


Evidence Based on an Integrative Analysis of Multi-Omics Data on METTL7A as a Molecular Marker in Pan-Cancer

Abstract
:1. Introduction
2. Materials and Methods
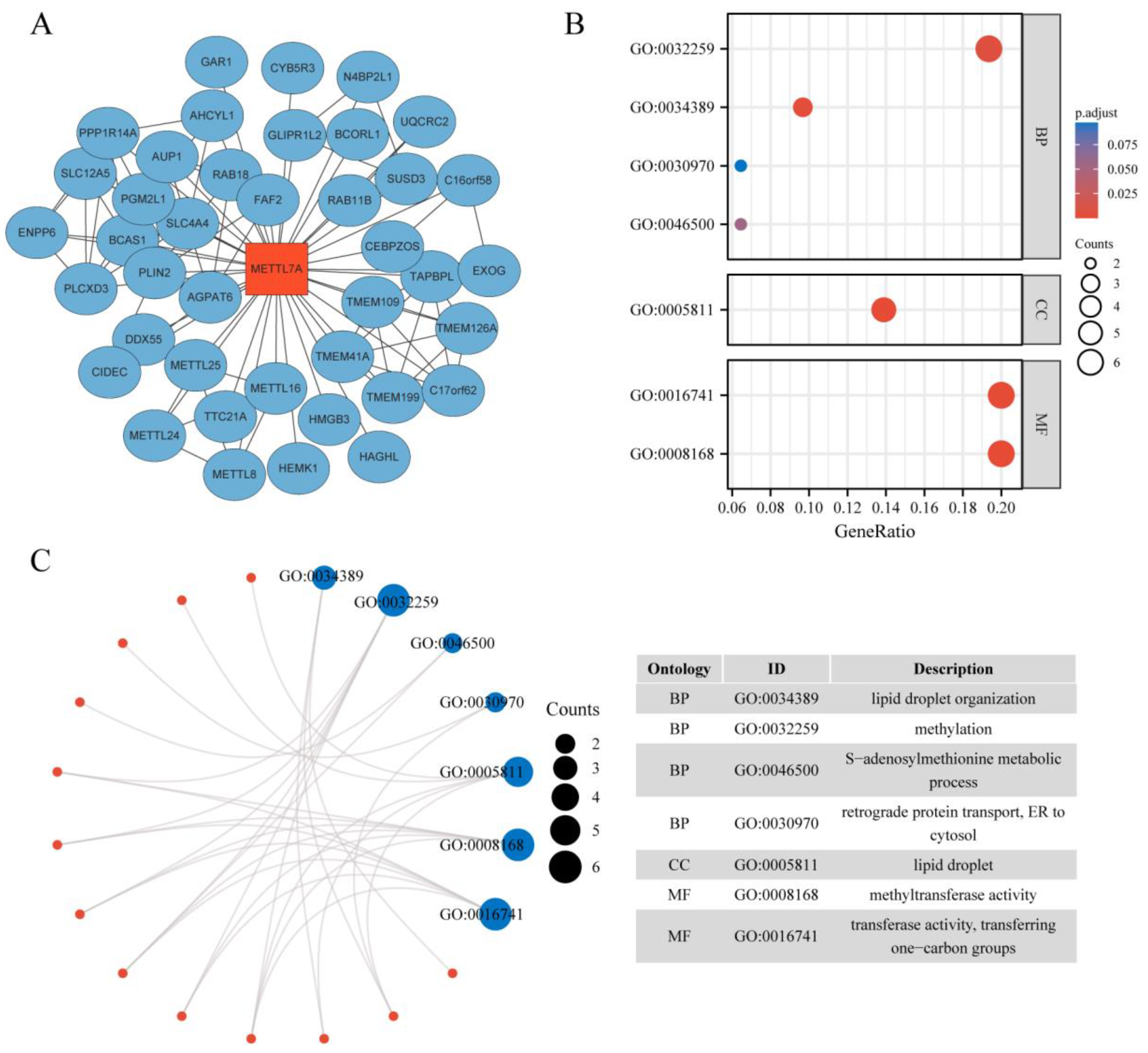
2.1. Protein–Protein Interaction Network and Enrichment Analysis of METTL7A-Binding Proteins
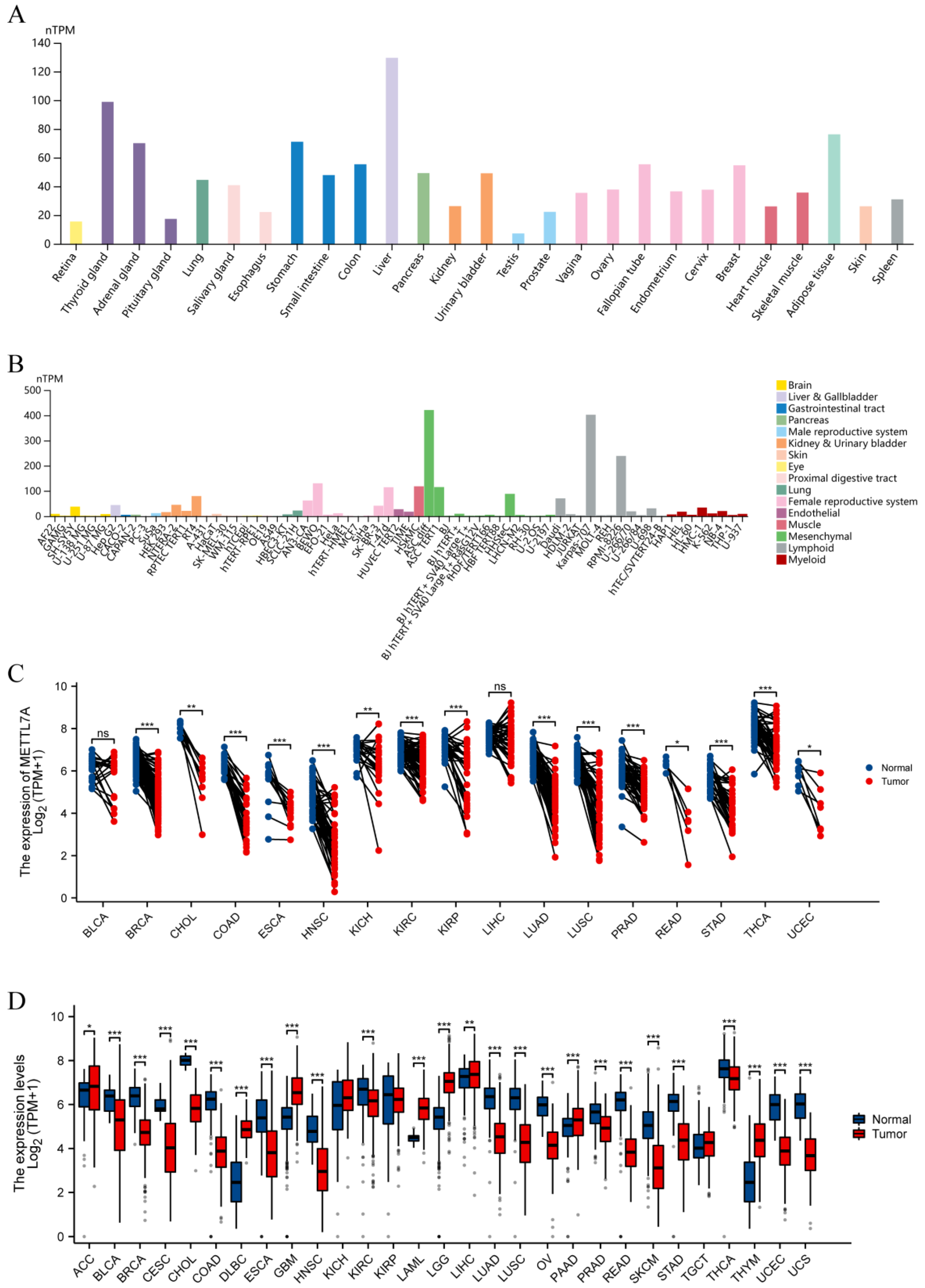
2.2. METTL7A Expression Analysis
2.3. METTL7A Expression in Subtypes of Cancers
2.4. Diagnostic Value and Prognostic Value Analysis in Pan-Cancer
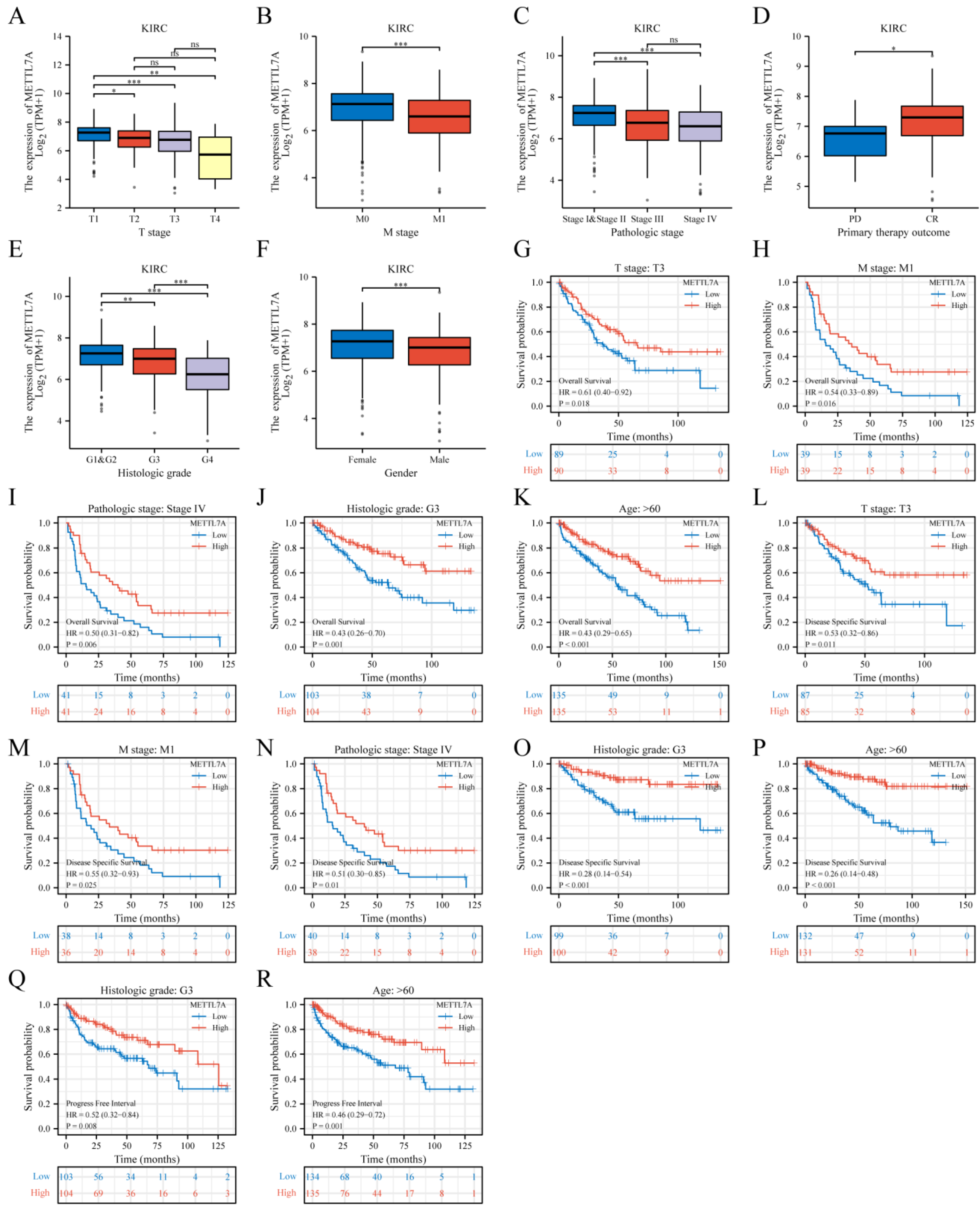
2.5. METTL7A Expression and Prognostic Value Analysis in Various Clinical Subgroups of KIRC
2.6. Immune Infiltration and Co-Expression Gene Analysis of METTL7A in KIRC
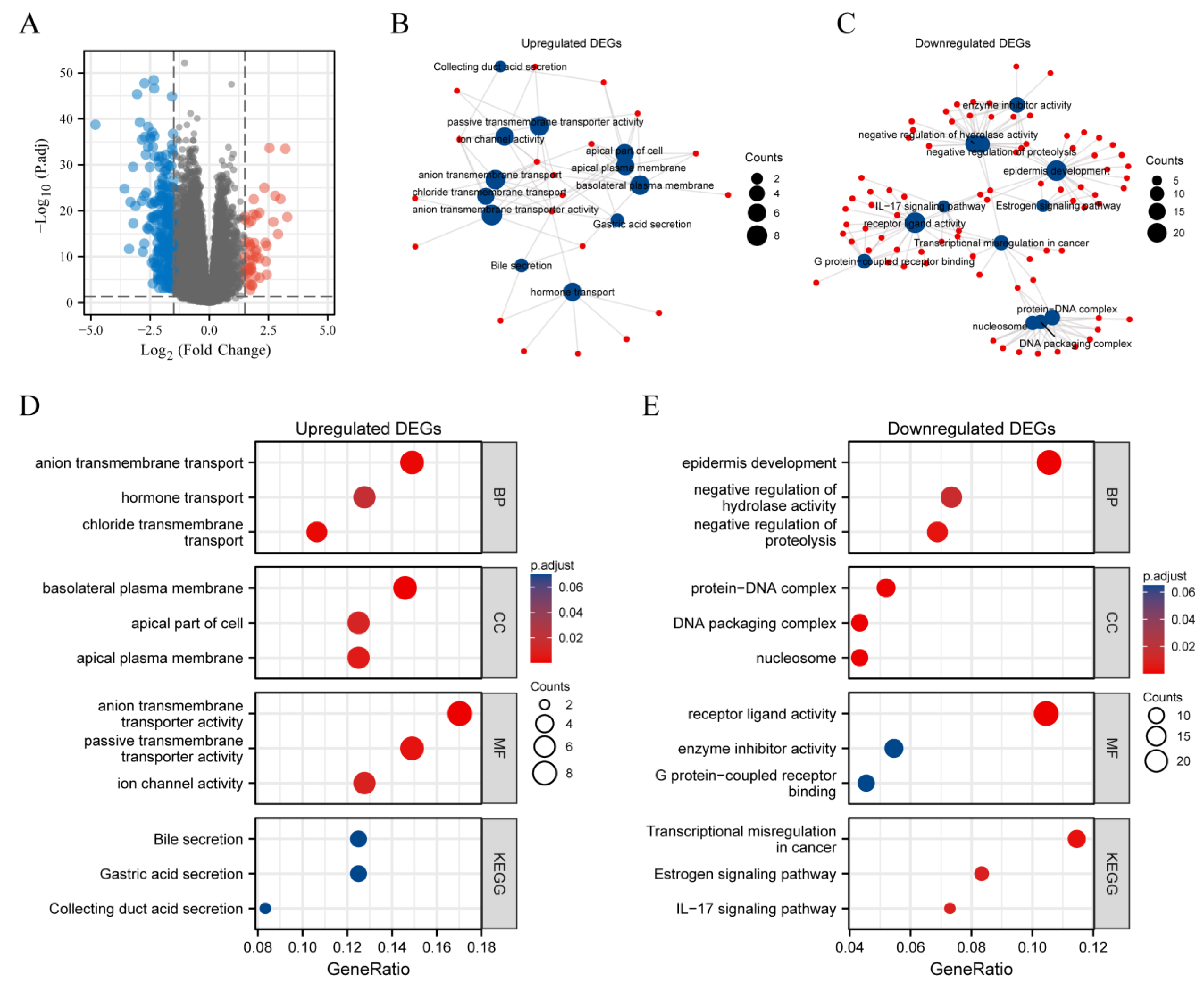
2.7. DEGs between METTL7A High Expression and Low Expression Groups in KIRC
3. Results
3.1. PPI Network and GO Enrichment Analysis of METTL7A-Binding Proteins
3.2. METTL7A Expression Analysis
3.3. METTL7A Expression in Molecular or Immune Subtype of Cancers
3.4. Diagnostic Value of METTL7A in Pan-Cancer
3.5. Prognostic Value of METTL7A in Pan-Cancer
3.6. METTL7A Expression and Prognostic Value Analysis in Various Clinical Subgroups of KIRC
3.7. Immune Infiltration and Co-Expression Gene Analysis of METTL7A in KIRC
3.8. DEGs between METTL7A High and Low Expression Groups in KIRC
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, Z.X.; Li, L.M.; Sun, H.L.; Liu, S.M. Link Between m6A Modification and Cancers. Front. Bioeng. Biotechnol. 2018, 6, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Wu, R.; Ming, L. The role of m6A RNA methylation in cancer. Biomed. Pharmacother. 