (MMPs)
4.2. The CAF Secretome Contributes to Prostate Cancer Growth and Metastatic Progression
4.2.1. CAF-Mediated Oncogenic Signaling
4.2.2. CAF Regulation of Angiogenesis
4.2.3. CAFs Mediate Metastatic Potential
4.2.4. CAFs and TME Immunomodulation
5. CAFs Contribute to Therapeutic Resistance in Prostate Cancer
5.1. CAFs and Therapeutic Resistance to Androgen/AR-Directed Therapy
5.2. CAFs and Chemoresistance
5.3. Immune Checkpoint Inhibitor Resistance
6. Targeting and Reprogramming of CAFs
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Rycaj, K.; Li, H.; Zhou, J.; Chen, X.; Tang, D.G. Cellular determinants and microenvironmental regulation of prostate cancer metastasis. Semin. Cancer Biol. 2017, 44, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Ritch, C.R.; Cookson, M.S. Advances in the management of castration resistant prostate cancer. BMJ 2016, 355, i4405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosso, D.; Pagliuca, M.; Sonpavde, G.; Pond, G.; Lucarelli, G.; Rossetti, S.; Facchini, G.; Scagliarini, S.; Carteni, G.; Daniele, B.; et al. PSA declines and survival in patients with metastatic castration-resistant prostate cancer treated with enzalutamide: A retrospective case-report study. Medicine 2017, 96, e6817. [Google Scholar] [CrossRef]
- Eder, T.; Weber, A.; Neuwirt, H.; Grünbacher, G.; Ploner, C.; Klocker, H.; Sampson, N.; Eder, I.E. Cancer-Associated Fibroblasts Modify the Response of Prostate Cancer Cells to Androgen and Anti-Androgens in Three-Dimensional Spheroid Culture. Int. J. Mol. Sci. 2016, 17, 1458. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Placencio-Hickok, V.R.; Madhav, A.; Haldar, S.; Tripathi, M.; Billet, S.; Mishra, R.; Smith, B.; Rohena-Rivera, K.; Agarwal, P.; et al. Heterogeneous cancer-associated fibroblast population potentiates neuroendocrine differentiation and castrate resistance in a CD105-dependent manner. Oncogene 2019, 38, 716–730. [Google Scholar] [CrossRef]
- ChallaSivaKanaka, S.; Vickman, R.E.; Kakarla, M.; Hayward, S.W.; Franco, O.E. Fibroblast heterogeneity in prostate carcinogenesis. Cancer Lett. 2022, 525, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Thies, K.A.; Lefler, J.E.; Leone, G.; Ostrowski, M.C. PTEN in the Stroma. Cold Spring Harb. Perspect. Med. 2019, 9, a036111. [Google Scholar] [CrossRef]
- Polyak, K.; Haviv, I.; Campbell, I.G. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009, 25, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Paraiso, K.H.T.; Smalley, K.S.M. Fibroblast-mediated drug resistance in cancer. Biochem. Pharmacol. 2013, 85, 1033–1041. [Google Scholar] [CrossRef]
- Bonollo, F.; Thalmann, G.N.; Kruithof-de Julio, M.; Karkampouna, S. The Role of Cancer-Associated Fibroblasts in Prostate Cancer Tumorigenesis. Cancers 2020, 12, 1887. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.; Sainson, R.C. Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin. Cancer Biol. 2014, 25, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef]
- Hynes, R.O.; Naba, A. Overview of the matrisome--an inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [Green Version]
- Muhl, L.; Genové, G.; Leptidis, S.; Liu, J.; He, L.; Mocci, G.; Sun, Y.; Gustafsson, S.; Buyandelger, B.; Chivukula, I.V.; et al. Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat. Commun. 2020, 11, 3953. [Google Scholar] [CrossRef]
- Räsänen, K.; Vaheri, A. Activation of fibroblasts in cancer stroma. Exp. Cell Res. 2010, 316, 2713–2722. [Google Scholar] [CrossRef]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Hensel, J.; Secondini, C.; Wetterwald, A.; Schwaninger, R.; Fleischmann, A.; Raffelsberger, W.; Poch, O.; Delorenzi, M.; Temanni, R.; et al. The Molecular Signature of the Stroma Response in Prostate Cancer-Induced Osteoblastic Bone Metastasis Highlights Expansion of Hematopoietic and Prostate Epithelial Stem Cell Niches. PLoS ONE 2014, 9, e114530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calon, A.; Tauriello, D.V.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Henry, G.H.; Malewska, A.; Joseph, D.B.; Malladi, V.S.; Lee, J.; Torrealba, J.; Mauck, R.J.; Gahan, J.C.; Raj, G.V.; Roehrborn, C.G.; et al. A Cellular Anatomy of the Normal Adult Human Prostate and Prostatic Urethra. Cell Rep. 2018, 25, 3530–3542.e3535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuxhorn, J.A.; Ayala, G.E.; Smith, M.J.; Smith, V.C.; Dang, T.D.; Rowley, D.R. Reactive stroma in human prostate cancer: Induction of myofibroblast phenotype and extracellular matrix remodeling. Clin. Cancer Res. 2002, 8, 2912–2923. [Google Scholar]
- Vitale, G.; Caraglia, M.; Jung, V.; Kamradt, J.; Gentilini, D.; Di Martino, M.T.; Dicitore, A.; Abate, M.; Tagliaferri, P.; Itro, A.; et al. Molecular Characterization of Cancer Associated Fibroblasts in Prostate Cancer. Cancers 2022, 14, 2943. [Google Scholar] [CrossRef]
- Chung, L.W.; Cunha, G.R. Stromal-epithelial interactions: II. Regulation of prostatic growth by embryonic urogenital sinus mesenchyme. Prostate 1983, 4, 503–511. [Google Scholar] [CrossRef]
- Cunha, G.R.; Donjacour, A. Stromal-epithelial interactions in normal and abnormal prostatic development. Prog. Clin. Biol. Res. 1987, 239, 251–272. [Google Scholar]
- Welsh, M.; Moffat, L.; McNeilly, A.; Brownstein, D.; Saunders, P.T.K.; Sharpe, R.M.; Smith, L.B. Smooth Muscle Cell-Specific Knockout of Androgen Receptor: A New Model for Prostatic Disease. Endocrinology 2011, 152, 3541–3551. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Yeh, C.-R.; Niu, Y.; Chang, H.-C.; Tsai, Y.-C.; Moses, H.L.; Shyr, C.-R.; Chang, C.; Yeh, S. Altered prostate epithelial development in mice lacking the androgen receptor in stromal fibroblasts. Prostate 2012, 72, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Zhang, C.; Lin, C.-C.; Niu, Y.; Lai, K.-P.; Chang, H.-c.; Yeh, S.-D.; Chang, C.; Yeh, S. Altered prostate epithelial development and IGF-1 signal in mice lacking the androgen receptor in stromal smooth muscle cells. Prostate 2011, 71, 517–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef] [PubMed]
- Akhmetshina, A.; Palumbo, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat. Commun. 2012, 3, 735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Zhang, L.; Zhou, Z.; Kwon, O.-J.; Zhang, Y.; Nguyen, H.; Dumpit, R.; True, L.; Nelson, P.; Dong, B.; et al. Spatially Restricted Stromal Wnt Signaling Restrains Prostate Epithelial Progenitor Growth through Direct and Indirect Mechanisms. Cell Stem Cell 2019, 24, 753–768.e756. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhu, G.; Yang, Y.; Wang, F.; Xiao, Y.T.; Zhang, N.; Bian, X.; Zhu, Y.; Yu, Y.; Liu, F.; et al. Single-cell analysis reveals transcriptomic remodellings in distinct cell types that contribute to human prostate cancer progression. Nat. Cell Biol. 2021, 23, 87–98. [Google Scholar] [CrossRef]
- Kazlauskas, A. PDGFs and their receptors. Gene 2017, 614, 1–7. [Google Scholar] [CrossRef]
- Uezumi, A.; Fukada, S.; Yamamoto, N.; Ikemoto-Uezumi, M.; Nakatani, M.; Morita, M.; Yamaguchi, A.; Yamada, H.; Nishino, I.; Hamada, Y.; et al. Identification and characterization of PDGFRα+ mesenchymal progenitors in human skeletal muscle. Cell Death Dis. 2014, 5, e1186. [Google Scholar] [CrossRef] [Green Version]
- Gallini, R.; Lindblom, P.; Bondjers, C.; Betsholtz, C.; Andrae, J. PDGF-A and PDGF-B induces cardiac fibrosis in transgenic mice. Exp. Cell Res. 2016, 349, 282–290. [Google Scholar] [CrossRef]
- Olson, L.E.; Soriano, P. Increased PDGFRα Activation Disrupts Connective Tissue Development and Drives Systemic Fibrosis. Dev. Cell 2009, 16, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Gouveia, L.; Betsholtz, C.; Andrae, J. PDGF-A signaling is required for secondary alveolar septation and controls epithelial proliferation in the developing lung. Development 2018, 145, dev161976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano, P. The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development 1997, 124, 2691–2700. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Hampel, B.; Malisan, F.; Niederegger, H.; Testi, R.; Jansen-Dürr, P. Differential regulation of apoptotic cell death in senescent human cells. Exp. Gerontol. 2004, 39, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [Green Version]
