
Pharmacophore-Based Screening, Molecular Docking, and Dynamic Simulation of Fungal Metabolites as Inhibitors of Multi-Targets in Neurodegenerative Disorders
 , , ,
, , ,  , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Material and Methodologies
2.1. Tools Used for Computational Study
2.2. Preparation of Ligands
2.3. Preparation of Target Proteins
2.4. Pharmacophore Generation and Virtual Screening
2.5. Molecular Docking
2.6. Prediction of Physicochemical Properties and Toxicity Level
2.7. Molecular Dynamics (MD) Simulation
2.8. Calculations of Free Energy (Prime-MM/GBSA)
Sol_Lipo + ΔGSolv_GB + ΔGPacking + ΔGSelf−contact
3. Results and Discussion
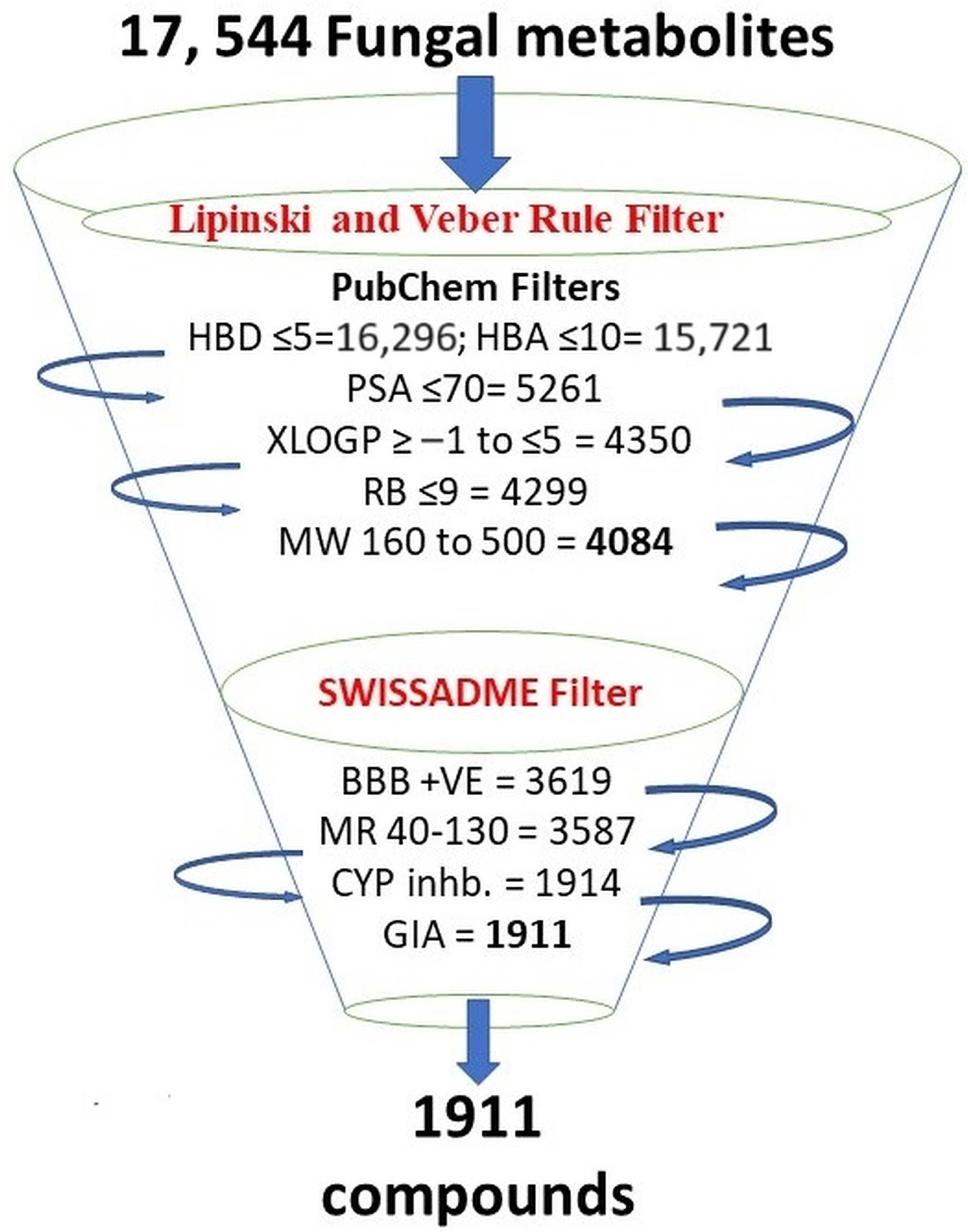
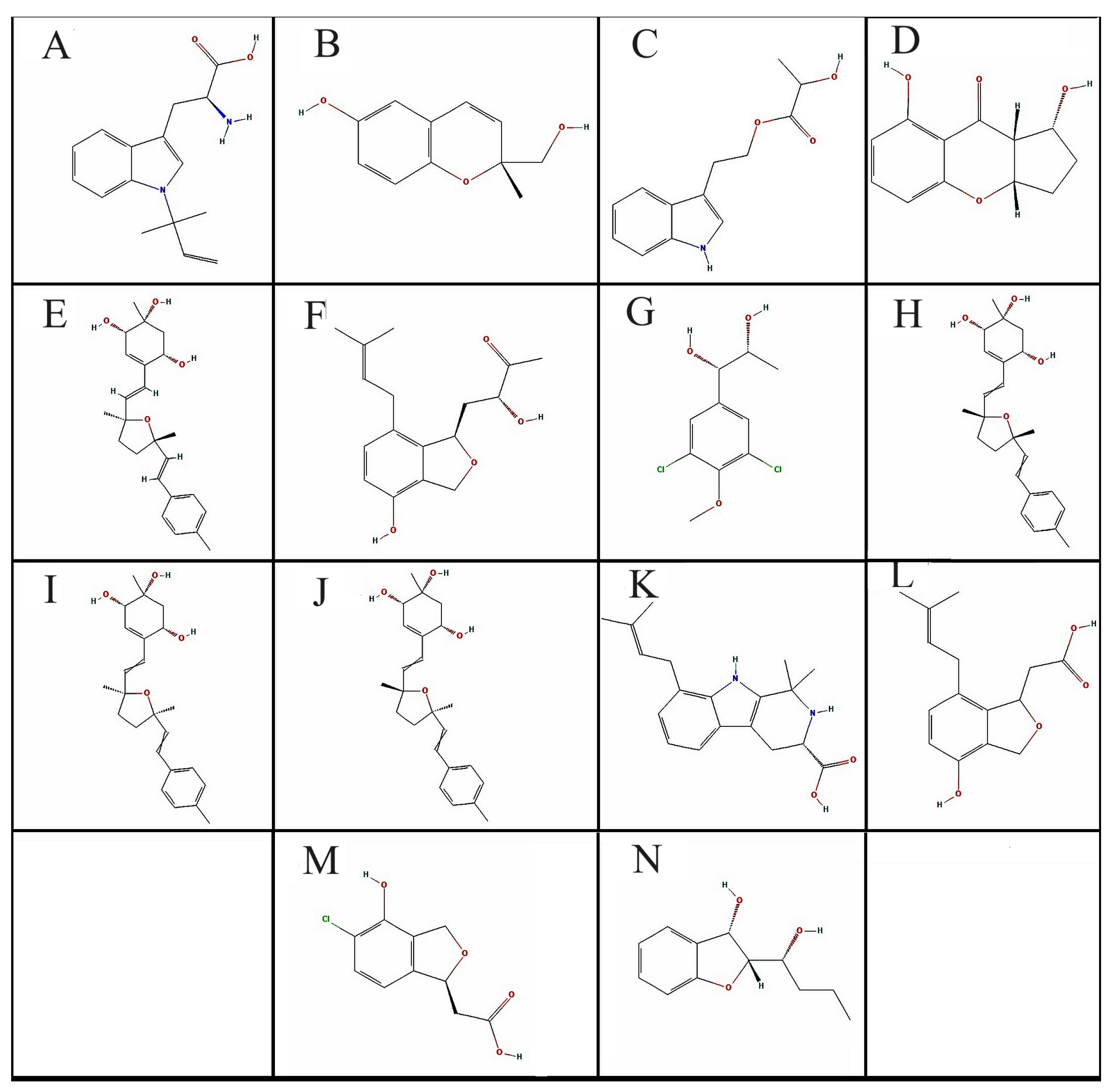
3.1. Criteria for Selecting Compounds during Retrieval
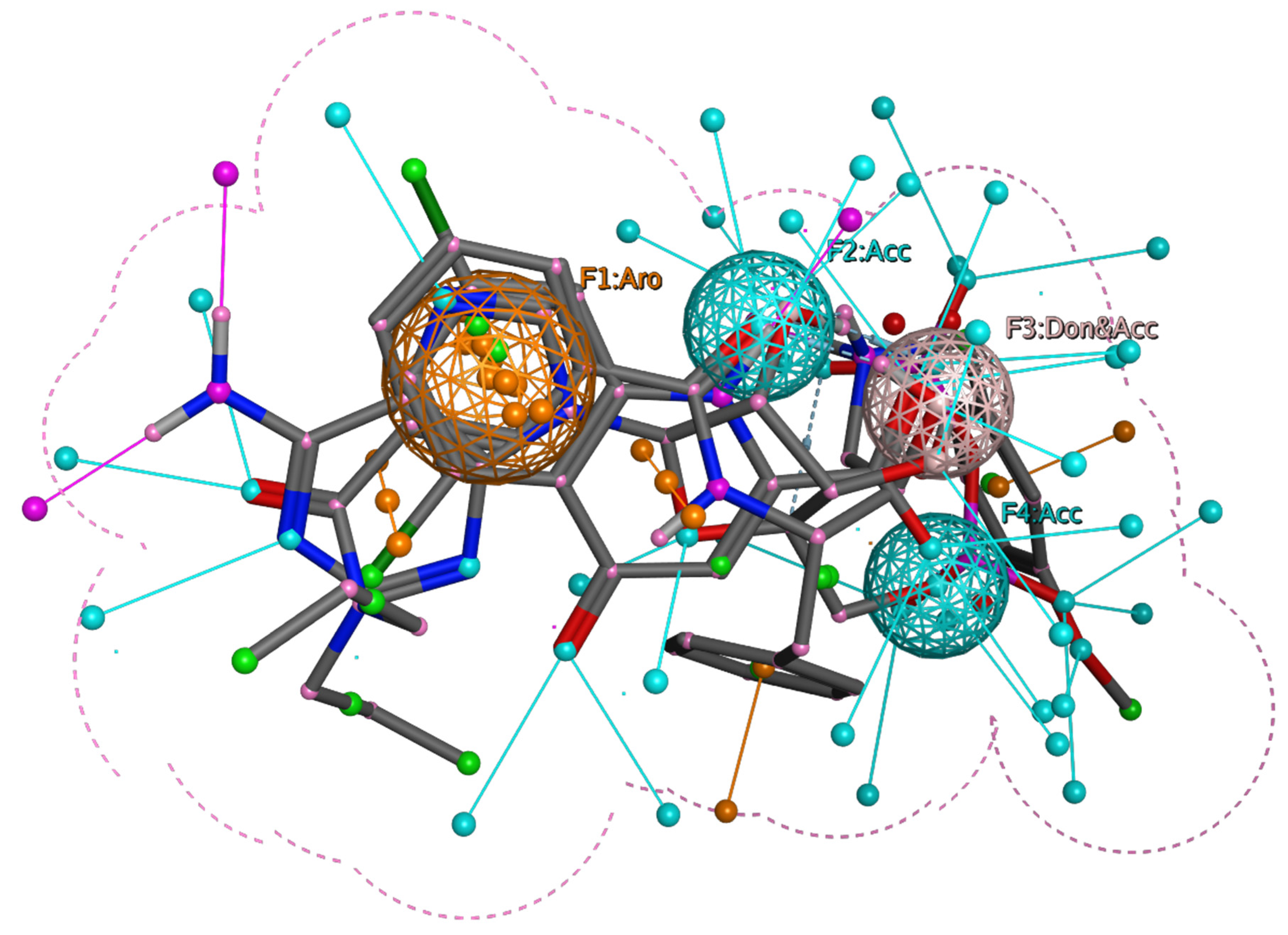
3.2. Pharmacophore Modeling and Screening of Compounds
3.3. Physicochemical and Pharmacokinetics Parameters
3.4. Toxicity Prediction
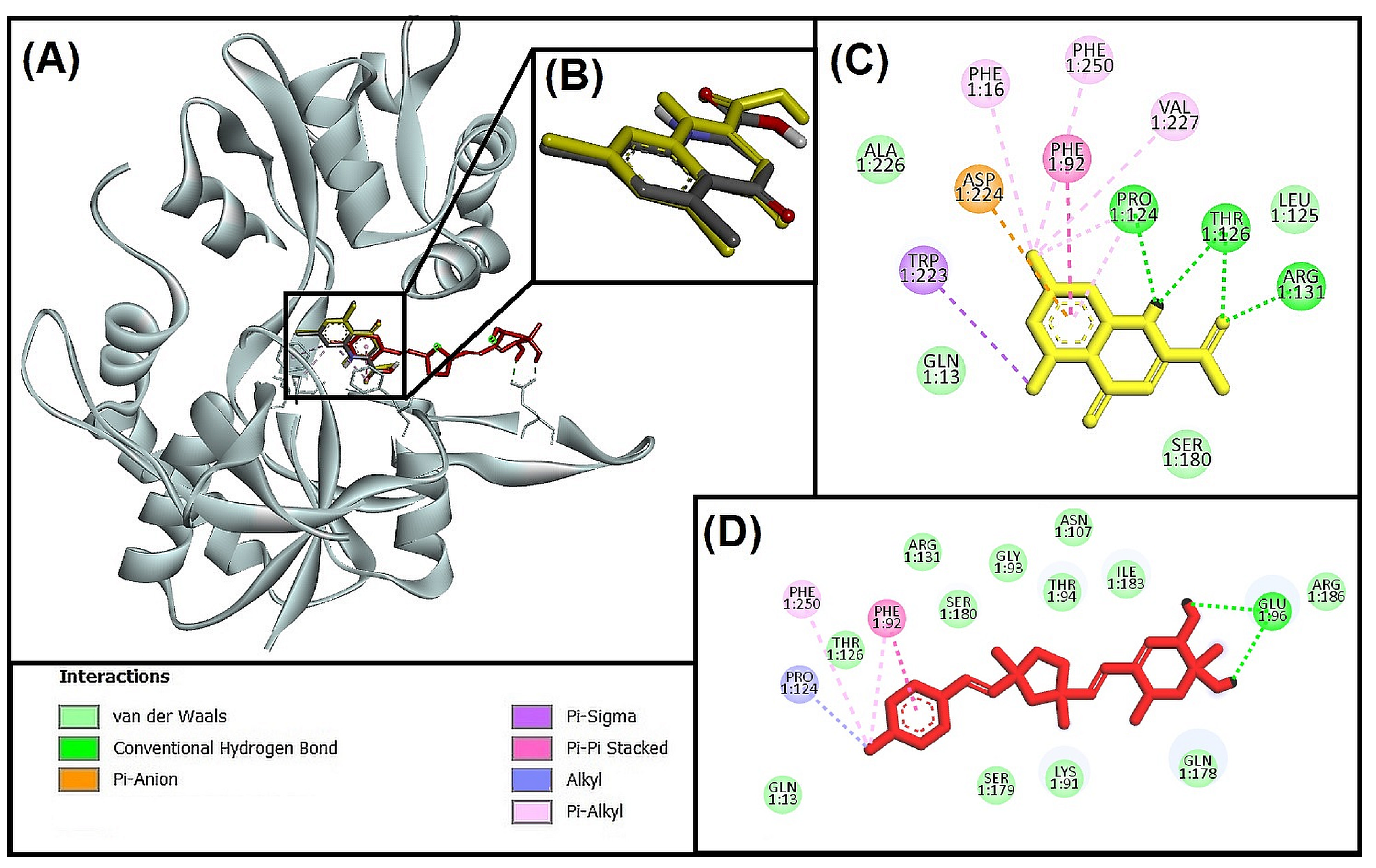