2019, 112, 108613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, S.W.; Zhang, S.Y.; Gao, S.J.; Chen, Y.; Huang, Y. m6A-express: Uncovering complex and condition-specific m6A regulation of gene expression. Nucleic Acids Res. 2021, 49, e116. [Google Scholar] [CrossRef]
- Qin, Y.; Li, L.; Luo, E.; Hou, J.; Yan, G.; Wang, D.; Qiao, Y.; Tang, C. Role of m6A RNA methylation in cardiovascular disease. Int. J. Mol. Med. 2020, 46, 1958–1972. [Google Scholar] [CrossRef] [PubMed]
- Ignatova, V.V.; Jansen, P.W.T.C.; Baltissen, M.P.; Vermeulen, M.; Schneider, R. The interactome of a family of potential methyltransferases in HeLa cells. Sci. Rep. 2019, 9, 6584. [Google Scholar] [CrossRef] [Green Version]
- Ying, X.; Liu, B.; Yuan, Z.; Huang, Y.; Chen, C.; Jiang, X.; Zhang, H.; Qi, D.; Yang, S.; Lin, S.; et al. METTL1-m7 G-EGFR/EFEMP1 axis promotes the bladder cancer development. Clin. Transl. Med. 2021, 11, e675. [Google Scholar] [CrossRef]
- Chen, Z.; Zhu, W.; Zhu, S.; Sun, K.; Liao, J.; Liu, H.; Dai, Z.; Han, H.; Ren, X.; Yang, Q.; et al. METTL1 promotes hepatocarcinogenesis via m7 G tRNA modification-dependent translation control. Clin. Transl. Med. 2021, 11, e661. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Yi, Y.; Yue, X.; Wu, X.; Zhu, M.; Chen, Y.; Peng, S.; Kuang, M.; Lin, S.; Peng, Z. Methyltransferase 1 is required for nonhomologous end-joining repair and renders hepatocellular carcinoma resistant to radiotherapy. Hepatology 2022, ahead of print. [Google Scholar] [CrossRef]
