

Pyrazolo-triazolo-pyrimidine Scaffold as a Molecular Passepartout for the Pan-Recognition of Human Adenosine Receptors


 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Computational Methodologies
2.1.1. Protein Preparation
2.1.2. Molecular Docking
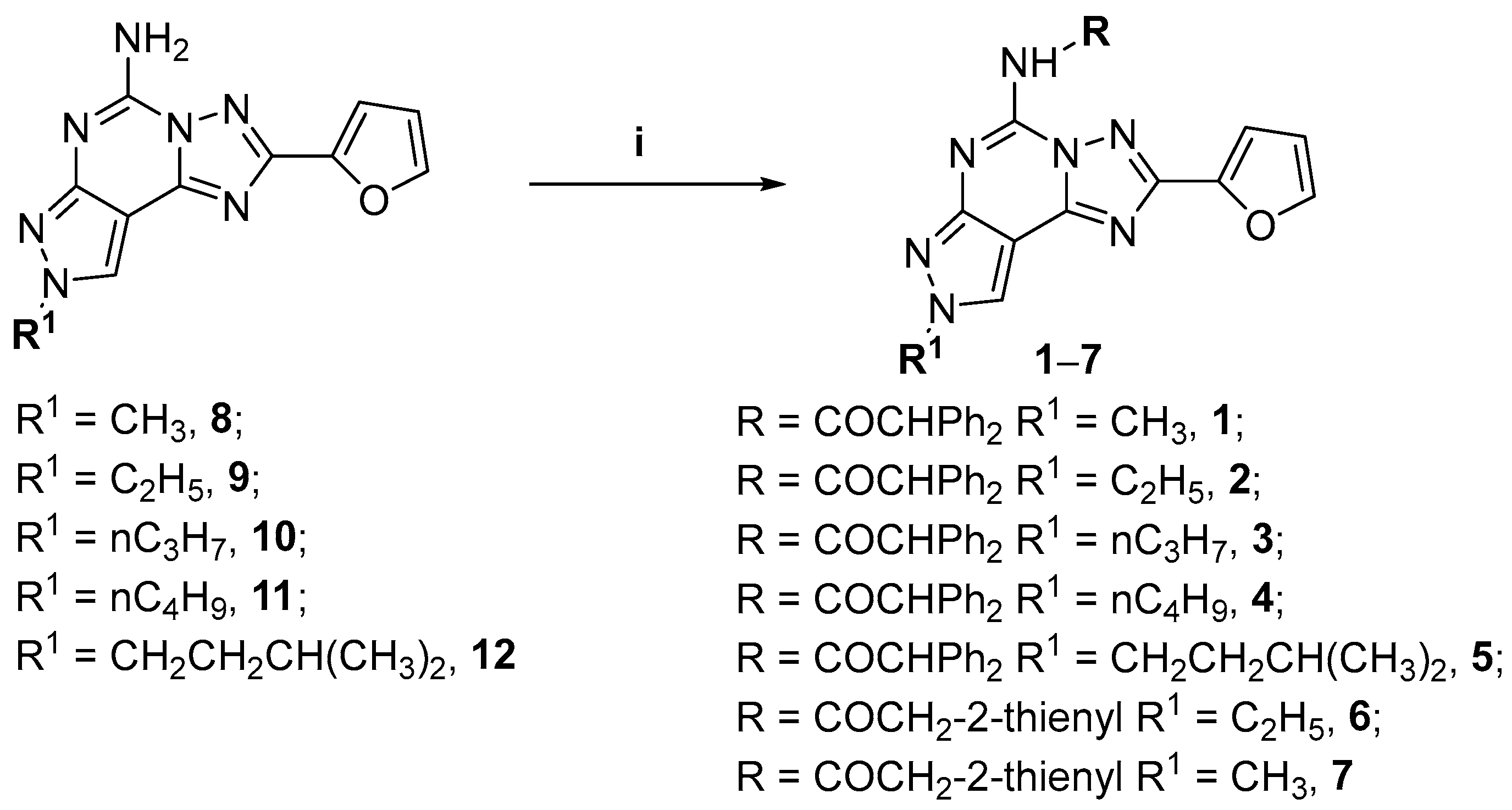
2.2. Chemistry
2.3. Biology
2.3.1. Binding at Human A1, A2A, A2B, and A3 Adenosine Receptors
2.3.2. Adenylyl Cyclase Activity in CHO Cells Expressing hA2B Receptors
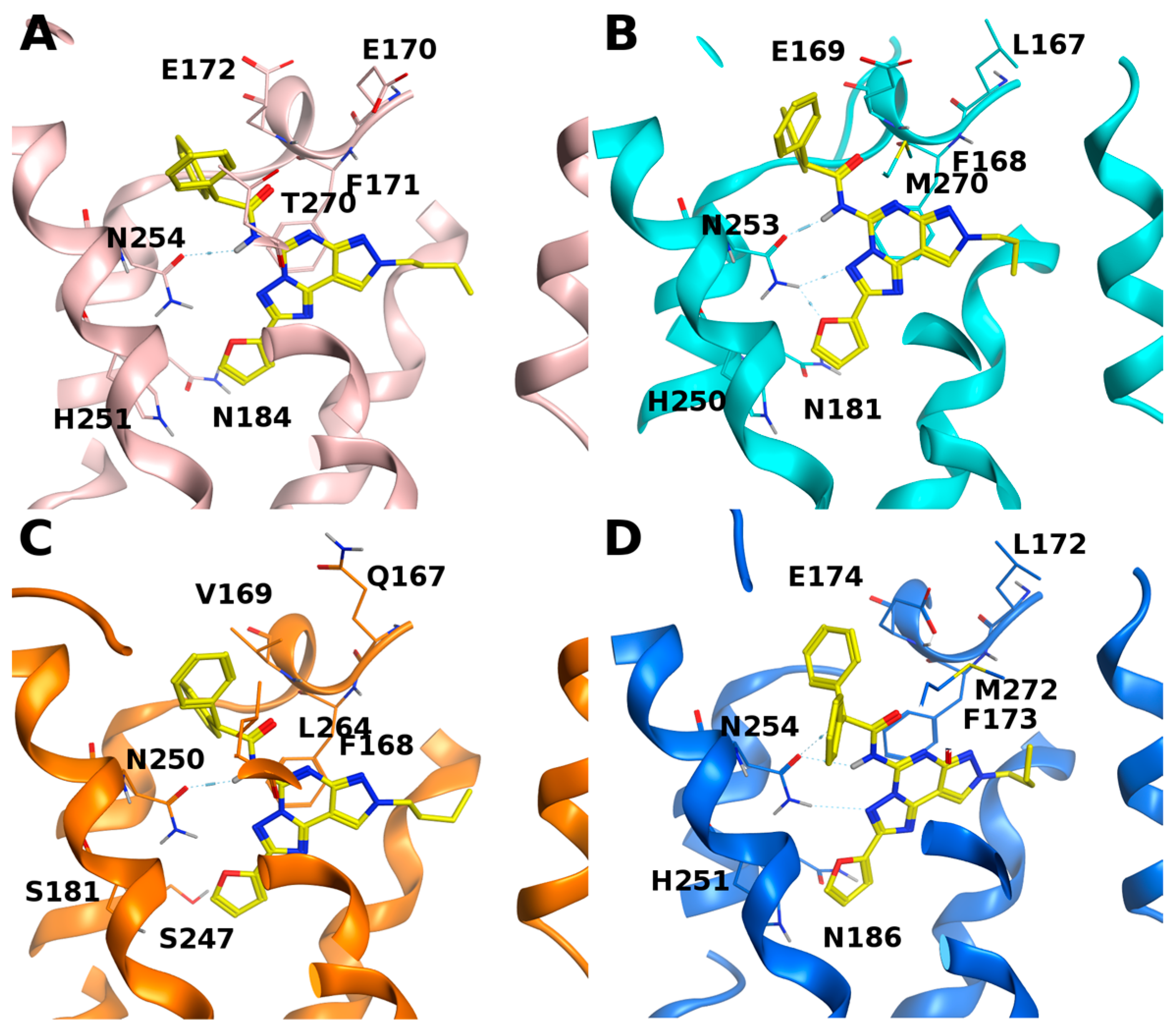
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. A basis for distinguishing two types of purinergic receptor. In Cell Membrane Receptors for Drugs and Hormones: A Multidisciplinary Approach; Bolis, L., Straub, R.W., Eds.; Raven Press: New York, NY, USA, 1978; pp. 107–118. [Google Scholar]
- IJzerman, A.P.; Jacobson, K.A.; Müller, C.E.; Cronstein, B.N.; Cunha, R.A. International Union of Basic and Clinical Pharmacology. CXII: Adenosine Receptors: A Further Update. Pharmacol. Rev. 2022, 74, 340–372. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, F.; Pasquini, S.; Contri, C.; Cappello, M.; Nigro, M.; Travagli, A.; Merighi, S.; Gessi, S.; Borea, P.A.; Varani, K. Pharmacology of Adenosine Receptors: Recent Advancements. Biomolecules 2023, 13, 1387. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Gao, Z.G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine receptors as drug targets—What are the challenges? Nat. Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef]
- Effendi, W.I.; Nagano, T.; Kobayashi, K.; Nishimura, Y. Focusing on Adenosine Receptors as a Potential Targeted Therapy in Human Diseases. Cells 2020, 9, 785. [Google Scholar] [CrossRef]
- Vincenzi, F.; Rotondo, J.C.; Pasquini, S.; Di Virgilio, F.; Varani, K.; Tognon, M. A3 Adenosine and P2X7 Purinergic Receptors as New Targets for an Innovative Pharmacological Therapy of Malignant Pleural Mesothelioma. Front. Oncol. 2021, 11, 679285. [Google Scholar] [CrossRef]
- Hammami, A.; Allard, D.; Allard, B.; Stagg, J. Targeting the adenosine pathway for cancer immunotherapy. Semin. Immunol. 2019, 42, 101304. [Google Scholar] [CrossRef]
