

DAEMDA: A Method with Dual-Channel Attention Encoding for miRNA–Disease Association Prediction


Abstract
:1. Introduction
- The encoder based on the transformer architecture is used to deeply and comprehensively explore the latent node features by fully exploiting the graph properties in the heterogeneous network constructed from multi-feature information so that the node feature embedding is obtained with richer semantic information.
- DAEMDA organically combines node embedding encoding obtained based on graph attention and self-attention encoders to obtain high-quality feature embedding combinations.
- DAEMDA can predict MDA end-to-end, outperforming baseline methods in multiple experiments on publicly available datasets and achieving excellent performance in case studies with more stringent validation criteria.
2. Materials and Methods
2.1. Experimental Data
2.1.1. Human miRNA–Disease Associations
2.1.2. Disease Semantic Similarity
2.1.3. MiRNA Functional Similarity
2.1.4. Gaussian Interaction Profile Kernel Similarity for miRNAs and Diseases
2.1.5. Aggregating Similarity Features and Constructing Complex Networks
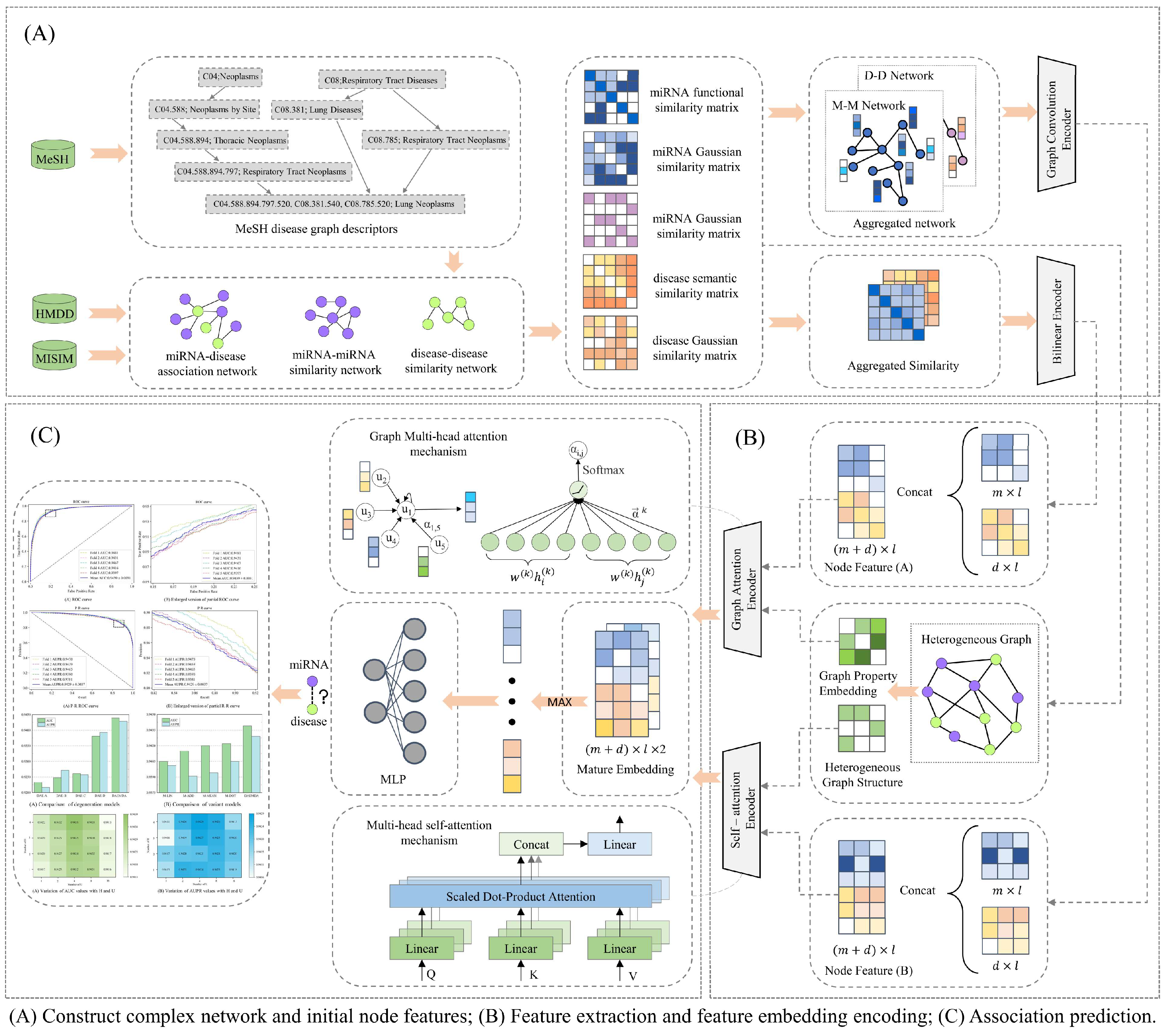
2.2. Model Framework
2.2.1. Graph Attention-Based Encoder
2.2.2. Self-Attention-Based Encoder
2.2.3. Predicting miRNA–Disease Associations
3. Results
3.1. Experimental Settings
3.1.1. Parameter Settings
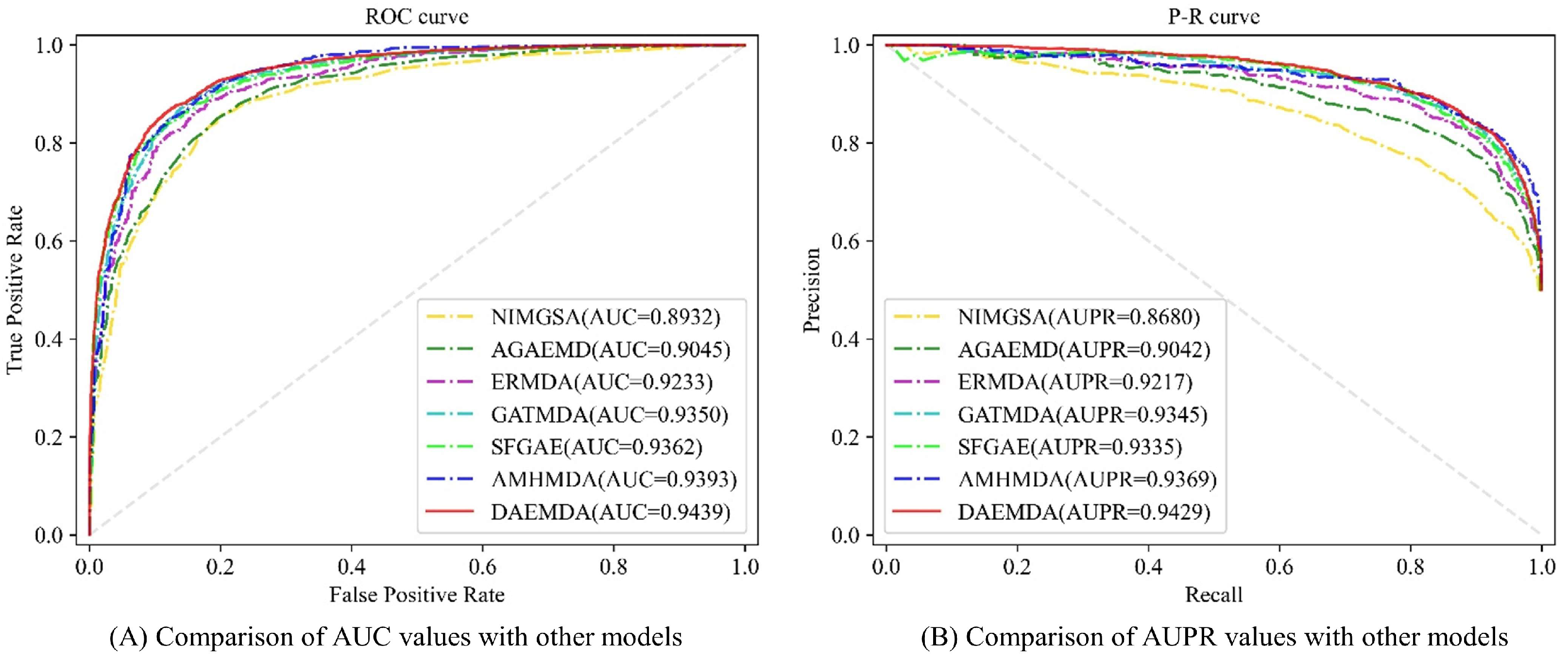
3.1.2. Baselines
- NIMGSA [31]: A neural inductive matrix completion-based method with graph autoencoders and self-attention mechanism for miRNA–disease association prediction.
- AGAEMD [20]: The authors considered the node-to-node attention profile in the heterogeneity graph and dynamically updated the miRNA functional similarity matrix during model iterations.
- ERMDA [32]: The authors utilize a resampling method to process the existing data and use integrated learning to introduce a soft voting method for the final prediction of the association.
- GATMDA [33]: A new computational method to discover unknown miRNA–disease associations based on a graph attention network with multi-source information, which effectively fuses linear and non-linear features.
- SFGAE [34]: Association prediction between two classes of nodes was accomplished by constructing miRNA self-embeddings and disease self-embeddings, independent of graph interactions between the two classes of graphs.
- AMHMDA [35]: GCN is used to extract multi-perspective node information from multi-similarity network species for constructing hypergraphs, and an attention mechanism is used to fuse features from hypergraph nodes for predicting MDA.
3.1.3. Experimental Environment
Hardware Equipment Used in This Study
- CPU: Intel Xeon Platinum 8255C, 2.50 GHz;
- GPU: RTX 2080Ti (11 GB), cuda12.0;
- Memory: 40 GB.