- Orellana, E.A.; Liu, Q.; Yankova, E.; Pirouz, M.; De Braekeleer, E.; Zhang, W.; Lim, J.; Aspris, D.; Sendinc, E.; Garyfallos, D.A.; et al. METTL1-mediated m7G modification of Arg-TCT tRNA drives oncogenic transformation. Mol. Cell 2021, 81, 3323–3338.e14. [Google Scholar] [CrossRef]
- Dai, Z.; Liu, H.; Liao, J.; Huang, C.; Ren, X.; Zhu, W.; Zhu, S.; Peng, B.; Li, S.; Lai, J.; et al. N7-Methylguanosine tRNA modification enhances oncogenic mRNA translation and promotes intrahepatic cholangiocarcinoma progression. Mol. Cell 2021, 81, 3339–3355.e8. [Google Scholar] [CrossRef]
- Zeng, C.; Huang, W.; Li, Y.; Weng, H. Roles of METTL3 in cancer: Mechanisms and therapeutic targeting. J. Hematol. Oncol. 2020, 13, 117. [Google Scholar] [CrossRef]
- He, J.; Zhou, M.; Yin, J.; Wan, J.; Chu, J.; Jia, J.; Sheng, J.; Wang, C.; Yin, H.; He, F. METTL3 restrains papillary thyroid cancer progression via m6A/c-Rel/IL-8-mediated neutrophil infiltration. Mol. Ther. 2021, 29, 1821–1837. [Google Scholar] [CrossRef]
- Liu, J.; Eckert, M.A.; Harada, B.T.; Liu, S.-M.; Lu, Z.; Yu, K.; Tienda, S.M.; Chryplewicz, A.; Zhu, A.C.; Yang, Y.; et al. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat. Cell Biol. 2018, 20, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Ying, Y.; Zhao, Y.; Mundy, D.I.; Zhu, M.; Anderson, R.G. Chinese hamster ovary K2 cell lipid droplets appear to be metabolic organelles involved in membrane traffic. J. Biol. Chem. 2004, 279, 3787–3792. [Google Scholar] [CrossRef] [Green Version]
- Zehmer, J.K.; Bartz, R.; Bisel, B.; Liu, P.; Seemann, J.; Anderson, R.G. Targeting sequences of UBXD8 and AAM-B reveal that the ER has a direct role in the emergence and regression of lipid droplets. J. Cell Sci. 2009, 122 Pt 20, 3694–3702. [Google Scholar] [CrossRef] [Green Version]