- Robins, S.P. Biochemistry and functional significance of collagen cross-linking. Biochem. Soc. Trans. 2007, 35, 849–852. [Google Scholar] [CrossRef] [Green Version]
- Graf, R.; Freyberg, M.; Kaiser, D.; Friedl, P. Mechanosensitive induction of apoptosis in fibroblasts is regulated by thrombospondin-1 and integrin associated protein (CD47). Apoptosis 2002, 7, 493–498. [Google Scholar] [CrossRef]
- Jurj, A.; Ionescu, C.; Berindan-Neagoe, I.; Braicu, C. The extracellular matrix alteration, implication in modulation of drug resistance mechanism: Friends or foes? J. Exp. Clin. Cancer Res. 2022, 41, 276. [Google Scholar] [CrossRef]
- Buechler, M.B.; Turley, S.J. A short field guide to fibroblast function in immunity. Semin. Immunol. 2018, 35, 48–58. [Google Scholar] [CrossRef]
- Newman, A.C.; Nakatsu, M.N.; Chou, W.; Gershon, P.D.; Hughes, C.C. The requirement for fibroblasts in angiogenesis: Fibroblast-derived matrix proteins are essential for endothelial cell lumen formation. Mol. Biol. Cell 2011, 22, 3791–3800. [Google Scholar] [CrossRef]
- Brizzi, M.F.; Tarone, G.; Defilippi, P. Extracellular matrix, integrins, and growth factors as tailors of the stem cell niche. Curr. Opin. Cell Biol. 2012, 24, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 2003, 200, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Gharaee-Kermani, M.; Kasina, S.; Moore, B.B.; Thomas, D.; Mehra, R.; Macoska, J.A. CXC-type chemokines promote myofibroblast phenoconversion and prostatic fibrosis. PLoS ONE 2012, 7, e49278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, B.; Andreeff, M.; Marini, F. The participation of mesenchymal stem cells in tumor stroma formation and their application as targeted-gene delivery vehicles. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2007; pp. 263–283. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Vickman, R.E.; Broman, M.M.; Lanman, N.A.; Franco, O.E.; Sudyanti, P.A.G.; Ni, Y.; Ji, Y.; Helfand, B.T.; Petkewicz, J.; Paterakos, M.C.; et al. Heterogeneity of human prostate carcinoma-associated fibroblasts implicates a role for subpopulations in myeloid cell recruitment. Prostate 2020, 80, 173–185. [Google Scholar] [CrossRef]
- Flavell, S.J.; Hou, T.Z.; Lax, S.; Filer, A.D.; Salmon, M.; Buckley, C.D. Fibroblasts as novel therapeutic targets in chronic inflammation. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S241–S246. [Google Scholar] [CrossRef] [Green Version]
- Schauer, I.G.; Rowley, D.R. The functional role of reactive stroma in benign prostatic hyperplasia. Differentiation 2011, 82, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Sharon, Y.; Alon, L.; Glanz, S.; Servais, C.; Erez, N. Isolation of normal and cancer-associated fibroblasts from fresh tissues by Fluorescence Activated Cell Sorting (FACS). J. Vis. Exp. 2013, 71, e4425. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef] [Green Version]
- Pietras, K.; Pahler, J.; Bergers, G.; Hanahan, D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008, 5, e19. [Google Scholar] [CrossRef] [PubMed]
- Saotome, T.; Inoue, H.; Fujimiya, M.; Fujiyama, Y.; Bamba, T. Morphological and immunocytochemical identification of periacinar fibroblast-like cells derived from human pancreatic acini. Pancreas 1997, 14, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Cong, M.; Park, T.J.; Scholten, D.; Brenner, D.A.; Kisseleva, T. Contribution of bone marrow-derived fibrocytes to liver fibrosis. Hepatobiliary Surg. Nutr. 2015, 4, 34–47. [Google Scholar] [CrossRef]
- Li, Y.; Li, C.X.; Ye, H.; Chen, F.; Melamed, J.; Peng, Y.; Liu, J.; Wang, Z.; Tsou, H.C.; Wei, J.; et al. Decrease in stromal androgen receptor associates with androgen-independent disease and promotes prostate cancer cell proliferation and invasion. J. Cell. Mol. Med. 2008, 12, 2790–2798. [Google Scholar] [CrossRef] [Green Version]
- Palethorpe, H.M.; Leach, D.A.; Need, E.F.; Drew, P.A.; Smith, E. Myofibroblast androgen receptor expression determines cell survival in co-cultures of myofibroblasts and prostate cancer cells in vitro. Oncotarget 2018, 9, 19100–19114. [Google Scholar] [CrossRef] [Green Version]
- Webber, M.M.; Trakul, N.; Thraves, P.S.; Bello-DeOcampo, D.; Chu, W.W.; Storto, P.D.; Huard, T.K.; Rhim, J.S.; Williams, D.E. A human prostatic stromal myofibroblast cell line WPMY-1: A model for stromal-epithelial interactions in prostatic neoplasia. Carcinogenesis 1999, 20, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Younesi, F.S.; Son, D.O.; Firmino, J.; Hinz, B. Myofibroblast Markers and Microscopy Detection Methods in Cell Culture and Histology. Methods Mol. Biol. 2021, 2299, 17–47. [Google Scholar] [CrossRef]
- Liu, A.Y.; True, L.D. Characterization of prostate cell types by CD cell surface molecules. Am. J. Pathol. 2002, 160, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Rakocevic, J.; Orlic, D.; Mitrovic-Ajtic, O.; Tomasevic, M.; Dobric, M.; Zlatic, N.; Milasinovic, D.; Stankovic, G.; Ostojić, M.; Labudovic-Borovic, M. Endothelial cell markers from clinician’s perspective. Exp. Mol. Pathol. 2017, 102, 303–313. [Google Scholar] [CrossRef]
- Augsten, M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef]
- Cortez, E.; Roswall, P.; Pietras, K. Functional subsets of mesenchymal cell types in the tumor microenvironment. Semin. Cancer Biol. 2014, 25, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Ishii, G.; Ochiai, A.; Neri, S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv. Drug Deliv. Rev. 2016, 99, 186–196. [Google Scholar] [CrossRef]
- Joshi, R.S.; Kanugula, S.S.; Sudhir, S.; Pereira, M.P.; Jain, S.; Aghi, M.K. The Role of Cancer-Associated Fibroblasts in Tumor Progression. Cancers 2021, 13, 1399. [Google Scholar] [CrossRef] [PubMed]
- Franco, O.E.; Jiang, M.; Strand, D.W.; Peacock, J.; Fernandez, S.; Jackson, R.S., 2nd; Revelo, M.P.; Bhowmick, N.A.; Hayward, S.W. Altered TGF-β signaling in a subpopulation of human stromal cells promotes prostatic carcinogenesis. Cancer Res. 2011, 71, 1272–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiskowski, M.A.; Jackson, R.S., 2nd; Banerjee, J.; Li, X.; Kang, M.; Iturregui, J.M.; Franco, O.E.; Hayward, S.W.; Bhowmick, N.A. Role for stromal heterogeneity in prostate tumorigenesis. Cancer Res. 2011, 71, 3459–3470. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Peehl, D.M. Tumor-promoting phenotype of CD90hi prostate cancer-associated fibroblasts. Prostate 2009, 69, 991–1000. [Google Scholar] [CrossRef] [Green Version]
- Mezawa, Y.; Orimo, A. The roles of tumor- and metastasis-promoting carcinoma-associated fibroblasts in human carcinomas. Cell Tissue Res. 2016, 365, 675–689. [Google Scholar] [CrossRef]
- Direkze, N.C.; Hodivala-Dilke, K.; Jeffery, R.; Hunt, T.; Poulsom, R.; Oukrif, D.; Alison, M.R.; Wright, N.A. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004, 64, 8492–8495. [Google Scholar] [CrossRef] [Green Version]
- Forino, M.; Torregrossa, R.; Ceol, M.; Murer, L.; Vella, M.D.; Prete, D.D.; D’Angelo, A.; Anglani, F. TGFβ1 induces epithelial–mesenchymal transition, but not myofibroblast transdifferentiation of human kidney tubular epithelial cells in primary culture. Int. J. Exp. Pathol. 2006, 87, 197–208. [Google Scholar] [CrossRef]
- Jung, Y.; Kim, J.K.; Shiozawa, Y.; Wang, J.; Mishra, A.; Joseph, J.; Berry, J.E.; McGee, S.; Lee, E.; Sun, H.; et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun 2013, 4, 1795. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; Diprete, B.; Betz, K.S.; et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Du, L.; Lin, L.; Wang, Y. Tumour-associated mesenchymal stem/stromal cells: Emerging therapeutic targets. Nat. Rev. Drug Discov. 2017, 16, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Worthley, D.L.; Si, Y.; Quante, M.; Churchill, M.; Mukherjee, S.; Wang, T.C. Bone marrow cells as precursors of the tumor stroma. Exp. Cell Res. 2013, 319, 1650–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Oft, M.; Akhurst, R.J.; Balmain, A. Metastasis is driven by sequential elevation of H-ras and Smad2 levels. Nat. Cell Biol. 2002, 4, 487–494. [Google Scholar] [CrossRef]
- Potenta, S.; Zeisberg, E.; Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 2008, 99, 1375–1379. [Google Scholar] [CrossRef] [Green Version]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef]