- Spinaci, A.; Buccioni, M.; Chang, C.; Dal Ben, D.; Francucci, B.; Lambertucci, C.; Volpini, R.; Marucci, G. Adenosine A2A Receptor Antagonists: Chemistry, SARs, and Therapeutic Potential. In Purinergic Receptors and Their Modulators. Topics in Medicinal Chemistry; Colotta, V., Supuran, C.T., Eds.; Springer: Cham, Switzerland, 2023; Volume 41. [Google Scholar]
- Coppi, E.; Cherchi, F.; Venturini, M.; Lucarini, E.; Corradetti, R.; Di Cesare Mannelli, L.; Ghelardini, C.; Pedata, F.; Pugliese, A. Therapeutic Potential of Highly Selective A3 Adenosine Receptor Ligands in the Central and Peripheral Nervous System. Molecules 2022, 27, 1890. [Google Scholar] [CrossRef]
- Gatta, F.; Del Giudice, M.; Borioni, A.; Borea, P.; Dionisotti, S.; Ongini, E. Synthesis of imidazo[1,2-c]pyrazolo[4,3-e]pyrimidines, pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidines and 1,2,4-triazolo[5,1-i]purines: New potent adenosine A2 receptor antagonists. Eur. J. Med. Chem. 1993, 28, 569–576. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Cacciari, B.; Romagnoli, R.; Spalluto, G.; Monopoli, A.; Ongini, E.; Varani, K.; Borea, P.A. 7-Substituted 5-Amino-2-(2-furyl)pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines as A2A Adenosine Receptor Antagonists: A Study on the Importance of Modifications at the Side Chain on the Activity and Solubility. J. Med. Chem. 2002, 45, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Redenti, S.; Ciancetta, A.; Pastorin, G.; Cacciari, B.; Moro, S.; Spalluto, G.; Federico, S. Pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidines and Structurally Simplified Analogs. Chemistry and SAR Profile as Adenosine Receptor Antagonists. Curr. Top. Med. Chem. 2016, 16, 3224–3257. [Google Scholar] [CrossRef] [PubMed]
- Prencipe, F.; Da Ros, T.; Cescon, E.; Grieco, I.; Persico, M.; Spalluto, G.; Federico, S. Adenosine receptor ligands, probes, and functional conjugates: A 20-year hystory of pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidines (PTP). In Purinergic Receptors and Their Modulators; Colotta, V., Supuran, C.T., Eds.; Springer: Cham, Switzerland, 2023; ISBN 978-3-031-39724-0. [Google Scholar]