3.5. Molecular Docking and Interactions Analysis
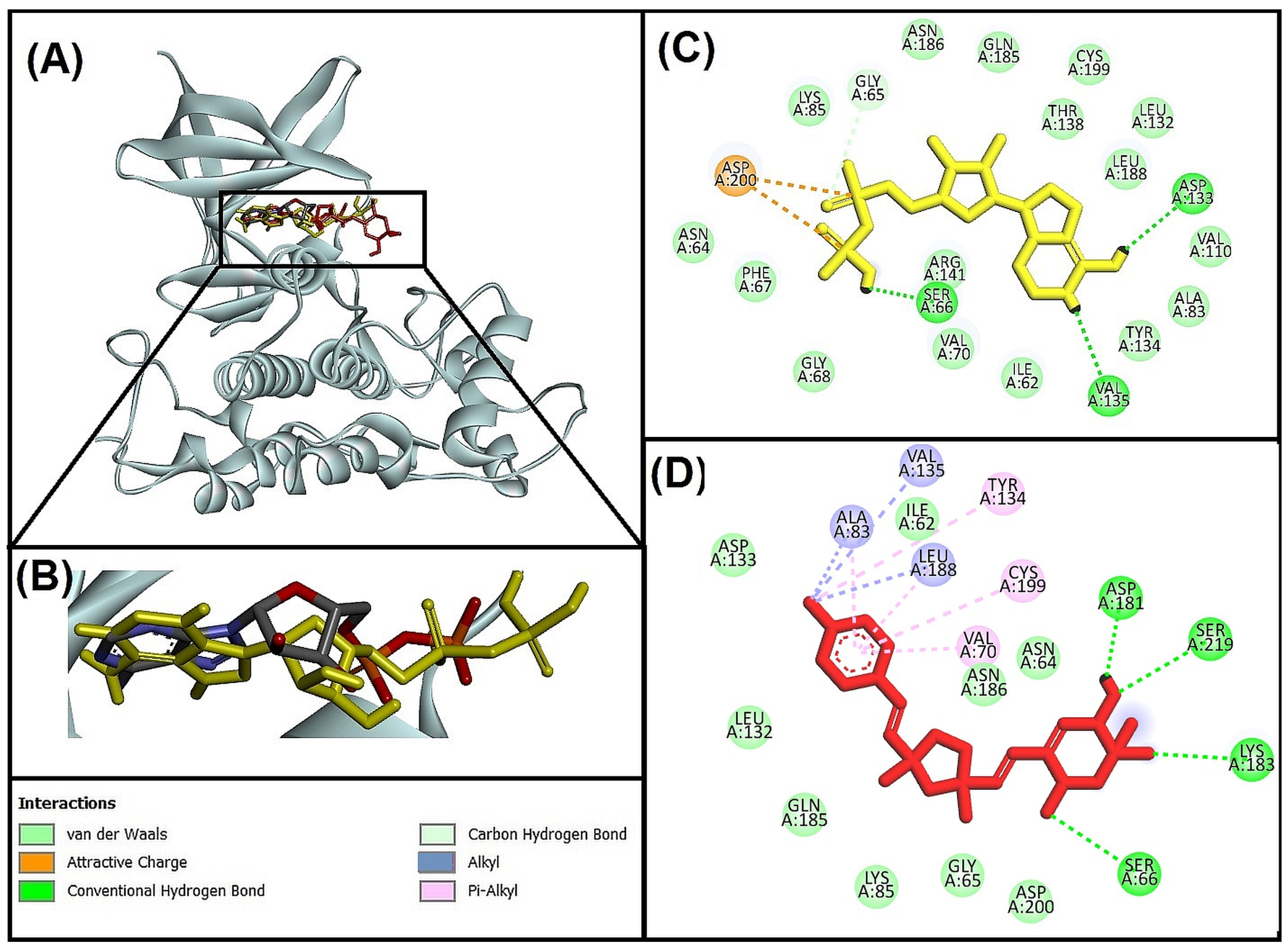
3.5.1. Molecular Interaction Analysis of Glycogen Synthase Kinase 3 Beta (GSK-3β) and Best-Hit Ligand
3.5.2. Molecular Interaction Analysis of N-methyl-D-Aspartate Receptor (NMDA) and Best-Hit Ligand
3.5.3. Molecular Interaction Analysis of Human Beta-Secretase (BACE-1) and Best Hit Ligand
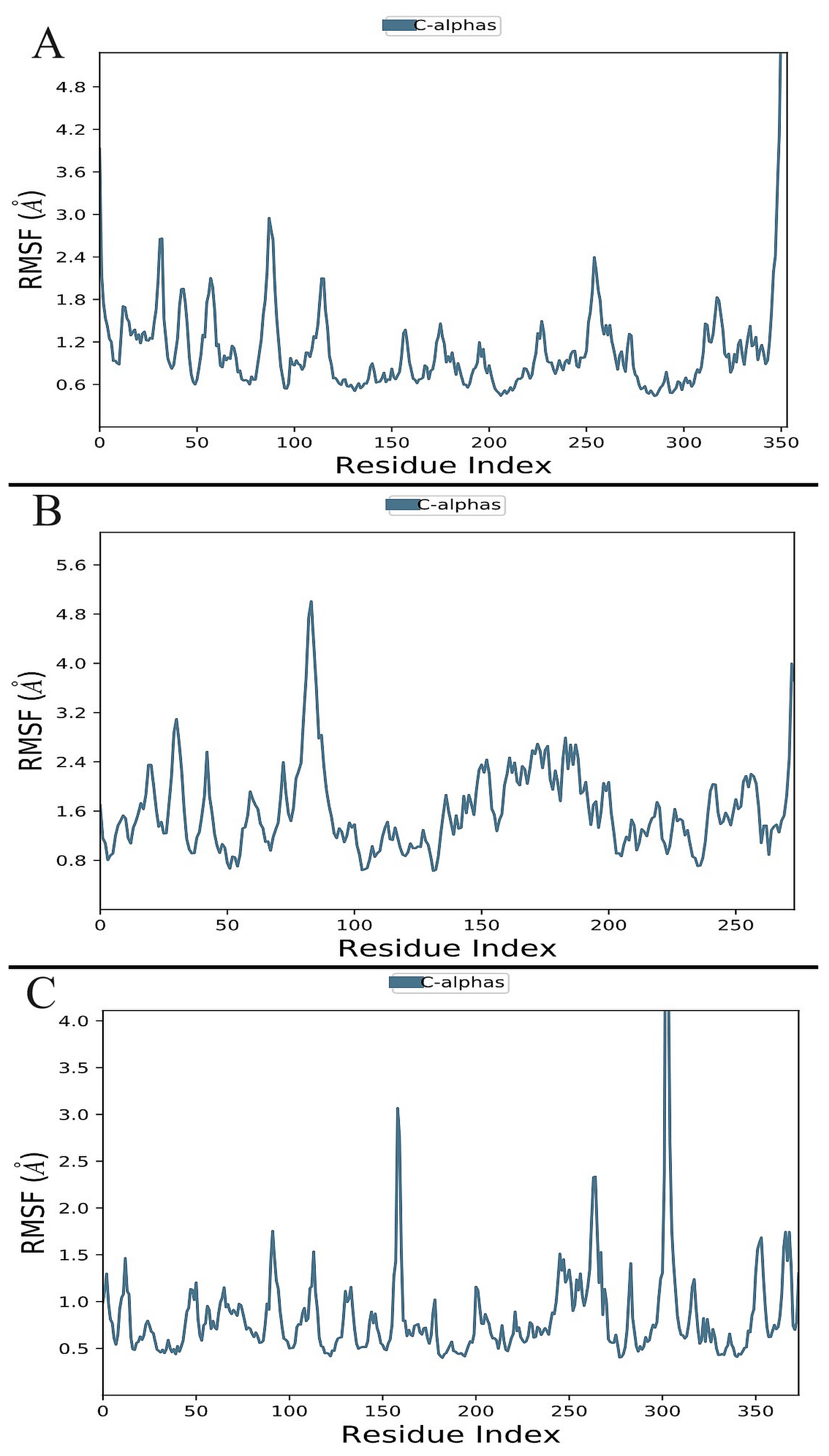
3.6. Molecular Dynamics Simulation
3.6.1. Analysis of Root-Mean-Square Deviation (RMSD) and RMSF