- Alexander, J.; Cukierman, E. Stromal dynamic reciprocity in cancer: Intricacies of fibroblastic-ECM interactions. Curr. Opin. Cell Biol. 2016, 42, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Tian, B.; Lessan, K.; Kahm, J.; Kleidon, J.; Henke, C. beta 1 integrin regulates fibroblast viability during collagen matrix contraction through a phosphatidylinositol 3-kinase/Akt/protein kinase B signaling pathway. J. Biol. Chem. 2002, 277, 24667–24675. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Xu, G.; Wu, M.; Zhang, Y.; Li, Q.; Liu, P.; Zhu, T.; Song, A.; Zhao, L.; Han, Z.; et al. Overexpression of vimentin contributes to prostate cancer invasion and metastasis via src regulation. Anticancer Res. 2008, 28, 327–334. [Google Scholar] [PubMed]
- O’Connell, J.T.; Sugimoto, H.; Cooke, V.G.; MacDonald, B.A.; Mehta, A.I.; LeBleu, V.S.; Dewar, R.; Rocha, R.M.; Brentani, R.R.; Resnick, M.B.; et al. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc. Natl. Acad. Sci. USA 2011, 108, 16002–16007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öhlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ustach, C.V.; Taube, M.E.; Hurst, N.J., Jr.; Bhagat, S.; Bonfil, R.D.; Cher, M.L.; Schuger, L.; Kim, H.-R.C. A Potential Oncogenic Activity of Platelet-Derived Growth Factor D in Prostate Cancer Progression. Cancer Res. 2004, 64, 1722–1729. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M.D.; Watt, F.M. Fibroblast heterogeneity: Implications for human disease. J. Clin. Investig. 2018, 128, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Donjacour, A.A.; Thomson, A.A.; Cunha, G.R. FGF-10 plays an essential role in the growth of the fetal prostate. Dev. Biol. 2003, 261, 39–54. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Luo, Y.; Kan, M.; McKeehan, W.L. Fibroblast growth factor-10. A second candidate stromal to epithelial cell andromedin in prostate. J. Biol. Chem. 1999, 274, 12827–12834. [Google Scholar] [CrossRef] [Green Version]
- Memarzadeh, S.; Xin, L.; Mulholland, D.J.; Mansukhani, A.; Wu, H.; Teitell, M.A.; Witte, O.N. Enhanced Paracrine FGF10 Expression Promotes Formation of Multifocal Prostate Adenocarcinoma and an Increase in Epithelial Androgen Receptor. Cancer Cell 2007, 12, 572–585. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [Green Version]
- Ishii, K.; Mizokami, A.; Tsunoda, T.; Iguchi, K.; Kato, M.; Hori, Y.; Arima, K.; Namiki, M.; Sugimura, Y. Heterogenous induction of carcinoma-associated fibroblast-like differentiation in normal human prostatic fibroblasts by co-culturing with prostate cancer cells. J. Cell. Biochem. 2011, 112, 3604–3611. [Google Scholar] [CrossRef] [PubMed]
- Orr, B.; Riddick, A.C.P.; Stewart, G.D.; Anderson, R.A.; Franco, O.E.; Hayward, S.W.; Thomson, A.A. Identification of stromally expressed molecules in the prostate by tag-profiling of cancer-associated fibroblasts, normal fibroblasts and fetal prostate. Oncogene 2012, 31, 1130–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Strand, D.W.; Rowley, D.R. Fibroblast growth factor-2 mediates transforming growth factor-β action in prostate cancer reactive stroma. Oncogene 2008, 27, 450–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turpin, A.; Duterque-Coquillaud, M.; Vieillard, M.-H. Bone Metastasis: Current State of Play. Transl. Oncol. 2020, 13, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Engl, T.; Relja, B.; Marian, D.; Blumenberg, C.; Müller, I.; Beecken, W.D.; Jones, J.; Ringel, E.M.; Bereiter-Hahn, J.; Jonas, D.; et al. CXCR4 chemokine receptor mediates prostate tumor cell adhesion through alpha5 and beta3 integrins. Neoplasia 2006, 8, 290–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejana, E.; Hirschi, K.K.; Simons, M. The molecular basis of endothelial cell plasticity. Nat. Commun. 2017, 8, 14361. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Sohn, Y.J.; Lee, R.; Yoo, H.J.; Kang, J.Y.; Choi, N.; Na, D.; Yeon, J.H. Cancer-Associated Fibroblasts Differentiated by Exosomes Isolated from Cancer Cells Promote Cancer Cell Invasion. Int. J. Mol. Sci. 2020, 21, 8153. [Google Scholar] [CrossRef]
- Yeon, J.H.; Jeong, H.E.; Seo, H.; Cho, S.; Kim, K.; Na, D.; Chung, S.; Park, J.; Choi, N.; Kang, J.Y. Cancer-derived exosomes trigger endothelial to mesenchymal transition followed by the induction of cancer-associated fibroblasts. Acta Biomater. 2018, 76, 146–153. [Google Scholar] [CrossRef]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef] [Green Version]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef] [Green Version]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Kurose, K.; Gilley, K.; Matsumoto, S.; Watson, P.H.; Zhou, X.P.; Eng, C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat. Genet. 2002, 32, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Saidou, J.; Watabe, K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. 2010, 15, 166–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Cheaito, K.A.; Bahmad, H.F.; Hadadeh, O.; Saleh, E.; Dagher, C.; Hammoud, M.S.; Shahait, M.; Mrad, Z.A.; Nassif, S.; Tawil, A.; et al. EMT Markers in Locally-Advanced Prostate Cancer: Predicting Recurrence? Front. Oncol. 2019, 9, 131. [Google Scholar] [CrossRef] [Green Version]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769. [Google Scholar] [CrossRef] [Green Version]
- Montanari, M.; Rossetti, S.; Cavaliere, C.; D’Aniello, C.; Malzone, M.G.; Vanacore, D.; Di Franco, R.; La Mantia, E.; Iovane, G.; Piscitelli, R.; et al. Epithelial-mesenchymal transition in prostate cancer: An overview. Oncotarget 2017, 8, 35376–35389. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Bera, S. CAF cellular glycolysis: Linking cancer cells with the microenvironment. Tumour Biol. 2016, 37, 8503–8514. [Google Scholar] [CrossRef]
- Avery, D.; Govindaraju, P.; Jacob, M.; Todd, L.; Monslow, J.; Puré, E. Extracellular matrix directs phenotypic heterogeneity of activated fibroblasts. Matrix Biol. 2018, 67, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.; Kyprianou, N. Mechanisms navigating the TGF-β pathway in prostate cancer. Asian J. Urol. 2015, 2, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzaei, S.; Paskeh, M.D.A.; Saghari, Y.; Zarrabi, A.; Hamblin, M.R.; Entezari, M.; Hashemi, M.; Aref, A.R.; Hushmandi, K.; Kumar, A.P.; et al. Transforming growth factor-beta (TGF-β) in prostate cancer: A dual function mediator? Int. J. Biol. Macromol. 2022, 206, 435–452. [Google Scholar] [CrossRef] [PubMed]
- Grubisha, M.J.; Cifuentes, M.E.; Hammes, S.R.; DeFranco, D.B. A Local Paracrine and Endocrine Network Involving TGFβ, Cox-2, ROS, and Estrogen Receptor β Influences Reactive Stromal Cell Regulation of Prostate Cancer Cell Motility. Mol. Endocrinol. 2012, 26, 940–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlides, S.; Tsirigos, A.; Migneco, G.; Whitaker-Menezes, D.; Chiavarina, B.; Flomenberg, N.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Pestell, R.G.; et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle 2010, 9, 3485–3505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Mazor, M.; Kawano, Y.; Walker, M.M.; Leung, H.Y.; Armstrong, K.; Waxman, J.; Kypta, R.M. Analysis of Wnt Gene Expression in Prostate Cancer: Mutual Inhibition by WNT11 and the Androgen Receptor. Cancer Res. 2004, 64, 7918–7926. [Google Scholar] [CrossRef] [Green Version]
- Avgustinova, A.; Iravani, M.; Robertson, D.; Fearns, A.; Gao, Q.; Klingbeil, P.; Hanby, A.M.; Speirs, V.; Sahai, E.; Calvo, F.; et al. Tumour cell-derived Wnt7a recruits and activates fibroblasts to promote tumour aggressiveness. Nat. Commun. 2016, 7, 10305. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Placencio, V.; Iturregui, J.M.; Uwamariya, C.; Sharif-Afshar, A.R.; Koyama, T.; Hayward, S.W.; Bhowmick, N.A. Prostate tumor progression is mediated by a paracrine TGF-beta/Wnt3a signaling axis. Oncogene 2008, 27, 7118–7130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegner, K.A.; Mueller, B.R.; Unterberger, C.J.; Avila, E.J.; Ruetten, H.; Turco, A.E.; Oakes, S.R.; Girardi, N.M.; Halberg, R.B.; Swanson, S.M.; et al. Prostate epithelial-specific expression of activated PI3K drives stromal collagen production and accumulation. J. Pathol. 2020, 250, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target. Ther. 2021, 6, 218. [Google Scholar] [CrossRef] [PubMed]