3.1.4. Evaluation Metrics
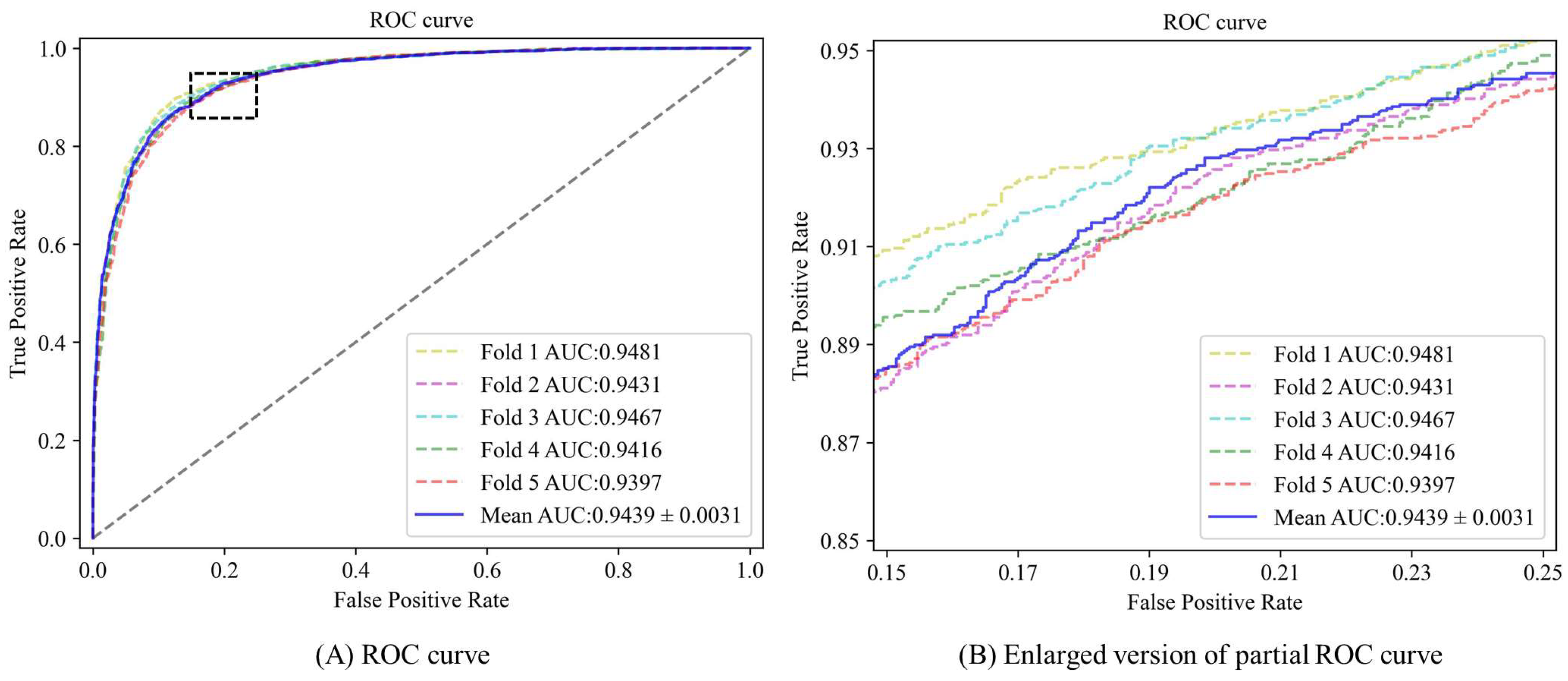
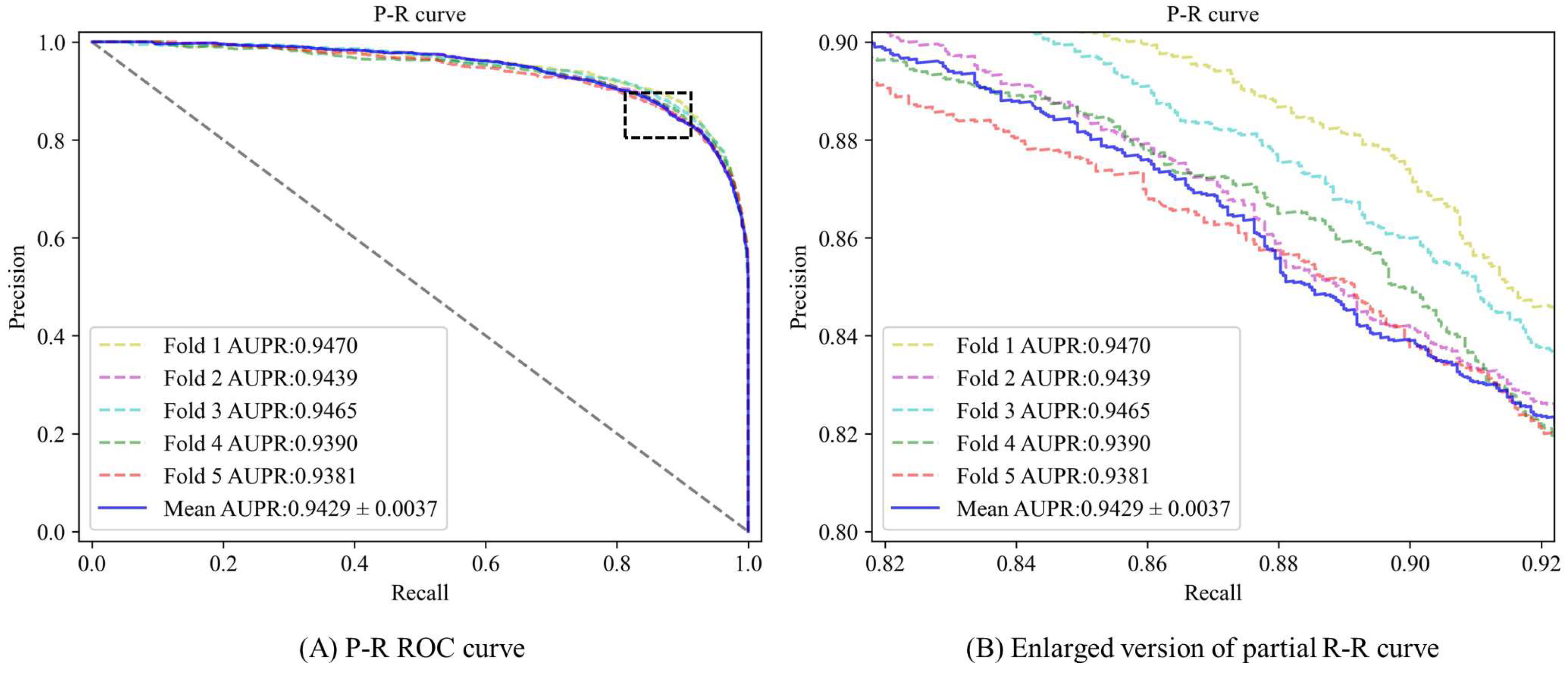
3.2. Performance Evaluation
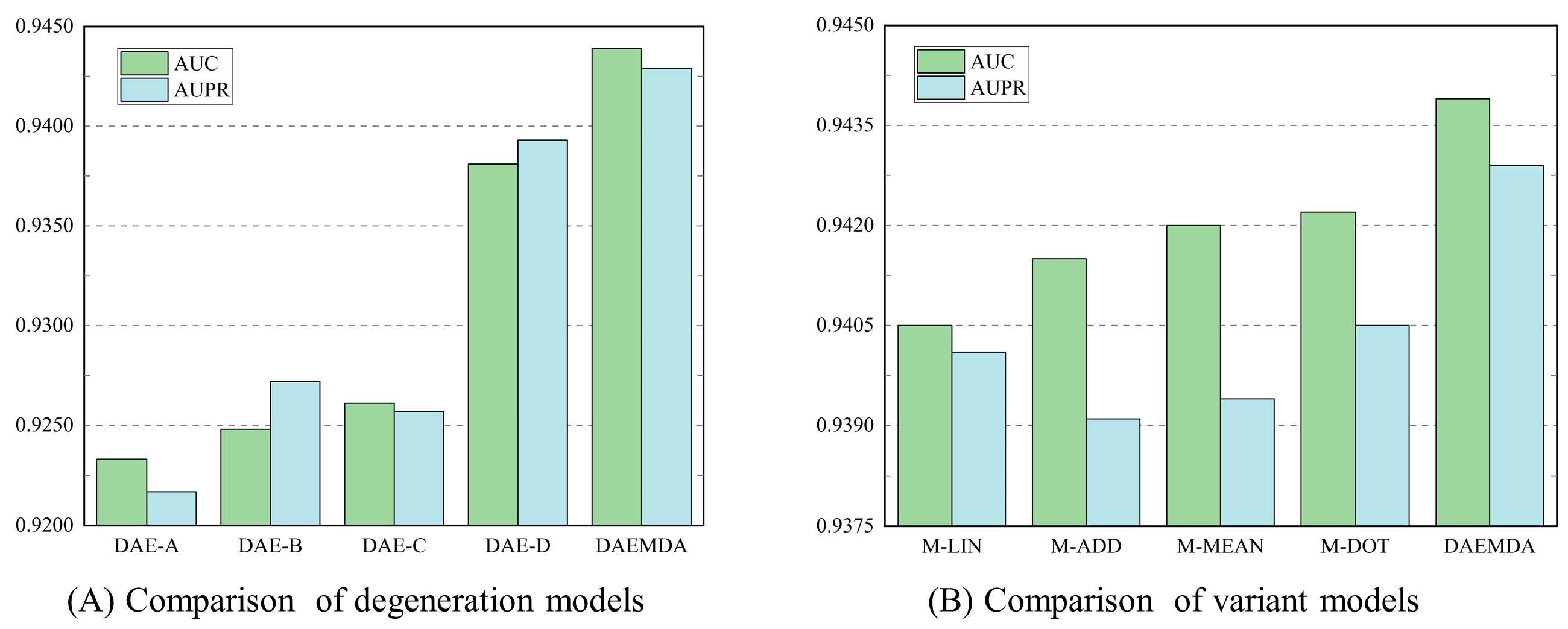
3.3. Ablation Experiment
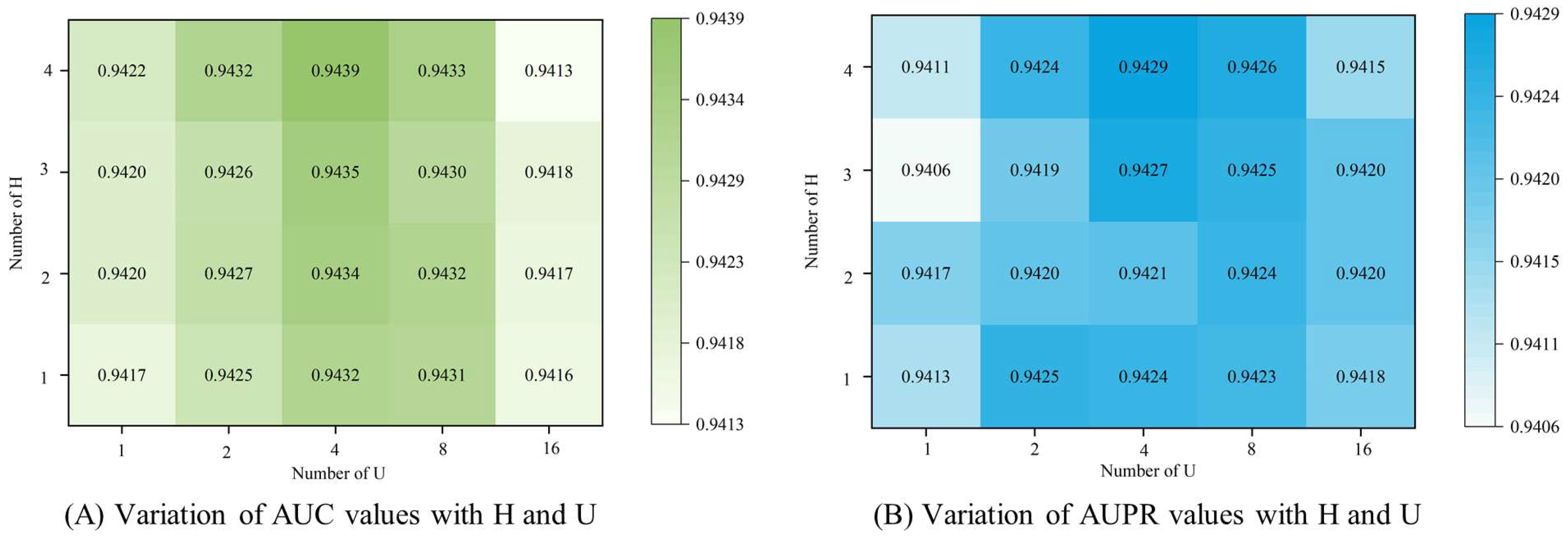
3.4. Parameter Analysis
3.4.1. Number of Attention Heads
3.4.2. Number of Feature Dimension
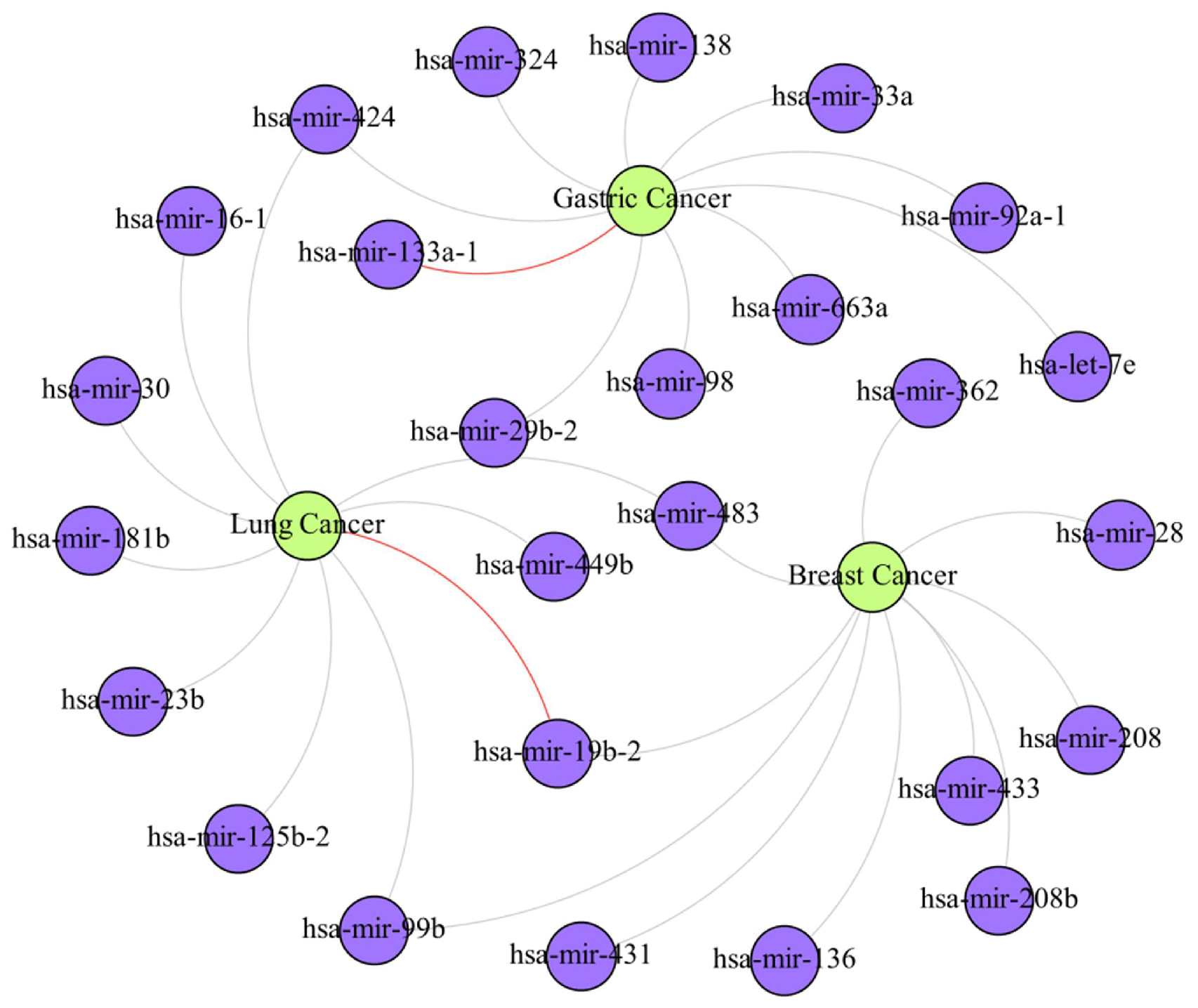
4. Case Study
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef]
- Li, Z.; Huang, H.; Chen, P.; He, M.; Li, Y.; Arnovitz, S.; Jiang, X.; He, C.; Hyjek, E.; Zhang, J.; et al. Publisher Correction: miR-196b directly targets both HOXA9/MEIS1 oncogenes and FAS tumour suppressor in MLL-rearranged leukaemia. Nat. Commun. 2018, 9, 16192. [Google Scholar] [CrossRef]
- Wilkening, S.; Bader, A. Quantitative real-time polymerase chain reaction: Methodical analysis and mathematical model. J. Biomol. Tech. 2004, 15, 107–111. [Google Scholar] [PubMed]
- Mendell, J.T.; Olson, E.N. MicroRNAs in stress signaling and human disease. Cell 2012, 148, 1172–1187. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Cui, F.; Lei, C.; Ma, S.; Huang, J.; Wang, X.; Qian, H.; Zhang, D.; Yang, Y. ATG7-mediated autophagy involves in miR-138-5p regulated self-renewal and invasion of lung cancer stem-like cells derived from A549 cells. Anticancer Drugs 2021, 32, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Liao, B.; Li, X.; Li, K. Network Consistency Projection for Human miRNA-Disease Associations Inference. Sci. Rep. 2016, 6, 36054. [Google Scholar] [CrossRef]
- Shi, H.; Zhang, G.; Zhou, M.; Cheng, L.; Yang, H.; Wang, J.; Sun, J.; Wang, Z. Integration of Multiple Genomic and Phenotype Data to Infer Novel miRNA-Disease Associations. PLoS ONE 2016, 11, e0148521. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, Z.C.; Xie, D.; Huang, D.S.; Zhao, Q.; Yan, G.Y.; You, Z.H. A novel computational model based on super-disease and miRNA for potential miRNA-disease association prediction. Mol. Biosyst. 2017, 13, 1202–1212. [Google Scholar] [CrossRef]