3.6.2. Secondary Structure Elements Analysis
3.6.3. Histogram for Molecular Interactions of Protein–Ligand Complexes
3.6.4. Analysis of Solvent-Accessible Surface Area (SASA) and Radius of Gyration (Rg)
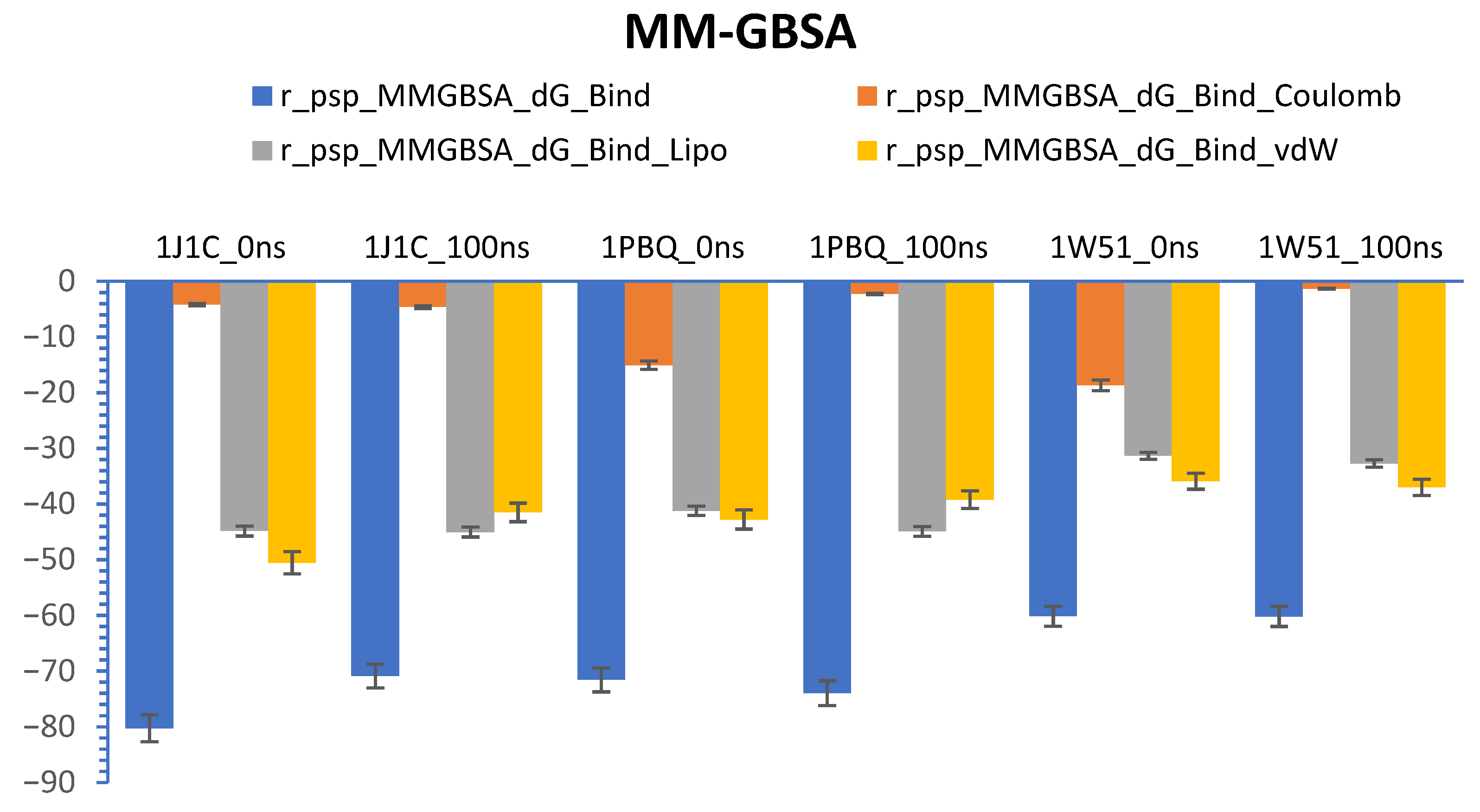
3.7. Calculations of Prime-MM/GBSA (Free Energy)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 30 August 2023).
- CDC. Global Health—Saudi Arabia. Available online: https://www.cdc.gov/globalhealth/countries/saudi_arabia/default.htm (accessed on 5 January 2022).