- Procopio, M.G.; Laszlo, C.; Al Labban, D.; Kim, D.E.; Bordignon, P.; Jo, S.H.; Goruppi, S.; Menietti, E.; Ostano, P.; Ala, U.; et al. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat. Cell Biol. 2015, 17, 1193–1204. [Google Scholar] [CrossRef] [Green Version]
- Strell, C.; Paulsson, J.; Jin, S.B.; Tobin, N.P.; Mezheyeuski, A.; Roswall, P.; Mutgan, C.; Mitsios, N.; Johansson, H.; Wickberg, S.M.; et al. Impact of Epithelial-Stromal Interactions on Peritumoral Fibroblasts in Ductal Carcinoma in Situ. J. Natl. Cancer Inst. 2019, 111, 983–995. [Google Scholar] [CrossRef]
- Wang, Y.M.; Wang, W.; Qiu, E.D. Osteosarcoma cells induce differentiation of mesenchymal stem cells into cancer associated fibroblasts through Notch and Akt signaling pathway. Int. J. Clin. Exp. Pathol. 2017, 10, 8479–8486. [Google Scholar]
- Katzmann, D.J.; Stefan, C.J.; Babst, M.; Emr, S.D. Vps27 recruits ESCRT machinery to endosomes during MVB sorting. J. Cell Biol. 2003, 162, 413–423. [Google Scholar] [CrossRef]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef]
- Taylor, D.D.; Gercel-Taylor, C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol. Oncol. 2008, 110, 13–21. [Google Scholar] [CrossRef]
- Webber, J.P.; Spary, L.K.; Sanders, A.J.; Chowdhury, R.; Jiang, W.G.; Steadman, R.; Wymant, J.; Jones, A.T.; Kynaston, H.; Mason, M.D.; et al. Differentiation of tumour-promoting stromal myofibroblasts by cancer exosomes. Oncogene 2015, 34, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ren, H.; Dai, B.; Li, J.; Shang, L.; Huang, J.; Shi, X. Hepatocellular carcinoma-derived exosomal miRNA-21 contributes to tumor progression by converting hepatocyte stellate cells to cancer-associated fibroblasts. J. Exp. Clin. Cancer Res. 2018, 37, 324. [Google Scholar] [CrossRef] [PubMed]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinstein-Achiasaf, L.; Morein, D.; Ben-Yaakov, H.; Liubomirski, Y.; Meshel, T.; Elbaz, E.; Dorot, O.; Pichinuk, E.; Gershovits, M.; Weil, M.; et al. Persistent Inflammatory Stimulation Drives the Conversion of MSCs to Inflammatory CAFs That Promote Pro-Metastatic Characteristics in Breast Cancer Cells. Cancers 2021, 13, 1472. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Mellone, M.; Hanley, C.J.; Thirdborough, S.; Mellows, T.; Garcia, E.; Woo, J.; Tod, J.; Frampton, S.; Jenei, V.; Moutasim, K.A.; et al. Induction of fibroblast senescence generates a non-fibrogenic myofibroblast phenotype that differentially impacts on cancer prognosis. Aging 2016, 9, 114–132. [Google Scholar] [CrossRef] [Green Version]
- Fordyce, C.; Fessenden, T.; Pickering, C.; Jung, J.; Singla, V.; Berman, H.; Tlsty, T. DNA Damage Drives an Activin A–Dependent Induction of Cyclooxygenase-2 in Premalignant Cells and Lesions. Cancer Prev. Res. 2010, 3, 190–201. [Google Scholar] [CrossRef] [Green Version]
- Fordyce, C.A.; Patten, K.T.; Fessenden, T.B.; DeFilippis, R.; Hwang, E.S.; Zhao, J.; Tlsty, T.D. Cell-extrinsic consequences of epithelial stress: Activation of protumorigenic tissue phenotypes. Breast Cancer Res. 2012, 14, R155. [Google Scholar] [CrossRef] [Green Version]
- Chiarugi, P.; Paoli, P.; Cirri, P. Tumor microenvironment and metabolism in prostate cancer. Semin. Oncol. 2014, 41, 267–280. [Google Scholar] [CrossRef]
- Vlaeminck-Guillem, V. Extracellular Vesicles in Prostate Cancer Carcinogenesis, Diagnosis, and Management. Front. Oncol. 2018, 8, 222. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.M., Jr.; Matheus, W.E.; Garcia, P.V.; Stopiglia, R.M.; Billis, A.; Ferreira, U.; Fávaro, W.J. Characterization of reactive stroma in prostate cancer: Involvement of growth factors, metalloproteinase matrix, sexual hormones receptors and prostatic stem cells. Int. Braz. J. Urol. 2015, 41, 849–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahriari, K.; Shen, F.; Worrede-Mahdi, A.; Liu, Q.; Gong, Y.; Garcia, F.U.; Fatatis, A. Cooperation among heterogeneous prostate cancer cells in the bone metastatic niche. Oncogene 2017, 36, 2846–2856. [Google Scholar] [CrossRef] [PubMed]
- Ao, M.; Franco, O.E.; Park, D.; Raman, D.; Williams, K.; Hayward, S.W. Cross-talk between Paracrine-Acting Cytokine and Chemokine Pathways Promotes Malignancy in Benign Human Prostatic Epithelium. Cancer Res. 2007, 67, 4244–4253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochette, A.; Boufaied, N.; Scarlata, E.; Hamel, L.; Brimo, F.; Whitaker, H.C.; Ramos-Montoya, A.; Neal, D.E.; Dragomir, A.; Aprikian, A.; et al. Asporin is a stromally expressed marker associated with prostate cancer progression. Br. J. Cancer 2017, 116, 775–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Sterling, J.A.; Fan, K.H.; Vessella, R.L.; Shyr, Y.; Hayward, S.W.; Matrisian, L.M.; Bhowmick, N.A. Loss of TGF-β responsiveness in prostate stromal cells alters chemokine levels and facilitates the development of mixed osteoblastic/osteolytic bone lesions. Mol. Cancer Res. 2012, 10, 494–503. [Google Scholar] [CrossRef] [Green Version]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [Green Version]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [Green Version]
- Finak, G.; Bertos, N.; Pepin, F.; Sadekova, S.; Souleimanova, M.; Zhao, H.; Chen, H.; Omeroglu, G.; Meterissian, S.; Omeroglu, A.; et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med. 2008, 14, 518–527. [Google Scholar] [CrossRef]
- Jungwirth, U.; van Weverwijk, A.; Evans, R.J.; Jenkins, L.; Vicente, D.; Alexander, J.; Gao, Q.; Haider, S.; Iravani, M.; Isacke, C.M. Impairment of a distinct cancer-associated fibroblast population limits tumour growth and metastasis. Nat. Commun. 2021, 12, 3516. [Google Scholar] [CrossRef]
- Chekenya, M.; Krakstad, C.; Svendsen, A.; Netland, I.A.; Staalesen, V.; Tysnes, B.B.; Selheim, F.; Wang, J.; Sakariassen, P.Ø.; Sandal, T.; et al. The progenitor cell marker NG2/MPG promotes chemoresistance by activation of integrin-dependent PI3K/Akt signaling. Oncogene 2008, 27, 5182–5194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e816. [Google Scholar] [CrossRef]
- Driskell, R.R.; Lichtenberger, B.M.; Hoste, E.; Kretzschmar, K.; Simons, B.D.; Charalambous, M.; Ferron, S.R.; Herault, Y.; Pavlovic, G.; Ferguson-Smith, A.C.; et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 2013, 504, 277–281. [Google Scholar] [CrossRef] [Green Version]
- Rinkevich, Y.; Walmsley, G.G.; Hu, M.S.; Maan, Z.N.; Newman, A.M.; Drukker, M.; Januszyk, M.; Krampitz, G.W.; Gurtner, G.C.; Lorenz, H.P.; et al. Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science 2015, 348, aaa2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.Y.; Roudier, M.P.; True, L.D. Heterogeneity in Primary and Metastatic Prostate Cancer as Defined by Cell Surface CD Profile. Am. J. Pathol. 2004, 165, 1543–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dakhova, O.; Ozen, M.; Creighton, C.J.; Li, R.; Ayala, G.; Rowley, D.; Ittmann, M. Global gene expression analysis of reactive stroma in prostate cancer. Clin. Cancer Res. 2009, 15, 3979–3989. [Google Scholar] [CrossRef] [Green Version]
- Eiro, N.; Fernandez-Gomez, J.; Sacristán, R.; Fernandez-Garcia, B.; Lobo, B.; Gonzalez-Suarez, J.; Quintas, A.; Escaf, S.; Vizoso, F.J. Stromal factors involved in human prostate cancer development, progression and castration resistance. J. Cancer Res. Clin. Oncol. 2017, 143, 351–359. [Google Scholar] [CrossRef]
- Nordby, Y.; Richardsen, E.; Rakaee, M.; Ness, N.; Donnem, T.; Patel, H.R.H.; Busund, L.-T.; Bremnes, R.M.; Andersen, S. High expression of PDGFR-β in prostate cancer stroma is independently associated with clinical and biochemical prostate cancer recurrence. Sci. Rep. 2017, 7, 43378. [Google Scholar] [CrossRef]
- Kisselbach, L.; Merges, M.; Bossie, A.; Boyd, A. CD90 Expression on human primary cells and elimination of contaminating fibroblasts from cell cultures. Cytotechnology 2009, 59, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Muguruma, Y.; Yahata, T.; Miyatake, H.; Sakai, D.; Mochida, J.; Hotta, T.; Ando, K. Expression of CD90 on keratinocyte stem/progenitor cells. Br. J. Dermatol. 2006, 154, 1062–1070. [Google Scholar] [CrossRef]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Céspedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Gucciardo, F.; Pirson, S.; Baudin, L.; Lebeau, A.; Noël, A. uPARAP/Endo180: A multifaceted protein of mesenchymal cells. Cell Mol. Life Sci. 2022, 79, 255. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josefsson, A.; Adamo, H.; Hammarsten, P.; Granfors, T.; Stattin, P.; Egevad, L.; Laurent, A.E.; Wikström, P.; Bergh, A. Prostate cancer increases hyaluronan in surrounding nonmalignant stroma, and this response is associated with tumor growth and an unfavorable outcome. Am. J. Pathol. 2011, 179, 1961–1968. [Google Scholar] [CrossRef]