- Niu, Y.W.; Liu, H.; Wang, G.H.; Yan, G.Y. Maximal entropy random walk on heterogenous network for MIRNA-disease Association prediction. Math. Biosci. 2018, 306, 1–9. [Google Scholar] [CrossRef]
- Chen, X.; Wang, L.; Qu, J.; Guan, N.N.; Li, J.Q. Predicting miRNA-disease association based on inductive matrix completion. Bioinformatics 2018, 34, 4256–4265. [Google Scholar] [CrossRef]
- Pasquier, C.; Gardès, J. Prediction of miRNA-disease associations with a vector space model. Sci. Rep. 2016, 6, 27036. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, Q.F.; Yan, G.Y. RKNNMDA: Ranking-based KNN for MiRNA-Disease Association prediction. RNA Biol. 2017, 14, 952–962. [Google Scholar] [CrossRef]
- Chen, X.; Huang, L.; Xie, D.; Zhao, Q. EGBMMDA: Extreme Gradient Boosting Machine for MiRNA-Disease Association prediction. Cell Death Dis. 2018, 9, 3. [Google Scholar] [CrossRef]
- Che, K.; Guo, M.; Wang, C.; Liu, X.; Chen, X. Predicting MiRNA-Disease Association by Latent Feature Extraction with Positive Samples. Genes 2019, 10, 80. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, Y.; Li, A.; Li, Z.; Liu, W.; Chen, Z. Bipartite Heterogeneous Network Method Based on Co-neighbor for MiRNA-Disease Association Prediction. Front. Genet. 2019, 10, 385. [Google Scholar] [CrossRef]
- Wang, L.; You, Z.H.; Chen, X.; Li, Y.M.; Dong, Y.N.; Li, L.P.; Zheng, K. LMTRDA: Using logistic model tree to predict MiRNA-disease associations by fusing multi-source information of sequences and similarities. PLoS Comput. Biol. 2019, 15, e1006865. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, X.; Yin, J. Adaptive boosting-based computational model for predicting potential miRNA-disease associations. Bioinformatics 2019, 35, 4730–4738. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Wu, Q.W.; Gao, Z.; Ni, J.C.; Zheng, C.H. MiRNA-disease association prediction via hypergraph learning based on high-dimensionality features. BMC Med. Inform. Decis. Mak. 2021, 21, 133. [Google Scholar] [CrossRef]