- Abeysinghe, A.A.D.T.; Deshapriya, R.D.U.S.; Udawatte, C. Alzheimer’s Disease; a Review of the Pathophysiological Basis and Therapeutic Interventions. Life Sci. 2020, 256, 117996. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Noh, G.O.; Kim, K. Behavioural and Psychological Symptoms of Dementia in Patients with Alzheimer’s Disease and Family Caregiver Burden: A Path Analysis. BMC Geriatr. 2021, 21, 160. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Schedin-Weiss, S.; Inoue, M.; Hromadkova, L.; Teranishi, Y.; Yamamoto, N.G.; Wiehager, B.; Bogdanovic, N.; Winblad, B.; Sandebring-Matton, A.; Frykman, S.; et al. Monoamine Oxidase B Is Elevated in Alzheimer Disease Neurons, Is Associated with γ-Secretase and Regulates Neuronal Amyloid β-Peptide Levels. Alzheimers Res. Ther. 2017, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Balaraman, Y.; Limaye, A.R.; Levey, A.I.; Srinivasan, S. Glycogen Synthase Kinase 3beta and Alzheimer’s Disease: Pathophysiological and Therapeutic Significance. Cell Mol. Life Sci. 2006, 63, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The Cholinergic System in the Pathophysiology and Treatment of Alzheimer’s Disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Dou, K.-X.; Tan, M.-S.; Tan, C.-C.; Cao, X.-P.; Hou, X.-H.; Guo, Q.-H.; Tan, L.; Mok, V.; Yu, J.-T. Comparative Safety and Effectiveness of Cholinesterase Inhibitors and Memantine for Alzheimer’s Disease: A Network Meta-Analysis of 41 Randomized Controlled Trials. Alzheimers Res. Ther. 2018, 10, 126. [Google Scholar] [CrossRef]
- Mani, S.; Jindal, D.; Chopra, H.; Jha, S.K.; Singh, S.K.; Ashraf, G.M.; Kamal, M.; Iqbal, D.; Chellappan, D.K.; Dey, A.; et al. ROCK2 Inhibition: A Futuristic Approach for the Management of Alzheimer’s Disease. Neurosci. Biobehav. Rev. 2022, 142, 104871. [Google Scholar] [CrossRef]
- Iqbal, D.; Rehman, M.T.; Bin Dukhyil, A.; Rizvi, S.M.D.; Al Ajmi, M.F.; Alshehri, B.M.; Banawas, S.; Khan, M.S.; Alturaiki, W.; Alsaweed, M. High-Throughput Screening and Molecular Dynamics Simulation of Natural Product-like Compounds against Alzheimer’s Disease through Multitarget Approach. Pharmaceuticals 2021, 14, 937. [Google Scholar] [CrossRef]
- Iqbal, D.; Rehman, M.T.; Alajmi, M.F.; Alsaweed, M.; Jamal, Q.M.S.; Alasiry, S.M.; Albaker, A.B.; Hamed, M.; Kamal, M.; Albadrani, H.M. Multitargeted Virtual Screening and Molecular Simulation of Natural Product-like Compounds against GSK3β, NMDA-Receptor, and BACE-1 for the Management of Alzheimer’s Disease. Pharmaceuticals 2023, 16, 622. [Google Scholar] [CrossRef]
- Iqbal, D.; Rizvi, S.M.D.; Rehman, M.T.; Khan, M.S.; Bin Dukhyil, A.; AlAjmi, M.F.; Alshehri, B.M.; Banawas, S.; Zia, Q.; Alsaweed, M.; et al. Soyasapogenol-B as a Potential Multitarget Therapeutic Agent for Neurodegenerative Disorders: Molecular Docking and Dynamics Study. Entropy 2022, 24, 593. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z. Monoamine Oxidase Inhibitors: Promising Therapeutic Agents for Alzheimer’s Disease (Review). Mol. Med. Rep. 2014, 9, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Llorens-Marítin, M.; Jurado, J.; Hernández, F.; Ávila, J. GSK-3β, a Pivotal Kinase in Alzheimer Disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar]
- Coman, H.; Nemeş, B. New Therapeutic Targets in Alzheimer’s Disease. Int. J. Gerontol. 2017, 11, 2–6. [Google Scholar] [CrossRef]
- Kumari, S.; Singh, A.; Singh, A.K.; Yadav, Y.; Bajpai, S.; Kumar, P.; Upadhyay, A.D.; Shekhar, S.; Dwivedi, S.; Dey, A.B.; et al. Circulatory GSK-3β: Blood-Based Biomarker and Therapeutic Target for Alzheimer’s Disease. J. Alzheimers Dis. 2022, 85, 249–260. [Google Scholar] [CrossRef]
- Pinky, P.; Pfitzer, J.; Senfeld, J.; Hong, H.; Bhattacharya, S.; Suppiramaniam, V.; Qureshi, I.; Reed, M. Recent Insights on Glutamatergic Dysfunction in Alzheimer’s Disease and Therapeutic Implications. Neurosci. 2022, 22, 461–471. [Google Scholar] [CrossRef]
- Schneider, L.S.; Mangialasche, F.; Andreasen, N.; Feldman, H.; Giacobini, E.; Jones, R.; Mantua, V.; Mecocci, P.; Pani, L.; Winblad, B.; et al. Clinical Trials and Late-Stage Drug Development for Alzheimer’s Disease: An Appraisal from 1984 to 2014. J. Intern Med. 2014, 275, 251–283. [Google Scholar] [CrossRef]
- Huang, L.-K.; Chao, S.-P.; Hu, C.-J. Clinical Trials of New Drugs for Alzheimer Disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef]
- Ibrahim, M.M.; Gabr, M.T. Multitarget Therapeutic Strategies for Alzheimer’s Disease. Neural Regen. Res. 2019, 14, 437–440. [Google Scholar] [CrossRef]
- Maramai, S.; Benchekroun, M.; Gabr, M.T.; Yahiaoui, S. Multitarget Therapeutic Strategies for Alzheimer’s Disease: Review on Emerging Target Combinations. Biomed. Res. Int. 2020, 2020, 5120230. [Google Scholar] [CrossRef]
- Jana, A.; Bhattacharjee, A.; Das, S.S.; Srivastava, A.; Choudhury, A.; Bhattacharjee, R.; De, S.; Perveen, A.; Iqbal, D.; Gupta, P.K.; et al. Molecular Insights into Therapeutic Potentials of Hybrid Compounds Targeting Alzheimer’s Disease. Mol. Neurobiol. 2022, 59, 3512–3528. [Google Scholar] [CrossRef] [PubMed]
- Clyde, A. Ultrahigh Throughput Protein-Ligand Docking with Deep Learning. Methods Mol. Biol. 2022, 2390, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Ugale, V.G.; Bari, S.B. Identification of Potential Gly/NMDA Receptor Antagonists by Cheminformatics Approach: A Combination of Pharmacophore Modelling, Virtual Screening and Molecular Docking Studies. SAR QSAR Env. Res. 2016, 27, 125–145. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, X.; Lin, X. A Review on Applications of Computational Methods in Drug Screening and Design. Molecules 2020, 25, E1375. [Google Scholar] [CrossRef]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.M.; Govender, T.; Naicker, T.; Kruger, H.G. Current Trends in Computer Aided Drug Design and a Highlight of Drugs Discovered via Computational Techniques: A Review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef]