- Nissen, N.I.; Karsdal, M.; Willumsen, N. Collagens and Cancer associated fibroblasts in the reactive stroma and its relation to Cancer biology. J. Exp. Clin. Cancer Res. 2019, 38, 115. [Google Scholar] [CrossRef] [Green Version]
- Rozario, T.; DeSimone, D.W. The extracellular matrix in development and morphogenesis: A dynamic view. Dev. Biol. 2010, 341, 126–140. [Google Scholar] [CrossRef] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Payne, S.L.; Hendrix, M.J.; Kirschmann, D.A. Paradoxical roles for lysyl oxidases in cancer--a prospect. J. Cell Biochem. 2007, 101, 1338–1354. [Google Scholar] [CrossRef]
- Rodríguez, C.; Rodríguez-Sinovas, A.; Martínez-González, J. Lysyl oxidase as a potential therapeutic target. Drug News Perspect. 2008, 21, 218–224. [Google Scholar] [CrossRef]
- Kakkad, S.M.; Solaiyappan, M.; O’Rourke, B.; Stasinopoulos, I.; Ackerstaff, E.; Raman, V.; Bhujwalla, Z.M.; Glunde, K. Hypoxic tumor microenvironments reduce collagen I fiber density. Neoplasia 2010, 12, 608–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leeming, D.J.; Koizumi, M.; Qvist, P.; Barkholt, V.; Zhang, C.; Henriksen, K.; Byrjalsen, I.; Karsdal, M.A. Serum N-Terminal Propeptide of Collagen Type I is Associated with the Number of Bone Metastases in Breast and Prostate Cancer and Correlates to Other Bone Related Markers. Biomark Cancer 2011, 3, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penet, M.-F.; Kakkad, S.; Pathak, A.P.; Krishnamachary, B.; Mironchik, Y.; Raman, V.; Solaiyappan, M.; Bhujwalla, Z.M. Structure and Function of a Prostate Cancer Dissemination–Permissive Extracellular Matrix. Clin. Cancer Res. 2017, 23, 2245–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Leight, J.L.; Drain, A.P.; Weaver, V.M. Extracellular Matrix Remodeling and Stiffening Modulate Tumor Phenotype and Treatment Response. Annu. Rev. Cancer Biol. 2017, 1, 313–334. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Levesque, C.; Nelson, P.S. Cellular Constituents of the Prostate Stroma: Key Contributors to Prostate Cancer Progression and Therapy Resistance. Cold Spring Harb. Perspect. Med. 2018, 8, a030510. [Google Scholar] [CrossRef]
- Midwood, K.S.; Orend, G. The role of tenascin-C in tissue injury and tumorigenesis. J. Cell Commun. Signal. 2009, 3, 287–310. [Google Scholar] [CrossRef] [Green Version]
- Ishii, K.; Imanaka-Yoshida, K.; Yoshida, T.; Sugimura, Y. Role of stromal tenascin-C in mouse prostatic development and epithelial cell differentiation. Dev. Biol. 2008, 324, 310–319. [Google Scholar] [CrossRef] [Green Version]
- Chiquet, M.; Sarasa-Renedo, A.; Tunç-Civelek, V. Induction of tenascin-C by cyclic tensile strain versus growth factors: Distinct contributions by Rho/ROCK and MAPK signaling pathways. Biochim. Biophys. Acta 2004, 1693, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schauer, I.G.; Ressler, S.J.; Tuxhorn, J.A.; Dang, T.D.; Rowley, D.R. Elevated epithelial expression of interleukin-8 correlates with myofibroblast reactive stroma in benign prostatic hyperplasia. Urology 2008, 72, 205–213. [Google Scholar] [CrossRef] [PubMed]
- De Wever, O.; Nguyen, Q.D.; Van Hoorde, L.; Bracke, M.; Bruyneel, E.; Gespach, C.; Mareel, M. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004, 18, 1016–1018. [Google Scholar] [CrossRef] [PubMed]
- Trebaul, A.; Chan, E.K.; Midwood, K.S. Regulation of fibroblast migration by tenascin-C. Biochem. Soc. Trans. 2007, 35, 695–697. [Google Scholar] [CrossRef]
- Ni, W.-D.; Yang, Z.-T.; Cui, C.-A.; Cui, Y.; Fang, L.-Y.; Xuan, Y.-H. Tenascin-C is a potential cancer-associated fibroblasts marker and predicts poor prognosis in prostate cancer. Biochem. Biophys. Res. Commun. 2017, 486, 607–612. [Google Scholar] [CrossRef]
- Jiang, D.; Liang, J.; Noble, P.W. Hyaluronan in tissue injury and repair. Annu. Rev. Cell Dev. Biol. 2007, 23, 435–461. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Tao, D.; Zhang, P.; Liu, X.; Zhang, Y.; Cheng, J.; Yuan, H.; Liu, L.; Jiang, H. Hyaluronan synthase 2 expressed by cancer-associated fibroblasts promotes oral cancer invasion. J. Exp. Clin. Cancer Res. 2016, 35, 181. [Google Scholar] [CrossRef] [Green Version]
- Davidson, L.A.; Dzamba, B.D.; Keller, R.; Desimone, D.W. Live imaging of cell protrusive activity, and extracellular matrix assembly and remodeling during morphogenesis in the frog, Xenopus laevis. Dev. Dyn. 2008, 237, 2684–2692. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.; Hynes, R.; Kirschner, M. Temporal and spatial regulation of fibronectin in early Xenopus development. Cell 1984, 36, 729–740. [Google Scholar] [CrossRef]
- Attieh, Y.; Clark, A.G.; Grass, C.; Richon, S.; Pocard, M.; Mariani, P.; Elkhatib, N.; Betz, T.; Gurchenkov, B.; Vignjevic, D.M. Cancer-associated fibroblasts lead tumor invasion through integrin-β3-dependent fibronectin assembly. J. Cell Biol. 2017, 216, 3509–3520. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Chippada-Venkata, U.D.; Oh, W.K. Roles of matrix metalloproteinases and their natural inhibitors in prostate cancer progression. Cancers 2014, 6, 1298–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boire, A.; Covic, L.; Agarwal, A.; Jacques, S.; Sherifi, S.; Kuliopulos, A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 2005, 120, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Shareef, Z.; Kardooni, H.; Murillo-Garzón, V.; Domenici, G.; Stylianakis, E.; Steel, J.H.; Rabano, M.; Gorroño-Etxebarria, I.; Zabalza, I.; Vivanco, M.D.; et al. Protective effect of stromal Dickkopf-3 in prostate cancer: Opposing roles for TGFBI and ECM-1. Oncogene 2018, 37, 5305–5324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, D.; Al-Shareef, Z.; Gorroño-Etxebarria, I.; Atkins, S.; Turrell, F.; Chhetri, J.; Bengoa-Vergniory, N.; Zenzmaier, C.; Berger, P.; Waxman, J.; et al. Dickkopf-3 regulates prostate epithelial cell acinar morphogenesis and prostate cancer cell invasion by limiting TGF-β-dependent activation of matrix metalloproteases. Carcinogenesis 2016, 37, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, N.P.; Reyes, E.; Tapia, P.; Badínez, L.; Orellana, N. Differential expression of matrix metalloproteinase-2 expression in disseminated tumor cells and micrometastasis in bone marrow of patients with nonmetastatic and metastatic prostate cancer: Theoretical considerations and clinical implications-an immunocytochemical study. Bone Marrow Res. 2012, 2012, 259351. [Google Scholar] [CrossRef] [Green Version]
- Kajiwara, S.; Ishii, K.; Sasaki, T.; Kato, M.; Nishikawa, K.; Kanda, H.; Arima, K.; Watanabe, M.; Sugimura, Y. Castration-induced stromal remodeling disrupts the reconstituted prostate epithelial structure. Lab. Investig. 2020, 100, 670–681. [Google Scholar] [CrossRef]
- Sampson, N.; Brunner, E.; Weber, A.; Puhr, M.; Schäfer, G.; Szyndralewiez, C.; Klocker, H. Inhibition of Nox4-dependent ROS signaling attenuates prostate fibroblast activation and abrogates stromal-mediated protumorigenic interactions. Int. J. Cancer 2018, 143, 383–395. [Google Scholar] [CrossRef] [Green Version]
- Sampson, N.; Koziel, R.; Zenzmaier, C.; Bubendorf, L.; Plas, E.; Jansen-Dürr, P.; Berger, P. ROS signaling by NOX4 drives fibroblast-to-myofibroblast differentiation in the diseased prostatic stroma. Mol. Endocrinol. 2011, 25, 503–515. [Google Scholar] [CrossRef] [Green Version]
- Kwabi-Addo, B.; Ozen, M.; Ittmann, M. The role of fibroblast growth factors and their receptors in prostate cancer. Endocr. Relat. Cancer 2004, 11, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Polnaszek, N.; Kwabi-Addo, B.; Peterson, L.E.; Ozen, M.; Greenberg, N.M.; Ortega, S.; Basilico, C.; Ittmann, M. Fibroblast growth factor 2 promotes tumor progression in an autochthonous mouse model of prostate cancer. Cancer Res. 2003, 63, 5754–5760. [Google Scholar] [PubMed]