- Zehmer, J.K.; Bartz, R.; Liu, P.; Anderson, R.G. Identification of a novel N-terminal hydrophobic sequence that targets proteins to lipid droplets. J. Cell Sci. 2008, 121, 1852–1860. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; He, J.; Bach, D.-H.; Huang, Y.-H.; Li, Z.; Liu, H.; Lin, P.; Yang, J. Induction of m6A methylation in adipocyte exosomal LncRNAs mediates myeloma drug resistance. J. Exp. Clin. Cancer Res. 2022, 41, 4. [Google Scholar] [CrossRef] [PubMed]
- Jun, F.; Peng, Z.; Zhang, Y.; Shi, D. Quantitative proteomic analysis identifies novel regulators of methotrexate resistance in choriocarcinoma. Gynecol. Oncol. 2020, 157, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Ma, H.; Zhou, Y. Bioinformatics analysis of microarray data to identify the candidate biomarkers of lung adenocarcinoma. PeerJ 2019, 7, e7313. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Liu, Y.; Han, Z.; Tian, R. Identification of potential gene signatures associated with osteosarcoma by integrated bioinformatics analysis. PeerJ 2021, 9, e11496. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhu, Y.; Li, X.; Zhang, X.; Yu, B. Identification of potential biomarkers and metabolic pathways based on integration of metabolomic and transcriptomic data in the development of breast cancer. Arch. Gynecol. Obstet. 2021, 303, 1599–1606. [Google Scholar] [CrossRef]