- Iqbal, D.; Dukhyil, A.B.; Khan, M.S. Geno-Protective, Free Radical Scavenging and Antimicrobial Potential of Hyptis Suaveolens Methanolic Fraction: An In-Vitro Study. J. Pharm. Res. Int. 2021, 46–57. [Google Scholar] [CrossRef]
- Iqbal, D.; Khan, A.; A Ansari, I.; Khan, M.S. Investigating The Role of Novel Bioactive Compound from Ficus Virens Ait on Cigarette Smoke Induced Oxidative Stress and Hyperlipidemia in Rats. Iran. J. Pharm. Res. 2017, 16, 1089–1103. [Google Scholar]
- Jahan, S.; Redhu, N.S.; Siddiqui, A.J.; Iqbal, D.; Khan, J.; Banawas, S.; Alaidarous, M.; Alshehri, B.; Mir, S.A.; Adnan, M.; et al. Nobiletin as a Neuroprotectant against NMDA Receptors: An In Silico Approach. Pharmaceutics 2022, 14, 1123. [Google Scholar] [CrossRef]
- Alsagaby, S.A.; Iqbal, D.; Ahmad, I.; Patel, H.; Mir, S.A.; Madkhali, Y.A.; Oyouni, A.A.A.; Hawsawi, Y.M.; Alhumaydhi, F.A.; Alshehri, B.; et al. In Silico Investigations Identified Butyl Xanalterate to Competently Target CK2α (CSNK2A1) for Therapy of Chronic Lymphocytic Leukemia. Sci. Rep. 2022, 12, 17648. [Google Scholar] [CrossRef]
- Sawikr, Y.; Yarla, N.S.; Peluso, I.; Kamal, M.A.; Aliev, G.; Bishayee, A. Neuroinflammation in Alzheimer’s Disease: The Preventive and Therapeutic Potential of Polyphenolic Nutraceuticals. Adv. Protein Chem. Struct. Biol. 2017, 108, 33–57. [Google Scholar] [CrossRef]
- Bucciantini, M.; Leri, M.; Scuto, M.; Ontario, M.; Trovato Salinaro, A.; Calabrese, E.J.; Calabrese, V.; Stefani, M. Xenohormesis Underlyes the Anti-Aging and Healthy Properties of Olive Polyphenols. Mech. Ageing Dev. 2022, 202, 111620. [Google Scholar] [CrossRef] [PubMed]
- Jangra, A.; Gola, P.; Singh, J.; Gond, P.; Ghosh, S.; Rachamalla, M.; Dey, A.; Iqbal, D.; Kamal, M.; Sachdeva, P.; et al. Emergence of Taurine as a Therapeutic Agent for Neurological Disorders. Neural Regen. Res. 2024, 19, 62. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, D.; Khan, M.S.; Waiz, M.; Rehman, M.T.; Alaidarous, M.; Jamal, A.; Alothaim, A.S.; AlAjmi, M.F.; Alshehri, B.M.; Banawas, S.; et al. Exploring the Binding Pattern of Geraniol with Acetylcholinesterase through In Silico Docking, Molecular Dynamics Simulation, and In Vitro Enzyme Inhibition Kinetics Studies. Cells 2021, 10, 3533. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Chhibber, M.; Singh, I.P. Fungal Bioactive Compounds in Pharmaceutical Research and Development. Curr. Bioact. Compd. 2019, 15, 211–231. [Google Scholar] [CrossRef]
- Keller, N.P. Fungal Secondary Metabolism: Regulation, Function and Drug Discovery. Nat. Rev. Microbiol. 2019, 17, 167–180. [Google Scholar] [CrossRef]
- Abdel-Hadi, A.; Iqbal, D.; Alharbi, R.; Jahan, S.; Darwish, O.; Alshehri, B.; Banawas, S.; Palanisamy, M.; Ismail, A.; Aldosari, S. Myco-Synthesis of Silver Nanoparticles and Their Bioactive Role against Pathogenic Microbes. Biology 2023, 12, 661. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful New Tools for Exploring 3D Structures of Biological Macromolecules for Basic and Applied Research and Education in Fundamental Biology, Biomedicine, Biotechnology, Bioengineering and Energy Sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B. PubChem 2023 Update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. In Chemical Biology: Methods and Protocols; Hempel, J.E., Williams, C.H., Hong, C.C., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2015; pp. 243–250. ISBN 978-1-4939-2269-7. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- BIOVIA. BIOVIA Discovery Studio. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-discovery-studio/ (accessed on 30 October 2021).
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11 November 2006; Association for Computing Machinery: New York, NY, USA; p. 84. [Google Scholar]
- Release, S. 1: Desmond Molecular Dynamics System, DE Shaw Research, New York, NY, 2019; Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2019. [Google Scholar]
- Prasathkumar, M.; Raja, K.; Vasanth, K.; Khusro, A.; Sadhasivam, S.; Sahibzada, M.U.K.; Gawwad, M.R.A.; Al Farraj, D.A.; Elshikh, M.S. Phytochemical Screening and in Vitro Antibacterial, Antioxidant, Anti-Inflammatory, Anti-Diabetic, and Wound Healing Attributes of Senna Auriculata (L.) Roxb. Leaves. Arab. J. Chem. 2021, 14, 103345. [Google Scholar] [CrossRef]
- Aoki, M.; Yokota, T.; Sugiura, I.; Sasaki, C.; Hasegawa, T.; Okumura, C.; Ishiguro, K.; Kohno, T.; Sugio, S.; Matsuzaki, T. Structural Insight into Nucleotide Recognition in Tau-Protein Kinase I/Glycogen Synthase Kinase 3β. Acta Cryst. D 2004, 60, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Vuillard, L.; Cleasby, A.; Murray, C.W.; Yon, J. Apo and Inhibitor Complex Structures of BACE (β-Secretase). J. Mol. Biol. 2004, 343, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Gouaux, E. Mechanisms of Activation, Inhibition and Specificity: Crystal Structures of the NMDA Receptor NR1 Ligand-Binding Core. EMBO J. 2003, 22, 2873–2885. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE)|MOEsaic|PSILO. Available online: https://www.chemcomp.com/Products.htm (accessed on 30 August 2023).