- Choi, S.C.; Kim, S.J.; Choi, J.H.; Park, C.Y.; Shim, W.J.; Lim, D.S. Fibroblast growth factor-2 and -4 promote the proliferation of bone marrow mesenchymal stem cells by the activation of the PI3K-Akt and ERK1/2 signaling pathways. Stem Cells Dev. 2008, 17, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Kouhara, H.; Hadari, Y.R.; Spivak-Kroizman, T.; Schilling, J.; Bar-Sagi, D.; Lax, I.; Schlessinger, J. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 1997, 89, 693–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astin, J.W.; Batson, J.; Kadir, S.; Charlet, J.; Persad, R.A.; Gillatt, D.; Oxley, J.D.; Nobes, C.D. Competition amongst Eph receptors regulates contact inhibition of locomotion and invasiveness in prostate cancer cells. Nat. Cell Biol. 2010, 12, 1194–1204. [Google Scholar] [CrossRef]
- Li, S.; Wu, Z.; Ma, P.; Xu, Y.; Chen, Y.; Wang, H.; He, P.; Kang, Z.; Yin, L.; Zhao, Y.; et al. Ligand-dependent EphA7 signaling inhibits prostate tumor growth and progression. Cell Death Dis. 2017, 8, e3122. [Google Scholar] [CrossRef] [Green Version]
- Salem, A.F.; Gambini, L.; Billet, S.; Sun, Y.; Oshiro, H.; Zhao, M.; Hoffman, R.M.; Bhowmick, N.A.; Pellecchia, M. Prostate Cancer Metastases Are Strongly Inhibited by Agonistic Epha2 Ligands in an Orthotopic Mouse Model. Cancers 2020, 12, 2854. [Google Scholar] [CrossRef]
- Josson, S.; Gururajan, M.; Sung, S.Y.; Hu, P.; Shao, C.; Zhau, H.E.; Liu, C.; Lichterman, J.; Duan, P.; Li, Q.; et al. Stromal fibroblast-derived miR-409 promotes epithelial-to-mesenchymal transition and prostate tumorigenesis. Oncogene 2015, 34, 2690–2699. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [Green Version]
- Lanning, B.; Webber, J.; Uysal-Onganer, P.; Jiang, W.G.; Clayton, A.; Dart, D.A. Prostate Cancer Cell Extracellular Vesicles Increase Mineralisation of Bone Osteoblast Precursor Cells in an In Vitro Model. Biology 2021, 10, 318. [Google Scholar] [CrossRef]
- Ramirez-Garrastacho, M.; Bajo-Santos, C.; Line, A.; Martens-Uzunova, E.S.; de la Fuente, J.M.; Moros, M.; Soekmadji, C.; Tasken, K.A.; Llorente, A. Extracellular vesicles as a source of prostate cancer biomarkers in liquid biopsies: A decade of research. Br. J. Cancer 2022, 126, 331–350. [Google Scholar] [CrossRef]
- Sadovska, L.; Zayakin, P.; Bajo-Santos, C.; Endzeliņš, E.; Auders, J.; Keiša, L.; Jansons, J.; Lietuvietis, V.; Linē, A. Effects of urinary extracellular vesicles from prostate cancer patients on the transcriptomes of cancer-associated and normal fibroblasts. BMC Cancer 2022, 22, 1055. [Google Scholar] [CrossRef] [PubMed]
- Shephard, A.P.; Giles, P.; Mbengue, M.; Alraies, A.; Spary, L.K.; Kynaston, H.; Gurney, M.J.; Falcón-Pérez, J.M.; Royo, F.; Tabi, Z.; et al. Stroma-derived extracellular vesicle mRNA signatures inform histological nature of prostate cancer. J. Extracell. Vesicles 2021, 10, e12150. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; Carter, D.R.F.; Clayton, A.; Lambert, D.W.; Raposo, G.; Vader, P. Challenges and directions in studying cell–cell communication by extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2022, 23, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.; Stone, T.C.; Katilius, E.; Smith, B.C.; Gordon, B.; Mason, M.D.; Tabi, Z.; Brewis, I.A.; Clayton, A. Proteomics Analysis of Cancer Exosomes Using a Novel Modified Aptamer-based Array (SOMAscanTM) Platform. Mol. Cell. Proteom. 2014, 13, 1050–1064. [Google Scholar] [CrossRef] [Green Version]
- Culig, Z.; Puhr, M. Interleukin-6 and prostate cancer: Current developments and unsolved questions. Mol. Cell Endocrinol. 2018, 462, 25–30. [Google Scholar] [CrossRef]
- Ishii, K.; Sasaki, T.; Iguchi, K.; Kajiwara, S.; Kato, M.; Kanda, H.; Hirokawa, Y.; Arima, K.; Mizokami, A.; Sugimura, Y. Interleukin-6 induces VEGF secretion from prostate cancer cells in a manner independent of androgen receptor activation. Prostate 2018, 78, 849–856. [Google Scholar] [CrossRef]
- Yang, L.; Wang, L.; Lin, H.K.; Kan, P.Y.; Xie, S.; Tsai, M.Y.; Wang, P.H.; Chen, Y.T.; Chang, C. Interleukin-6 differentially regulates androgen receptor transactivation via PI3K-Akt, STAT3, and MAPK, three distinct signal pathways in prostate cancer cells. Biochem. Biophys. Res. Commun. 2003, 305, 462–469. [Google Scholar] [CrossRef]
- Paland, N.; Kamer, I.; Kogan-Sakin, I.; Madar, S.; Goldfinger, N.; Rotter, V. Differential influence of normal and cancer-associated fibroblasts on the growth of human epithelial cells in an in vitro cocultivation model of prostate cancer. Mol. Cancer Res. 2009, 7, 1212–1223. [Google Scholar] [CrossRef] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Ziaee, S.; Chu, G.C.; Huang, J.M.; Sieh, S.; Chung, L.W. Prostate cancer metastasis: Roles of recruitment and reprogramming, cell signal network and three-dimensional growth characteristics. Transl. Androl. Urol. 2015, 4, 438–454. [Google Scholar] [CrossRef]
- Cirri, P.; Chiarugi, P. Cancer-associated-fibroblasts and tumour cells: A diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2012, 31, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Duda, D.G.; Duyverman, A.M.; Kohno, M.; Snuderl, M.; Steller, E.J.; Fukumura, D.; Jain, R.K. Malignant cells facilitate lung metastasis by bringing their own soil. Proc. Natl. Acad. Sci. USA 2010, 107, 21677–21682. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langley, R.R.; Fidler, I.J. The seed and soil hypothesis revisited--the role of tumor-stroma interactions in metastasis to different organs. Int. J. Cancer 2011, 128, 2527–2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Shen, P.; Lee, Y.-C.; Pan, J.; Song, J.H.; Pan, T.; Lin, S.-C.; Liang, X.; Wang, G.; Panaretakis, T.; et al. Multiple pathways coordinating reprogramming of endothelial cells into osteoblasts by BMP4. iScience 2021, 24, 102388. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.Y.; Wu, J.Q.; He, Z.H.; He, M.F.; Sun, H.B. Cancer-associated fibroblast regulate proliferation and migration of prostate cancer cells through TGF-β signaling pathway. Life Sci. 2019, 235, 116791. [Google Scholar] [CrossRef]
- Eikesdal, H.P.; Becker, L.M.; Teng, Y.; Kizu, A.; Carstens, J.L.; Kanasaki, K.; Sugimoto, H.; LeBleu, V.S.; Kalluri, R. BMP7 Signaling in TGFBR2-Deficient Stromal Cells Provokes Epithelial Carcinogenesis. Mol. Cancer Res. 2018, 16, 1568–1578. [Google Scholar] [CrossRef] [Green Version]
- Feng, D.; Shi, X.; Xiong, Q.; Zhang, F.; Li, D.; Wei, W.; Yang, L. A Ferroptosis-Related Gene Prognostic Index Associated With Biochemical Recurrence and Radiation Resistance for Patients With Prostate Cancer Undergoing Radical Radiotherapy. Front. Cell Dev. Biol. 2022, 10, 803766. [Google Scholar] [CrossRef]
- Saleem, M.; Adhami, V.M.; Ahmad, N.; Gupta, S.; Mukhtar, H. Prognostic significance of metastasis-associated protein S100A4 (Mts1) in prostate cancer progression and chemoprevention regimens in an autochthonous mouse model. Clin. Cancer Res. 2005, 11, 147–153. [Google Scholar] [CrossRef]
- Ferrer, F.A.; Miller, L.J.; Andrawis, R.I.; Kurtzman, S.H.; Albertsen, P.C.; Laudone, V.P.; Kreutzer, D.L. Vascular Endothelial Growth Factor (VEGF) Expression in Human Prostate Cancer: In Situ and in Vitro Expression of VEGF by Human Prostate Cancer Cells. J. Urol. 1997, 157, 2329–2333. [Google Scholar] [CrossRef]
- San Martin, R.; Pathak, R.; Jain, A.; Jung, S.Y.; Hilsenbeck, S.G.; Piña-Barba, M.C.; Sikora, A.G.; Pienta, K.J.; Rowley, D.R. Tenascin-C and Integrin α9 Mediate Interactions of Prostate Cancer with the Bone Microenvironment. Cancer Res. 2017, 77, 5977–5988. [Google Scholar] [CrossRef] [Green Version]
- De Boeck, A.; Hendrix, A.; Maynard, D.; Van Bockstal, M.; Daniëls, A.; Pauwels, P.; Gespach, C.; Bracke, M.; De Wever, O. Differential secretome analysis of cancer-associated fibroblasts and bone marrow-derived precursors to identify microenvironmental regulators of colon cancer progression. Proteomics 2013, 13, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, L.; Jungwirth, U.; Avgustinova, A.; Iravani, M.; Mills, A.; Haider, S.; Harper, J.; Isacke, C.M. Cancer-Associated Fibroblasts Suppress CD8+ T-cell Infiltration and Confer Resistance to Immune-Checkpoint Blockade. Cancer Res. 2022, 82, 2904–2917. [Google Scholar] [CrossRef] [PubMed]