- Zhou, S.; Shen, Y.; Zheng, M.; Wang, L.; Che, R.; Hu, W.; Li, P. DNA methylation of METTL7A gene body regulates its transcriptional level in thyroid cancer. Oncotarget 2017, 8, 34652–34660. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Vivian, J.; Rao, A.A.; Nothaft, F.A.; Ketchum, C.; Armstrong, J.; Novak, A.; Pfeil, J.; Narkizian, J.; Deran, A.D.; Musselman-Brown, A.; et al. Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol. 2017, 35, 314–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ru, B.; Wong, C.N.; Tong, Y.; Zhong, J.Y.; Zhong, S.S.W.; Wu, W.C.; Chu, K.C.; Wong, C.Y.; Lau, C.Y.; Chen, I.; et al. TISIDB: An integrated repository portal for tumor-immune system interactions. Bioinformatics 2019, 35, 4200–4202. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lichtenberg, T.M.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Campeanu, I.J.; Jiang, Y.; Liu, L.; Pilecki, M.; Najor, A.; Cobani, E.; Manning, M.; Zhang, X.M.; Yang, Z.-Q. Multi-omics integration of methyltransferase-like protein family reveals clinical outcomes and functional signatures in human cancer. Sci. Rep. 2021, 11, 14784. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Kim, J.Y.; Kim, T.K.; Park, S.Y.; Im, G.I. Methyltransferase-like protein 7A (METTL7A) promotes cell survival and osteogenic differentiation under metabolic stress. Cell Death Discov. 2021, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Briata, P.; Caputo, L.; Zapparoli, E.; Marcaccini, E.; Passalacqua, M.; Brondolo, L.; Bordo, D.; Rossi, A.; Nicoletti, C.; Bucci, G.; et al. LncRNA EPR-induced METTL7A1 modulates target gene translation. Nucleic Acids Res. 2022, 50, 7608–7622. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Song, Y.; Chan, T.H.M.; Yang, H.; Lin, C.H.; Tay, D.J.T.; Hong, H.; Tang, S.J.; Tan, K.T.; Huang, X.X.; et al. An RNA editing/dsRNA binding-independent gene regulatory mechanism of ADARs and its clinical implication in cancer. Nucleic Acids Res. 2017, 45, 10436–10451. [Google Scholar] [CrossRef] [Green Version]