- Glukhova, A.; Thal, D.M.; Nguyen, A.T.; Vecchio, E.A.; Jörg, M.; Scammells, P.J.; May, L.T.; Sexton, P.M.; Christopoulos, A. Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 2017, 168, 867–877. [Google Scholar] [CrossRef]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; Ijzerman, A.P.; et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group ULC. Molecular Operating Environment (MOE), version 2022.02; Chemical Computing Group ULC: Montreal, QC, Canada, 2023; Available online: https://www.chemcomp.com/ (accessed on 15 July 2023).
- Katritch, V.; Fenalti, G.; Abola, E.E.; Roth, B.L.; Cherezov, V.; Stevens, R.C. Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 2014, 39, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Margiotta, E.; Deganutti, G.; Moro, S. Could the presence of sodium ion influence the accuracy and precision of the ligand-posing in the human A2A adenosine receptor orthosteric binding site using a molecular docking approach? Insights from Dockbench. J. Comput. Aided Mol. Des. 2018, 32, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Receptor Molecular Biology; Methods in Neurosciences; Elsevier: Amsterdam, The Netherlands, 1995; Volume 25, pp. 366–428. [Google Scholar]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Michielan, L.; Bolcato, C.; Federico, S.; Cacciari, B.; Bacilieri, M.; Klotz, K.-N.; Kachler, S.; Pastorin, G.; Cardin, R.; Sperduti, A.; et al. Combining selectivity and affinity predictions using an integrated Support Vector Machine (SVM) approach: An alternative tool to discriminate between the human adenosine A2A and A3 receptor pyrazolo-triazolo-pyrimidine antagonists binding sites. Bioorg. Med. Chem. 2009, 17, 5259–5274. [Google Scholar] [CrossRef]
- Michielan, L.; Stephanie, F.; Terfloth, L.; Hristozov, D.; Cacciari, B.; Klotz, K.; Spalluto, G.; Gasteiger, J.; Moro, S. Exploring Potency and Selectivity Receptor Antagonist Profiles Using a Multilabel Classification Approach: The Human Adenosine Receptors as a Key Study. J. Chem. Inf. Model. 2009, 49, 2820–2836. [Google Scholar] [CrossRef]