- Cassim, S.; Pouyssegur, J. Tumor Microenvironment: A Metabolic Player that Shapes the Immune Response. Int. J. Mol. Sci. 2020, 21, 157. [Google Scholar] [CrossRef] [Green Version]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-de-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Stultz, J.; Fong, L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 697–717. [Google Scholar] [CrossRef]
- Davidsson, S.; Ohlson, A.-L.; Andersson, S.-O.; Fall, K.; Meisner, A.; Fiorentino, M.; Andrén, O.; Rider, J.R. CD4 helper T cells, CD8 cytotoxic T cells, and FOXP3+ regulatory T cells with respect to lethal prostate cancer. Mod. Pathol. 2013, 26, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Fox, S.B.; Launchbury, R.; Bates, G.J.; Han, C.; Shaida, N.; Malone, P.R.; Harris, A.L.; Banham, A.H. The number of regulatory T cells in prostate cancer is associated with the androgen receptor and hypoxia-inducible factor (HIF)-2α but not HIF-1α. Prostate 2007, 67, 623–629. [Google Scholar] [CrossRef]
- Ghahremanifard, P.; Chanda, A.; Bonni, S.; Bose, P. TGF-β Mediated Immune Evasion in Cancer—Spotlight on Cancer-Associated Fibroblasts. Cancers 2020, 12, 3650. [Google Scholar] [CrossRef]
- Boudadi, K.; Antonarakis, E.S. Resistance to Novel Antiandrogen Therapies in Metastatic Castration-Resistant Prostate Cancer. Clin. Med. Insights Oncol. 2016, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Jiang, X.; Liang, X.; Jiang, G. Molecular and cellular mechanisms of castration resistant prostate cancer. Oncol. Lett. 2018, 15, 6063–6076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koushyar, S.; Meniel, V.S.; Phesse, T.J.; Pearson, H.B. Exploring the Wnt Pathway as a Therapeutic Target for Prostate Cancer. Biomolecules 2022, 12, 309. [Google Scholar] [CrossRef]
- Wadosky, K.M.; Koochekpour, S. Molecular mechanisms underlying resistance to androgen deprivation therapy in prostate cancer. Oncotarget 2016, 7, 64447–64470. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Wang, Y.Y.; Li, D.; Wang, C.; Wang, S.Y.; Shao, S.H.; Zhu, Z.Y.; Zhao, J.; Zhang, Y.; Ruan, Y.; et al. LMO2 upregulation due to AR deactivation in cancer-associated fibroblasts induces non-cell-autonomous growth of prostate cancer after androgen deprivation. Cancer Lett. 2021, 503, 138–150. [Google Scholar] [CrossRef]
- Cioni, B.; Nevedomskaya, E.; Melis, M.H.M.; van Burgsteden, J.; Stelloo, S.; Hodel, E.; Spinozzi, D.; de Jong, J.; van der Poel, H.; de Boer, J.P.; et al. Loss of androgen receptor signaling in prostate cancer-associated fibroblasts (CAFs) promotes CCL2- and CXCL8-mediated cancer cell migration. Mol. Oncol. 2018, 12, 1308–1323. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Haldar, S.; Placencio, V.; Madhav, A.; Rohena-Rivera, K.; Agarwal, P.; Duong, F.; Angara, B.; Tripathi, M.; Liu, Z.; et al. Stromal epigenetic alterations drive metabolic and neuroendocrine prostate cancer reprogramming. J. Clin. Investig. 2018, 128, 4472–4484. [Google Scholar] [CrossRef] [Green Version]
- Neuwirt, H.; Bouchal, J.; Kharaishvili, G.; Ploner, C.; Jöhrer, K.; Pitterl, F.; Weber, A.; Klocker, H.; Eder, I.E. Cancer-associated fibroblasts promote prostate tumor growth and progression through upregulation of cholesterol and steroid biosynthesis. Cell Commun. Signal. 2020, 18, 11. [Google Scholar] [CrossRef] [Green Version]
- Cioni, B.; Zwart, W.; Bergman, A.M. Androgen receptor moonlighting in the prostate cancer microenvironment. Endocr. Relat. Cancer 2018, 25, R331–R349. [Google Scholar] [CrossRef]
- Niu, Y.; Altuwaijri, S.; Yeh, S.; Lai, K.-P.; Yu, S.; Chuang, K.-H.; Huang, S.-P.; Lardy, H.; Chang, C. Targeting the stromal androgen receptor in primary prostate tumors at earlier stages. Proc. Natl. Acad. Sci. USA 2008, 105, 12188–12193. [Google Scholar] [CrossRef] [Green Version]
- Leach, D.A.; Need, E.F.; Toivanen, R.; Trotta, A.P.; Palethorpe, H.M.; Tamblyn, D.J.; Kopsaftis, T.; England, G.M.; Smith, E.; Drew, P.A.; et al. Stromal androgen receptor regulates the composition of the microenvironment to influence prostate cancer outcome. Oncotarget 2015, 6, 16135–16150. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.-i.; Hosokawa, H.; Koizumi, M.; Endo, Y.; Yahata, T.; Ando, K.; Hozumi, K. LMO2 is essential to maintain the ability of progenitors to differentiate into T-cell lineage in mice. eLife 2021, 10, e68227. [Google Scholar] [CrossRef] [PubMed]
- Ricciardelli, C.; Choong, C.S.; Buchanan, G.; Vivekanandan, S.; Neufing, P.; Stahl, J.; Marshall, V.R.; Horsfall, D.J.; Tilley, W.D. Androgen receptor levels in prostate cancer epithelial and peritumoral stromal cells identify non-organ confined disease. Prostate 2005, 63, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Jha, R.; Melamed, J.; Shapiro, E.; Hayward, S.W.; Lee, P. Stromal androgen receptor in prostate development and cancer. Am. J. Pathol. 2014, 184, 2598–2607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wikström, P.; Marusic, J.; Stattin, P.; Bergh, A. Low stroma androgen receptor level in normal and tumor prostate tissue is related to poor outcome in prostate cancer patients. Prostate 2009, 69, 799–809. [Google Scholar] [CrossRef]
- Foley, C.; Mitsiades, N. Moving Beyond the Androgen Receptor (AR): Targeting AR-Interacting Proteins to Treat Prostate Cancer. Horm. Cancer 2016, 7, 84–103. [Google Scholar] [CrossRef] [Green Version]
- Leach, D.A.; Buchanan, G. Stromal Androgen Receptor in Prostate Cancer Development and Progression. Cancers 2017, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Leach, D.A.; Panagopoulos, V.; Nash, C.; Bevan, C.; Thomson, A.A.; Selth, L.A.; Buchanan, G. Cell-lineage specificity and role of AP-1 in the prostate fibroblast androgen receptor cistrome. Mol. Cell Endocrinol. 2017, 439, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhao, J.; Ding, M.; Su, Y.; Cui, D.; Jiang, C.; Zhao, S.; Jia, G.; Wang, X.; Ruan, Y.; et al. Loss of exosomal miR-146a-5p from cancer-associated fibroblasts after androgen deprivation therapy contributes to prostate cancer metastasis. J. Exp. Clin. Cancer Res. 2020, 39, 282. [Google Scholar] [CrossRef]
- Iacona, J.R.; Lutz, C.S. miR-146a-5p: Expression, regulation, and functions in cancer. WIREs RNA 2019, 10, e1533. [Google Scholar] [CrossRef]
- Aggarwal, R.R.; Feng, F.Y.; Small, E.J. Emerging Categories of Disease in Advanced Prostate Cancer and Their Therapeutic Implications. Oncology 2017, 31, 467–474. [Google Scholar]
- Sumanasuriya, S.; De Bono, J. Treatment of Advanced Prostate Cancer-A Review of Current Therapies and Future Promise. Cold Spring Harb. Perspect. Med. 2018, 8, a030635. [Google Scholar] [CrossRef]
- Ammirante, M.; Shalapour, S.; Kang, Y.; Jamieson, C.A.; Karin, M. Tissue injury and hypoxia promote malignant progression of prostate cancer by inducing CXCL13 expression in tumor myofibroblasts. Proc. Natl. Acad. Sci. USA 2014, 111, 14776–14781. [Google Scholar] [CrossRef] [Green Version]
- Ammirante, M.; Luo, J.L.; Grivennikov, S.; Nedospasov, S.; Karin, M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 2010, 464, 302–305. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat. Med. 2012, 18, 1359–1368. [Google Scholar] [CrossRef] [Green Version]
- Cheteh, E.H.; Augsten, M.; Rundqvist, H.; Bianchi, J.; Sarne, V.; Egevad, L.; Bykov, V.J.N.; Östman, A.; Wiman, K.G. Human cancer-associated fibroblasts enhance glutathione levels and antagonize drug-induced prostate cancer cell death. Cell Death Dis. 2017, 8, e2848. [Google Scholar] [CrossRef] [Green Version]
- Gladson, C.L.; Welch, D.R. New insights into the role of CXCR4 in prostate cancer metastasis. Cancer Biol. Ther. 2008, 7, 1849–1851. [Google Scholar] [CrossRef] [Green Version]
- Hooijberg, J.H.; Pinedo, H.M.; Vrasdonk, C.; Priebe, W.; Lankelma, J.; Broxterman, H.J. The effect of glutathione on the ATPase activity of MRP1 in its natural membranes. FEBS Lett. 2000, 469, 47–51. [Google Scholar] [CrossRef] [Green Version]
- Joyce, H.; McCann, A.; Clynes, M.; Larkin, A. Influence of multidrug resistance and drug transport proteins on chemotherapy drug metabolism. Expert Opin. Drug Metab. Toxicol. 2015, 11, 795–809. [Google Scholar] [CrossRef]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014. [Google Scholar] [CrossRef] [Green Version]