- Ding, Y.; Tian, L.P.; Lei, X.; Liao, B.; Wu, F.X. Variational graph auto-encoders for miRNA-disease association prediction. Methods 2021, 192, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Fang, J.; Sun, Y.; Xie, G.; Lin, Z.; Gu, G. Predicting miRNA-Disease Associations via Node-Level Attention Graph Auto-Encoder. IEEE/ACM Trans. Comput. Biol. Bioinform. 2023, 20, 1308–1318. [Google Scholar] [CrossRef]
- Lou, Z.; Cheng, Z.; Li, H.; Teng, Z.; Liu, Y.; Tian, Z. Predicting miRNA-disease associations via learning multimodal networks and fusing mixed neighborhood information. Brief. Bioinform. 2022, 23, bbac159. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Luo, J.; Shen, C.; Lai, Z. Multi-view Multichannel Attention Graph Convolutional Network for miRNA-disease association prediction. Brief. Bioinform. 2021, 22, bbab174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, Z.; Wang, X.; Meng, Z.; Cui, W. MHTAN-DTI: Metapath-based hierarchical transformer and attention network for drug-target interaction prediction. Brief. Bioinform. 2023, 24, bbad079. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, Z.; Wang, K.; Gao, X.; Wang, G. End-to-end interpretable disease-gene association prediction. Brief. Bioinform. 2023, 24, bbad118. [Google Scholar] [CrossRef]
- Gu, P.; Wu, T.; Zou, M.; Pan, Y.; Guo, J.; Xiahou, J.; Peng, X.; Li, H.; Ma, J.; Zhang, L. Multi-Head Self-Attention Model for Classification of Temporal Lobe Epilepsy Subtypes. Front. Physiol. 2020, 11, 604764. [Google Scholar] [CrossRef]
- Ying, C.; Cai, T.; Luo, S.; Zheng, S.; Ke, G.; He, D.; Shen, Y.; Liu, T.-Y. Do Transformers Really Perform Bad for Graph Representation? arXiv 2021, arXiv:2106.05234. [Google Scholar] [CrossRef]