- Rasheed, M.A.; Iqbal, M.N.; Saddick, S.; Ali, I.; Khan, F.S.; Kanwal, S.; Ahmed, D.; Ibrahim, M.; Afzal, U.; Awais, M. Identification of Lead Compounds against Scm (Fms10) in Enterococcus Faecium Using Computer Aided Drug Designing. Life 2021, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, B.V.; Rammohan, A.; Babu, T.M.; Zheng, G.Y.; Chen, W.; Rajendra, W.; Zyryanov, G.V.; Gu, W. Molecular Insight into Isoform Specific Inhibition of PI3K-α and PKC-η with Dietary Agents through an Ensemble Pharmacophore and Docking Studies. Sci. Rep. 2021, 11, 12150. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies Using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]
- Hildebrand, P.W.; Rose, A.S.; Tiemann, J.K.S. Bringing Molecular Dynamics Simulation Data into View. Trends Biochem. Sci. 2019, 44, 902–913. [Google Scholar] [CrossRef]
- Maiorov, V.N.; Crippen, G.M. Significance of Root-Mean-Square Deviation in Comparing Three-Dimensional Structures of Globular Proteins. J. Mol. Biol. 1994, 235, 625–634. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Pollastri, M.P. Overview on the Rule of Five. Curr. Protoc. Pharmacol. 2010, 49, 9–12. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Ghose, A.K.; Herbertz, T.; Hudkins, R.L.; Dorsey, B.D.; Mallamo, J.P. Knowledge-Based, Central Nervous System (CNS) Lead Selection and Lead Optimization for CNS Drug Discovery. ACS Chem. Neurosci. 2011, 3, 50–68. [Google Scholar] [CrossRef] [PubMed]
- Kelder, J.; Grootenhuis, P.D.J.; Bayada, D.M.; Delbressine, L.P.C.; Ploemen, J.-P. Polar Molecular Surface as a Dominating Determinant for Oral Absorption and Brain Penetration of Drugs. Pharm. Res. 1999, 16, 1514–1519. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Yao, L.; Xu, L.; Xue, J.; Wei, X. Bisacremines A–D, Dimeric Acremines Produced by a Soil-Derived Acremonium Persicinum Strain. J. Nat. Prod. 2015, 78, 2161–2166. [Google Scholar] [CrossRef] [PubMed]
- Elangovan, N.D.; Dhanabalan, A.K.; Gunasekaran, K.; Kandimalla, R.; Sankarganesh, D. Screening of Potential Drug for Alzheimer’s Disease: A Computational Study with GSK-3 β Inhibition through Virtual Screening, Docking, and Molecular Dynamics Simulation. J. Biomol. Struct. Dyn. 2021, 39, 7065–7079. [Google Scholar] [CrossRef] [PubMed]
- Jabir, N.R.; Shakil, S.; Tabrez, S.; Khan, M.S.; Rehman, M.T.; Ahmed, B.A. In Silico Screening of Glycogen Synthase Kinase-3β Targeted Ligands against Acetylcholinesterase and Its Probable Relevance to Alzheimer’s Disease. J. Biomol. Struct. Dyn. 2021, 39, 5083–5092. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef]
- Leeson, P.D.; Iversen, L.L. The Glycine Site on the NMDA Receptor: Structure-Activity Relationships and Therapeutic Potential. J. Med. Chem. 1994, 37, 4053–4067. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Kumar, S. In Silico Analysis of Binding Interaction of Phytoconstituents with N-Methyl-D-Aspartate Receptor for Potential Therapeutic Use in Alzheimer’s Disease. Pharmacogn. Mag. 2018, 14, 638. [Google Scholar] [CrossRef]
- David, T.I.; Omotuyi, O.I.; Agboola, O.D.; Okonkwo, D.C.; Adelakun, N.S. Identification of Gly/NMDA Receptor Antagonist from Chromolaena Odorata’s Derived Compounds Using Induced Fit Docking and ADME Study. J. Biol. Eng. Res. Rev. 2019, 6, 19–26. [Google Scholar]
- Leri, M.; Bertolini, A.; Stefani, M.; Bucciantini, M. EVOO Polyphenols Relieve Synergistically Autophagy Dysregulation in a Cellular Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 7225. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, F.; Falletta, E.; Leri, M.; Angeloni, C.; Beghelli, D.; Giusti, L.; Milanesi, R.; Sampaio-Marques, B.; Ludovico, P.; Goppa, L.; et al. Anti-Aging and Neuroprotective Properties of Grifola Frondosa and Hericium Erinaceus Extracts. Nutrients 2022, 14, 4368. [Google Scholar] [CrossRef]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A. The β-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef]
- Sinha, S.; Anderson, J.P.; Barbour, R.; Basi, G.S.; Caccavello, R.; Davis, D.; Doan, M.; Dovey, H.F.; Frigon, N.; Hong, J. Purification and Cloning of Amyloid Precursor Protein β-Secretase from Human Brain. Nature 1999, 402, 537–540. [Google Scholar] [CrossRef]
- Adeniji, A.O.; Adams, P.W.; Mody, V.V. Chapter 7—Amyloid β Hypothesis in the Development of Therapeutic Agents for Alzheimer’s Disease. In Drug Discovery Approaches for the Treatment of Neurodegenerative Disorders; Adejare, A., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 109–143. ISBN 978-0-12-802810-0. [Google Scholar]
- Venugopal, C.; Demos, C.M.; Jagannatha Rao, K.; Pappolla, M.A.; Sambamurti, K. Beta-Secretase: Structure, Function, and Evolution. CNS Neurol. Disord. Drug Targets 2008, 7, 278–294. [Google Scholar] [CrossRef]
- Ullah, M.A.; Johora, F.T.; Sarkar, B.; Araf, Y.; Ahmed, N.; Nahar, A.N.; Akter, T. Computer-Assisted Evaluation of Plant-Derived β-Secretase Inhibitors in Alzheimer’s Disease. Egypt. J. Med. Hum. Genet. 2021, 22, 26. [Google Scholar] [CrossRef]
- Karplus, M. Molecular Dynamics Simulations of Biomolecules. Acc. Chem. Res. 2002, 35, 321–323. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Ausaf Ali, S.; Hassan, I.; Islam, A.; Ahmad, F. A Review of Methods Available to Estimate Solvent-Accessible Surface Areas of Soluble Proteins in the Folded and Unfolded States. Curr. Protein Pept. Sci. 2014, 15, 456–476. [Google Scholar]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert. Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds Code | PubChem CID | MW | #HA | #AHA | F-Csp3 | #RB | #HBA | #HBD | MR | TPSA | XLOGP3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 22216483 | R-N-DMAT | 272.34 | 20 | 9 | 0.31 | 5 | 3 | 2 | 80.9 | 68.25 | 0 |