- Klotz, K.-N.; Hessling, J.; Hegler, J.; Owman, C.; Kull, B.; Fredholm, B.B.; Lohse, M.J. Comparative pharmacology of human adenosine receptor subtypes—Characterization of stably transfected receptors in CHO cells. Naunyn Schmiedebergs Arch. Pharmacol. 1998, 357, 1–9. [Google Scholar] [CrossRef]
- De Lean, A.; Hancock, A.A.; Lefkowitz, R.J. Validation and statistical analysis of a computer modeling method for quantitative analysis of radioligand binding data for mixtures of pharmacological receptor subtypes. Mol. Pharmacol. 1982, 21, 5–16. [Google Scholar]
- Lohse, M.J.; Lenschow, V.; Schwabe, U. Two affinity states of Ri adenosine receptors in brain membranes. Analysis of guanine nucleotide and temperature effects on radioligand binding. Mol. Pharmacol. 1984, 26, 1–9. [Google Scholar]
- Baraldi, P.G.; Cacciari, B.; Romagnoli, R.; Spalluto, G.; Moro, S.; Klotz, K.N.; Leung, E.; Varani, K.; Gessi, S.; Merighi, S.; et al. Pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives as highly potent and selective human A3 adenosine receptor antagonists: Influence of the chain at the N8 pyrazole nitrogen. J. Med. Chem. 2000, 43, 4768–4780. [Google Scholar] [CrossRef]
- Klotz, K.N.; Cristalli, G.; Grifantini, M. Photoaffinity labeling of A1-adenosine receptors. J. Biol. Chem. 1985, 260, 14659–14664. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Weng, Y.; Xu, Y.; Lu, W.; Liu, W.; Liu, M.; Hua, T.; Song, G. Cryo-EM structure of the human adenosine A2B receptor-Gs signaling complex. Sci. Adv. 2022, 8, eadd3709. [Google Scholar] [CrossRef]
- Cai, H.; Xu, Y.; Guo, S.; He, X.; Sun, J.; Li, X.; Li, C.; Yin, W.; Cheng, X.; Jiang, H.; et al. Structures of adenosine receptor A2BR bound to endogenous and synthetic agonists. Cell Discov. 2022, 8, 140. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Wess, J.; van Rhee, A.M.; Schöneberg, T.; Jacobson, K.A. Site-directed mutagenesis identifies residues involved in ligand recognition in the human A2a adenosine receptor. J. Biol. Chem. 1995, 270, 13987–13997. [Google Scholar] [CrossRef]