- Huang, Z.; Shi, J.; Gao, Y.; Cui, C.; Zhang, S.; Li, J.; Zhou, Y.; Cui, Q. HMDD v3.0: A database for experimentally supported human microRNA-disease associations. Nucleic Acids Res. 2019, 47, D1013–D1017. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef]
- van Laarhoven, T.; Nabuurs, S.B.; Marchiori, E. Gaussian interaction profile kernels for predicting drug-target interaction. Bioinformatics 2011, 27, 3036–3043. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. Adv. Neural Inf. Process. Syst. 2017, 30. [Google Scholar]
- Jin, C.; Shi, Z.; Lin, K.; Zhang, H. Predicting miRNA-Disease Association Based on Neural Inductive Matrix Completion with Graph Autoencoders and Self-Attention Mechanism. Biomolecules 2022, 12, 64. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Wang, Z.; Liu, Z.; Duan, X.; Song, J.; Guo, M. Predicting miRNA-disease associations using an ensemble learning framework with resampling method. Brief. Bioinform. 2022, 23, bbab543. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Fang, T.; Zhang, Y.; Liang, C.; Xiao, Q.; Luo, J. Predicting miRNA-disease associations based on graph attention network with multi-source information. BMC Bioinform. 2022, 23, 244. [Google Scholar] [CrossRef]
- Ma, M.; Na, S.; Zhang, X.; Chen, C.; Xu, J. SFGAE: A self-feature-based graph autoencoder model for miRNA-disease associations prediction. Brief. Bioinform. 2022, 23, bbac340. [Google Scholar] [CrossRef]
- Ning, Q.; Zhao, Y.; Gao, J.; Chen, C.; Li, X.; Li, T.; Yin, M. AMHMDA: Attention aware multi-view similarity networks and hypergraph learning for miRNA-disease associations identification. Brief. Bioinform. 2023, 24, bbad094. [Google Scholar] [CrossRef]
- Xu, F.; Wang, Y.; Ling, Y.; Zhou, C.; Wang, H.; Teschendorff, A.E.; Zhao, Y.; Zhao, H.; He, Y.; Zhang, G.; et al. dbDEMC 3.0: Functional Exploration of Differentially Expressed miRNAs in Cancers of Human and Model Organisms. Genom. Proteom. Bioinform. 2022, 20, 446–454. [Google Scholar] [CrossRef]