| B | 46216805 | Daedalin A | 192.21 | 14 | 6 | 0.27 | 1 | 3 | 2 | 53.75 | 49.69 | 1.44 |
| C | 46880982 | See C in footer | 233.26 | 17 | 9 | 0.31 | 5 | 3 | 2 | 64.94 | 62.32 | 2.28 |
| D | 60166720 | Diaportheone B | 220.22 | 16 | 6 | 0.42 | 0 | 4 | 2 | 56.51 | 66.76 | 1.54 |
| E | 122187709 | Bisacremine A | 384.51 | 28 | 6 | 0.5 | 4 | 4 | 3 | 113.11 | 69.92 | 2.36 |
| F | 122206138 | Baccinol H | 290.35 | 21 | 6 | 0.47 | 5 | 4 | 2 | 81.68 | 66.76 | 2.21 |
| G | 139583580 | See G in footer | 251.11 | 15 | 6 | 0.4 | 3 | 3 | 2 | 59.86 | 49.69 | 2.05 |
| H | 139586224 | Bisacremine D | 384.51 | 28 | 6 | 0.5 | 4 | 4 | 3 | 113.11 | 69.92 | 2.36 |
| I | 139587420 | Bisacremine-C | 384.51 | 28 | 6 | 0.5 | 4 | 4 | 3 | 113.11 | 69.92 | 2.36 |
| J | 139587958 | Bisacremine B | 384.51 | 28 | 6 | 0.5 | 4 | 4 | 3 | 113.11 | 69.92 | 2.36 |
| K | 139588462 | Penipaline B | 312.41 | 23 | 9 | 0.42 | 3 | 3 | 3 | 97.44 | 65.12 | 1.35 |
| L | 139589365 | Emefuran D | 262.3 | 19 | 6 | 0.4 | 4 | 4 | 2 | 72.47 | 66.76 | 2.25 |
| M | 139591664 | See M in footer | 228.63 | 15 | 6 | 0.3 | 2 | 4 | 2 | 53.76 | 66.76 | 0.95 |
| N | 145720807 | Phexandiol B | 208.25 | 15 | 6 | 0.5 | 3 | 3 | 2 | 57.34 | 49.69 | 2.07 |
| Code | Hepatotoxicity (Probability) | Carcinogenicity (Probability) | Immunotoxicity (Probability) | Mutagenicity (Probability) | Cytotoxicity (Probability) | Predicted LD50 (mg/kg) |
|---|---|---|---|---|---|---|
| A | IA (0.68) | IA (0.70) | IA (0.99) | IA (0.74) | IA (0.79) | 225 |
| B | IA (0.80) | IA (0.59) | Active (0.75) | IA (0.64) | IA (0.69) | 500 |
| C | IA (0.57) | IA (0.66) | IA (0.96) | IA (0.77) | IA (0.80) | 3500 |
| D | IA (0.73) | IA (0.55) | IA (0.66) | IA (0.62) | IA (0.66) | 1060 |
| E | IA (0.78) | IA (0.56) | IA (0.69) | IA (0.72) | IA (0.75) | 5000 |
| F | IA (0.83) | IA (0.57) | Active (0.83) | IA (0.61) | IA (0.76) | 1500 |
| G | IA (0.73) | IA (0.57) | IA (0.69) | IA (0.75) | IA (0.68) | 1040 |
| H | IA (0.78) | IA (0.56) | IA (0.69) | IA (0.72) | IA (0.75) | 5000 |
| I | IA (0.78) | IA (0.56) | IA (0.69) | IA (0.72) | IA (0.75) | 5000 |
| J | IA (0.78) | IA (0.56) | IA (0.69) | IA (0.72) | IA (0.75) | 5000 |
| K | IA (0.63) | IA (0.70) | IA (0.86) | IA (0.72) | IA (0.70) | 550 |
| L | IA (0.79) | IA (0.60) | Active (0.69) | IA (0.61) | IA (0.77) | 1000 |
| M | IA (0.66) | IA (0.61) | IA (0.97) | IA (0.70) | IA (0.68) | 1500 |
| N | IA (0.71) | IA (0.68) | IA (0.96) | IA (0.62) | IA (0.81) | 1295 |
| Binding Energy (ΔG: Kcal/mol) | Binding Affinity (Ki: M−1) | |||||
|---|---|---|---|---|---|---|
| Code | GSK-3β (1J1C) | NMDA (1PBQ) | BACE-1 (1W51) | GSK-3β (1J1C) | NMDA (1PBQ) | BACE-1 (1W51) |
| A | −7.4 ± 0.2 | −7.2 ± 0.1 | −7.1 ± 0.1 | 2.7 × 105 | 1.9 × 105 | 1.6 × 105 |
| B | −6 ± 0.3 | −6.3 ± 0.1 | −6.5 ± 0.2 | 2.5 × 104 | 4.1 × 104 | 5.8 × 104 |
| C | −6.3 ± 0.1 | −7 ± 0.3 | −6.7 ± 0.1 | 4.1 × 104 | 1.4 × 105 | 8.2 × 104 |
| D | −7.2 ± 0.1 | −7.5 ± 0.1 | −6.8 ± 0.1 | 1.9 × 105 | 3.1 × 105 | 9.6 × 104 |
| E | −8.2 ± 0.2 | −8.6 ± 0.3 | −8.2 ± 0.2 | 1.0 × 106 | 2.0 × 106 | 1.0 × 106 |
| F | −7 ± 0.1 | −7.6 ± 0.1 | −7.6 ± 0.1 | 1.4 × 105 | 3.7 × 105 | 3.7 × 105 |
| G | −5.8 ± 0.1 | −5.5 ± 0.1 | −5.7 ± 0.1 | 1.8 × 104 | 1.1 × 104 | 1.5 × 104 |
| H | −8.6 ± 0.2 | −9.5 ± 0.2 | −9.3 ± 0.1 | 2.0 × 106 | 9.2 × 106 | 6.6 × 106 |
| I | −8.7 ± 0.2 | −9.5 ± 0.1 | −9.1 ± 0.2 | 2.4 × 106 | 9.2 × 106 | 4.7 × 106 |
| J | −7.6 ± 0.1 | −8.9 ± 0.2 | −9.3 ± 0.2 | 3.7 × 105 | 3.3 × 106 | 6.6 × 106 |
| K | −8 ± 0.1 | −8.5 ± 0.2 | −8.4 ± 0.1 | 7.3 × 105 | 1.7 × 106 | 1.4 × 106 |
| L | −6.9 ± 0.1 | −7.9 ± 0.2 | −6.9 ± 0.1 | 1.1 × 105 | 6.2 × 105 | 1.1 × 105 |
| M | −6.1 ± 0.1 | −7.1 ± 0.1 | −6.1 ± 0.1 | 2.9 × 104 | 1.6 × 105 | 2.9 × 104 |
| N | −6.3 ± 0.1 | −6.7 ± 0.1 | −6.6 ± 0.2 | 4.1 × 104 | 8.1 × 104 | 6.9 × 104 |
| NL | −6.8 ± 0.1 | −8.4 ± 0.2 | −7.8 ± 0.1 | 9.6 × 104 | 1.4 × 106 | 5.2 × 105 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iqbal, D.; Alsaweed, M.; Jamal, Q.M.S.; Asad, M.R.; Rizvi, S.M.D.; Rizvi, M.R.; Albadrani, H.M.; Hamed, M.; Jahan, S.; Alyenbaawi, H. Pharmacophore-Based Screening, Molecular Docking, and Dynamic Simulation of Fungal Metabolites as Inhibitors of Multi-Targets in Neurodegenerative Disorders. Biomolecules 2023, 13, 1613. https://doi.org/10.3390/biom13111613
Iqbal D, Alsaweed M, Jamal QMS, Asad MR, Rizvi SMD, Rizvi MR, Albadrani HM, Hamed M, Jahan S, Alyenbaawi H. Pharmacophore-Based Screening, Molecular Docking, and Dynamic Simulation of Fungal Metabolites as Inhibitors of Multi-Targets in Neurodegenerative Disorders. Biomolecules. 2023; 13(11):1613. https://doi.org/10.3390/biom13111613
Chicago/Turabian StyleIqbal, Danish, Mohammed Alsaweed, Qazi Mohammad Sajid Jamal, Mohammad Rehan Asad, Syed Mohd Danish Rizvi, Moattar Raza Rizvi, Hind Muteb Albadrani, Munerah Hamed, Sadaf Jahan, and Hadeel Alyenbaawi. 2023. "Pharmacophore-Based Screening, Molecular Docking, and Dynamic Simulation of Fungal Metabolites as Inhibitors of Multi-Targets in Neurodegenerative Disorders" Biomolecules 13, no. 11: 1613. https://doi.org/10.3390/biom13111613
APA StyleIqbal, D., Alsaweed, M., Jamal, Q. M. S., Asad, M. R., Rizvi, S. M. D., Rizvi, M. R., Albadrani, H. M., Hamed, M., Jahan, S., & Alyenbaawi, H. (2023). Pharmacophore-Based Screening, Molecular Docking, and Dynamic Simulation of Fungal Metabolites as Inhibitors of Multi-Targets in Neurodegenerative Disorders. Biomolecules, 13(11), 1613. https://doi.org/10.3390/biom13111613