- Gao, Z.-G.; Chen, A.; Barak, D.; Kim, S.-K.; Müller, C.E.; Jacobson, K.A. Identification by site-directed mutagenesis of residues involved in ligand recognition and activation of the human A3 adenosine receptor. J. Biol. Chem. 2002, 277, 19056–19063. [Google Scholar] [CrossRef]
- Peeters, M.C.; van Westen, G.J.P.; Li, Q.; Ijzerman, A.P. Importance of the extracellular loops in G protein-coupled receptors for ligand recognition and receptor activation. Trends Pharmacol. Sci. 2011, 32, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, M.; Wootten, D.; Conner, M.T.; Simms, J.; Kendrick, R.; Logan, R.T.; Poyner, D.R.; Barwell, J. Lifting the lid on GPCRs: The role of extracellular loops. Br. J. Pharmacol. 2012, 165, 1688–1703. [Google Scholar] [CrossRef]
- De Filippo, E.; Hinz, S.; Pellizzari, V.; Deganutti, G.; El-Tayeb, A.; Navarro, G.; Franco, R.; Moro, S.; Schiedel, A.C.; Müller, C.E. A2A and A2B adenosine receptors: The extracellular loop 2 determines high (A2A) or low affinity (A2B) for adenosine. Biochem. Pharmacol. 2020, 172, 113718. [Google Scholar] [CrossRef] [PubMed]
- Cuzzolin, A.; Sturlese, M.; Deganutti, G.; Salmaso, V.; Sabbadin, D.; Ciancetta, A.; Moro, S. Deciphering the Complexity of Ligand-Protein Recognition Pathways Using Supervised Molecular Dynamics (SuMD) Simulations. J. Chem. Inf. Model. 2016, 56, 687–705. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Compd | R | R 1 | hA1 (Ki nM) 1 | hA2A (Ki nM) 2 | hA2B (Ki nM) | hA2B (IC50 nM) 5 | hA3 (Ki nM) 6 |
|---|---|---|---|---|---|---|---|
| 1 | COCHPh2 | CH3 | 139 (82.7–235) | 216 (152–307) | 29.1 3 (22.8–37.1) | 363 (318–414) | 0.25 (0.15–0.41) |
| 2 | COCHPh2 | CH2CH3 | 156 (102–239) | 131 (127–135) | 18.3 3 (11.8–28.2) | 745 (482–1150) | 0.98 (0.54–1.75) |
| 3 | COCHPh2 | CH2CH2CH3 | 80.2 (49.7–129) | 46.5 (38.2–56.5) | 13.3 3 (5.71–30.8) | 491 (327–736) | 0.93 (0.69–1.25) |
| 4 | COCHPh2 | CH2CH2CH2CH3 | 129 (92.6–180) | 114 (81.3–159) | 11.5 3 (8.17–11.3) | 780 (650–936) | 1.20 (0.74–1.94) |