- Makhoul, C.; Gosavi, P.; Gleeson, P.A. The Golgi architecture and cell sensing. Biochem. Soc. Trans. 2018, 46, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, C.M.; Mellor, H. The tumor suppressor RhoBTB1 controls Golgi integrity and breast cancer cell invasion through METTL7B. BMC Cancer 2017, 17, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Yan, M.; Zhang, C.; Xue, J.; Zhang, Q.; Ma, S.; Guan, F.; Cao, W. Comprehensive analysis of the HOXA gene family identifies HOXA13 as a novel oncogenic gene in kidney renal clear cell carcinoma. J. Cancer Res. Clin. Oncol. 2020, 146, 1993–2006. [Google Scholar] [CrossRef]
- Klümper, N.; Ralser, D.J.; Bawden, E.G.; Landsberg, J.; Zarbl, R.; Kristiansen, G.; Toma, M.; Ritter, M.; Hölzel, M.; Ellinger, J.; et al. LAG3 (LAG-3, CD223) DNA methylation correlates with LAG3 expression by tumor and immune cells, immune cell infiltration, and overall survival in clear cell renal cell carcinoma. J. Immunother. Cancer 2020, 8, e000552. [Google Scholar] [CrossRef] [Green Version]
- Camacho, D.M.; Collins, K.M.; Powers, R.K.; Costello, J.C.; Collins, J.J. Next-Generation Machine Learning for Biological Networks. Cell 2018, 173, 1581–1592. [Google Scholar] [CrossRef] [Green Version]
- Poirion, O.B.; Jing, Z.; Chaudhary, K.; Huang, S.; Garmire, L.X. DeepProg: An ensemble of deep-learning and machine-learning models for prognosis prediction using multi-omics data. Genome Med. 2021, 13, 112. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Low Expression of METTL7A | High Expression of METTL7A | p |