- Zhang, J.X.; Song, W.; Chen, Z.H.; Wei, J.H.; Liao, Y.J.; Lei, J.; Hu, M.; Chen, G.Z.; Liao, B.; Lu, J.; et al. Prognostic and predictive value of a microRNA signature in stage II colon cancer: A microRNA expression analysis. Lancet Oncol. 2013, 14, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Umezu, T.; Tsuneyama, K.; Kanekura, K.; Hayakawa, M.; Tanahashi, T.; Kawano, M.; Taguchi, Y.H.; Toyoda, H.; Tamori, A.; Kuroda, M.; et al. Comprehensive analysis of liver and blood miRNA in precancerous conditions. Sci. Rep. 2020, 10, 21766. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Full Data | miRNA | Disease |
|---|---|---|---|
| # nodes | 1444 | 853 | 591 |
| # edges | 12,446 | - | - |
| # density | 0.0247 | - | - |
| # degree | 17.238 | 14.591 | 21.059 |
| # Ave_cen | 0.0119 | 0.0101 | 0.0146 |
| Testing Set | Accuracy | F1-Score | Precision | Recall |
|---|---|---|---|---|
| 1 | 0.8737 | 0.8682 | 0.9076 | 0.8321 |
| 2 | 0.8777 | 0.8763 | 0.8862 | 0.8666 |
| 3 | 0.8797 | 0.8817 | 0.8671 | 0.8967 |
| 4 | 0.8773 | 0.8763 | 0.8830 | 0.8698 |
| 5 | 0.8638 | 0.8706 | 0.8294 | 0.9160 |
| Mean | 0.8744 ± 0.0057 | 0.8746 ± 0.0048 | 0.8747 ± 0.0261 | 0.8763 ± 0.0286 |
| True Labels | Predicted Labels | |
|---|---|---|
| Yes MDA | No MDA | |
| Yes MDA | TP = 2187 | FN = 338 |
| No MDA | FP = 302 | TN = 2151 |
| Method | AUC | AUPR |
|---|---|---|
| NIMGSA | 0.8932 | 0.8680 |
| AGAEMD | 0.9045 | 0.9042 |
| ERMDA | 0.9233 | 0.9217 |
| GATMDA | 0.9350 | 0.9345 |
| SFGAE | 0.9362 | 0.9335 |
| AMHMDA | 0.9393 | 0.9369 |
| DAEMDA | 0.9439 | 0.9429 |
| Method | DAE-A | DAE-B | DAE-C | DAE-D | DAEMDA |
|---|---|---|---|---|---|
| AUC | 0.9233 | 0.9248 | 0.9261 | 0.9381 | 0.9439 |
| AUPR | 0.9217 | 0.9272 | 0.9257 | 0.9393 | 0.9429 |
| Method | M-LIN | M-DOT | M-MEAN | M-ADD | DAEMDA |
|---|---|---|---|---|---|
| AUC | 0.9405 | 0.9415 | 0.9420 | 0.9422 | 0.9439 |
| AUPR | 0.9401 | 0.9391 | 0.9394 | 0.9405 | 0.9429 |
| Cancer | Top 10 Prediction | |||||
|---|---|---|---|---|---|---|