| 5 | COCHPh2 | CH2CH2CH(CH3)2 | 441 (281–692) | 159 (136–188) | n.d. | >10,000 | 5.86 (3.34–10.3) |
| 6 | COCH2-2-thienyl | CH2CH3 | 148 (116–189) | 15.9 (8.24–30.8) | n.d. | >10,000 | 196 (160–241) |
| 7 | COCH2-2-thienyl | CH3 | 444 (390–505) | 56 (26.7–117) | n.d. | >10,000 | 5.26 (3.70–7.47) |
| 8 | H | CH3 | 101 (81–127) | 2.80 (2.40–3.55) | 90 4 (81–101) | n.d. | 300 (265–339) |
| 9 | H | CH2CH3 | 5.00 (4.05–6.20) | 1.95 (1.70–2.10) | 65 4 (56–75) | n.d. | 331 (285–385) |
| 10 | H | CH2CH2CH3 | 10 (7–14) | 2.51 (1.90–3.37) | 39 4 (35–45) | n.d. | 408 (364–460) |
| 11 | H | CH2CH2CH2CH3 | 14 (11–17) | 1.60 (1.4–2.1) | 53 4 (40–69) | n.d. | 600 (525–691) |
| 12 | H | CH2CH2CH(CH3)2 | 2.00 (1.72–2.36) | 0.78 (0.60–1.00) | 9.1 4 (7.4–11.3) | n.d. | 700 (664–738) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salmaso, V.; Persico, M.; Da Ros, T.; Spalluto, G.; Kachler, S.; Klotz, K.-N.; Moro, S.; Federico, S. Pyrazolo-triazolo-pyrimidine Scaffold as a Molecular Passepartout for the Pan-Recognition of Human Adenosine Receptors. Biomolecules 2023, 13, 1610. https://doi.org/10.3390/biom13111610
Salmaso V, Persico M, Da Ros T, Spalluto G, Kachler S, Klotz K-N, Moro S, Federico S. Pyrazolo-triazolo-pyrimidine Scaffold as a Molecular Passepartout for the Pan-Recognition of Human Adenosine Receptors. Biomolecules. 2023; 13(11):1610. https://doi.org/10.3390/biom13111610
Chicago/Turabian StyleSalmaso, Veronica, Margherita Persico, Tatiana Da Ros, Giampiero Spalluto, Sonja Kachler, Karl-Norbert Klotz, Stefano Moro, and Stephanie Federico. 2023. "Pyrazolo-triazolo-pyrimidine Scaffold as a Molecular Passepartout for the Pan-Recognition of Human Adenosine Receptors" Biomolecules 13, no. 11: 1610. https://doi.org/10.3390/biom13111610
APA StyleSalmaso, V., Persico, M., Da Ros, T., Spalluto, G., Kachler, S., Klotz, K. -N., Moro, S., & Federico, S. (2023). Pyrazolo-triazolo-pyrimidine Scaffold as a Molecular Passepartout for the Pan-Recognition of Human Adenosine Receptors. Biomolecules, 13(11), 1610. https://doi.org/10.3390/biom13111610