- Shan, G.; Gu, J.; Zhou, D.; Li, L.; Cheng, W.; Wang, Y.; Tang, T.; Wang, X. Cancer-associated fibroblast-secreted exosomal miR-423-5p promotes chemotherapy resistance in prostate cancer by targeting GREM2 through the TGF-β signaling pathway. Exp. Mol. Med. 2020, 52, 1809–1822. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef]
- Tsuyada, A.; Chow, A.; Wu, J.; Somlo, G.; Chu, P.; Loera, S.; Luu, T.; Li, A.X.; Wu, X.; Ye, W.; et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012, 72, 2768–2779. [Google Scholar] [CrossRef] [Green Version]
- Versantvoort, C.H.M.; Broxterman, H.J.; Bagrij, T.; Scheper, R.J.; Twentyman, P.R. Regulation by glutathione of drug transport in multidrug-resistant human lung tumour cell lines overexpressing multidrug resistance-associated protein. Br. J. Cancer 1995, 72, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Zlotnik, A. Chemokines and cancer. Int. J. Cancer 2006, 119, 2026–2029. [Google Scholar] [CrossRef]
- Li, G.; Satyamoorthy, K.; Herlyn, M. N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res. 2001, 61, 3819–3825. [Google Scholar]
- Domanska, U.M.; Timmer-Bosscha, H.; Nagengast, W.B.; Oude Munnink, T.H.; Kruizinga, R.C.; Ananias, H.J.; Kliphuis, N.M.; Huls, G.; De Vries, E.G.; de Jong, I.J.; et al. CXCR4 inhibition with AMD3100 sensitizes prostate cancer to docetaxel chemotherapy. Neoplasia 2012, 14, 709–718. [Google Scholar] [CrossRef] [Green Version]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venning, F.A.; Wullkopf, L.; Erler, J.T. Targeting ECM Disrupts Cancer Progression. Front. Oncol. 2015, 5, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel Iii, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef] [Green Version]
- Kharaziha, P.; Rodriguez, P.; Li, Q.; Rundqvist, H.; Björklund, A.C.; Augsten, M.; Ullén, A.; Egevad, L.; Wiklund, P.; Nilsson, S.; et al. Targeting of distinct signaling cascades and cancer-associated fibroblasts define the efficacy of Sorafenib against prostate cancer cells. Cell Death Dis. 2012, 3, e262. [Google Scholar] [CrossRef] [Green Version]
- Shen, T.; Li, Y.; Zhu, S.; Yu, J.; Zhang, B.; Chen, X.; Zhang, Z.; Ma, Y.; Niu, Y.; Shang, Z. YAP1 plays a key role of the conversion of normal fibroblasts into cancer-associated fibroblasts that contribute to prostate cancer progression. J. Exp. Clin. Cancer Res. 2020, 39, 36. [Google Scholar] [CrossRef]
- Kuser-Abali, G.; Alptekin, A.; Lewis, M.; Garraway, I.P.; Cinar, B. YAP1 and AR interactions contribute to the switch from androgen-dependent to castration-resistant growth in prostate cancer. Nat. Commun. 2015, 6, 8126. [Google Scholar] [CrossRef] [Green Version]
- Lang, J.; Zhao, X.; Qi, Y.; Zhang, Y.; Han, X.; Ding, Y.; Guan, J.; Ji, T.; Zhao, Y.; Nie, G. Reshaping Prostate Tumor Microenvironment to Suppress Metastasis via Cancer-Associated Fibroblast Inactivation with Peptide-Assembly-Based Nanosystem. ACS Nano 2019, 13, 12357–12371. [Google Scholar] [CrossRef]
- Aigner, A.; Renneberg, H.; Bojunga, J.; Apel, J.; Nelson, P.S.; Czubayko, F. Ribozyme-targeting of a secreted FGF-binding protein (FGF-BP) inhibits proliferation of prostate cancer cells in vitro and in vivo. Oncogene 2002, 21, 5733–5742. [Google Scholar] [CrossRef] [Green Version]
- Herbert, C.; Schieborr, U.; Saxena, K.; Juraszek, J.; De Smet, F.; Alcouffe, C.; Bianciotto, M.; Saladino, G.; Sibrac, D.; Kudlinzki, D.; et al. Molecular Mechanism of SSR128129E, an Extracellularly Acting, Small-Molecule, Allosteric Inhibitor of FGF Receptor Signaling. Cancer Cell 2016, 30, 176–178. [Google Scholar] [CrossRef]
- Okada-Ban, M.; Thiery, J.P.; Jouanneau, J. Fibroblast growth factor-2. Int. J. Biochem. Cell Biol. 2000, 32, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.; Kim, D.H.; Lee, Y.H.; Jang, J.H.; Surh, Y.J. Fibroblast growth factor-2, derived from cancer-associated fibroblasts, stimulates growth and progression of human breast cancer cells via FGFR1 signaling. Mol. Carcinog. 2020, 59, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Otero, N.; Clinch, A.B.; Hope, J.; Wang, W.; Reinhart-King, C.A.; King, M.R. Cancer associated fibroblasts confer shear resistance to circulating tumor cells during prostate cancer metastatic progression. Oncotarget 2020, 11, 1037–1050. [Google Scholar] [CrossRef]
- Rettig, W.J.; Garin-Chesa, P.; Beresford, H.R.; Oettgen, H.F.; Melamed, M.R.; Old, L.J. Cell-surface glycoproteins of human sarcomas: Differential expression in normal and malignant tissues and cultured cells. Proc. Natl. Acad. Sci. USA 1988, 85, 3110–3114. [Google Scholar] [CrossRef] [Green Version]
- LeBeau, A.M.; Brennen, W.N.; Aggarwal, S.; Denmeade, S.R. Targeting the cancer stroma with a fibroblast activation protein-activated promelittin protoxin. Mol. Cancer Ther. 2009, 8, 1378–1386. [Google Scholar] [CrossRef] [Green Version]
- Scott, A.M.; Wiseman, G.; Welt, S.; Adjei, A.; Lee, F.T.; Hopkins, W.; Divgi, C.R.; Hanson, L.H.; Mitchell, P.; Gansen, D.N.; et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res. 2003, 9, 1639–1647. [Google Scholar] [PubMed]
- Brennen, W.N.; Rosen, D.M.; Wang, H.; Isaacs, J.T.; Denmeade, S.R. Targeting carcinoma-associated fibroblasts within the tumor stroma with a fibroblast activation protein-activated prodrug. J. Natl. Cancer Inst. 2012, 104, 1320–1334. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, C.F.; van Putten, S.M.; Lund, I.K.; Melander, M.C.; Nørregaard, K.S.; Jürgensen, H.J.; Reckzeh, K.; Christensen, K.R.; Ingvarsen, S.Z.; Gårdsvoll, H.; et al. The collagen receptor uPARAP/Endo180 as a novel target for antibody-drug conjugate mediated treatment of mesenchymal and leukemic cancers. Oncotarget 2017, 8, 44605–44624. [Google Scholar] [CrossRef]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Tateishi, K.; Kudo, Y.; Hoshikawa, M.; Tanaka, M.; Nakatsuka, T.; Fujiwara, H.; Miyabayashi, K.; Takahashi, R.; Tanaka, Y.; et al. Stromal remodeling by the BET bromodomain inhibitor JQ1 suppresses the progression of human pancreatic cancer. Oncotarget 2016, 7, 61469–61484. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Newby, J.M.; Lin, C.M.; Zhang, L.; Xu, F.; Kim, W.Y.; Forest, M.G.; Lai, S.K.; Milowsky, M.I.; Wobker, S.E.; et al. The Binding Site Barrier Elicited by Tumor-Associated Fibroblasts Interferes Disposition of Nanoparticles in Stroma-Vessel Type Tumors. ACS Nano 2016, 10, 9243–9258. [Google Scholar] [CrossRef]
- Nishihara, H. Human pathological basis of blood vessels and stromal tissue for nanotechnology. Adv. Drug Deliv. Rev. 2014, 74, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Sluka, P.; Davis, I.D. Cell mates: Paracrine and stromal targets for prostate cancer therapy. Nat. Rev. Urol. 2013, 10, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.; Wang, L.S.; Scholler, J.; Monslow, J.; Avery, D.; Newick, K.; O’Brien, S.; Evans, R.A.; Bajor, D.J.; Clendenin, C.; et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res. 2015, 75, 2800–2810. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ertl, H.C. Depletion of FAP+ cells reduces immunosuppressive cells and improves metabolism and functions CD8+ T cells within tumors. Oncotarget 2016, 7, 23282–23299. [Google Scholar] [CrossRef] [Green Version]
- Roberts, E.W.; Deonarine, A.; Jones, J.O.; Denton, A.E.; Feig, C.; Lyons, S.K.; Espeli, M.; Kraman, M.; McKenna, B.; Wells, R.J.; et al. Depletion of stromal cells expressing fibroblast activation protein-α from skeletal muscle and bone marrow results in cachexia and anemia. J. Exp. Med. 2013, 210, 1137–1151. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Owen, J.S.; Clayton, A.; Pearson, H.B. Cancer-Associated Fibroblast Heterogeneity, Activation and Function: Implications for Prostate Cancer. Biomolecules 2023, 13, 67. https://doi.org/10.3390/biom13010067
Owen JS, Clayton A, Pearson HB. Cancer-Associated Fibroblast Heterogeneity, Activation and Function: Implications for Prostate Cancer. Biomolecules. 2023; 13(1):67. https://doi.org/10.3390/biom13010067
Chicago/Turabian StyleOwen, Jasmine S., Aled Clayton, and Helen B. Pearson. 2023. "Cancer-Associated Fibroblast Heterogeneity, Activation and Function: Implications for Prostate Cancer" Biomolecules 13, no. 1: 67. https://doi.org/10.3390/biom13010067
APA StyleOwen, J. S., Clayton, A., & Pearson, H. B. (2023). Cancer-Associated Fibroblast Heterogeneity, Activation and Function: Implications for Prostate Cancer. Biomolecules, 13(1), 67. https://doi.org/10.3390/biom13010067