| Rank | miRNA | Evidence | Rank | miRNA | Evidence | |
| Breast Cancer | 1 | hsa-mir-28 | dbDEMC | 6 | hsa-mir-362 | dbDEMC |
| 2 | hsa-mir-483 | dbDEMC | 7 | hsa-mir-208 | dbDEMC | |
| 3 | hsa-mir-99b | dbDEMC | 8 | hsa-mir-19b-2 | dbDEMC | |
| 4 | hsa-mir-136 | dbDEMC | 9 | hsa-mir-433 | dbDEMC | |
| 5 | hsa-mir-431 | dbDEMC | 10 | hsa-mir-208b | dbDEMC | |
| Gastric Cancer | 1 | hsa-mir-29b-2 | dbDEMC | 6 | hsa-mir-92a-1 | dbDEMC |
| 2 | hsa-let-7e | dbDEMC | 7 | hsa-mir-98 | dbDEMC | |
| 3 | hsa-mir-33a | dbDEMC | 8 | hsa-mir-324 | dbDEMC | |
| 4 | hsa-mir-424 | dbDEMC | 9 | hsa-mir-138 | dbDEMC | |
| 5 | hsa-mir-133a-1 | Unconfirmed | 10 | hsa-mir-663a | dbDEMC | |
| Lung Cancer | 1 | hsa-mir-424 | dbDEMC | 6 | hsa-mir-99b | dbDEMC |
| 2 | hsa-mir-125b-2 | dbDEMC | 7 | hsa-mir-30 | dbDEMC | |
| 3 | hsa-mir-181b | dbDEMC | 8 | hsa-mir-483 | dbDEMC | |
| 4 | hsa-mir-23b | dbDEMC | 9 | hsa-mir-449b | dbDEMC | |
| 5 | hsa-mir-19b-2 | Unconfirmed | 10 | hsa-mir-16-1 | dbDEMC | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, B.; Sun, W.; Xu, D.; Wang, G.; Zhang, T. DAEMDA: A Method with Dual-Channel Attention Encoding for miRNA–Disease Association Prediction. Biomolecules 2023, 13, 1514. https://doi.org/10.3390/biom13101514
Dong B, Sun W, Xu D, Wang G, Zhang T. DAEMDA: A Method with Dual-Channel Attention Encoding for miRNA–Disease Association Prediction. Biomolecules. 2023; 13(10):1514. https://doi.org/10.3390/biom13101514
Chicago/Turabian StyleDong, Benzhi, Weidong Sun, Dali Xu, Guohua Wang, and Tianjiao Zhang. 2023. "DAEMDA: A Method with Dual-Channel Attention Encoding for miRNA–Disease Association Prediction" Biomolecules 13, no. 10: 1514. https://doi.org/10.3390/biom13101514
APA StyleDong, B., Sun, W., Xu, D., Wang, G., & Zhang, T. (2023). DAEMDA: A Method with Dual-Channel Attention Encoding for miRNA–Disease Association Prediction. Biomolecules, 13(10), 1514. https://doi.org/10.3390/biom13101514