|---|---|---|---|
| n | 269 | 270 | |
| T stage | n (%) | <0.001 | |
| T1 | 108 (20%) | 170 (31.5%) | |
| T2 | 38 (7.1%) | 33 (6.1%) | |
| T3 | 115 (21.3%) | 64 (11.9%) | |
| T4 | 8 (1.5%) | 3 (0.6%) | |
| M stage | n (%) | <0.001 | |
| M0 | 202 (39.9%) | 226 (44.7%) | |
| M1 | 54 (10.7%) | 24 (4.7%) | |
| Pathologic stage | n (%) | <0.001 | |
| Stage I | 105 (19.6%) | 167 (31.2%) | |
| Stage II | 29 (5.4%) | 30 (5.6%) | |
| Stage III | 77 (14.4%) | 46 (8.6%) | |
| Stage IV | 56 (10.4%) | 26 (4.9%) | |
| Primary therapy outcome | n (%) | 0.177 | |
| PD | 8 (5.4%) | 3 (2%) | |
| CR | 52 (35.4%) | 76 (51.7%) | |
| Histologic grade | n (%) | <0.001 | |
| G1 | 3 (0.6%) | 11 (2.1%) | |
| G2 | 97 (18.3%) | 138 (26%) | |
| G3 | 107 (20.2%) | 100 (18.8%) | |
| G4 | 60 (11.3%) | 15 (2.8%) | |
| Gender | n (%) | 0.025 | |
| Female | 80 (14.8%) | 106 (19.7%) | |
| Male | 189 (35.1%) | 164 (30.4%) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Chen, Y.; Shen, T. Evidence Based on an Integrative Analysis of Multi-Omics Data on METTL7A as a Molecular Marker in Pan-Cancer. Biomolecules 2023, 13, 195. https://doi.org/10.3390/biom13020195
Liu Z, Chen Y, Shen T. Evidence Based on an Integrative Analysis of Multi-Omics Data on METTL7A as a Molecular Marker in Pan-Cancer. Biomolecules. 2023; 13(2):195. https://doi.org/10.3390/biom13020195
Chicago/Turabian StyleLiu, Zikai, Yiqun Chen, and Tong Shen. 2023. "Evidence Based on an Integrative Analysis of Multi-Omics Data on METTL7A as a Molecular Marker in Pan-Cancer" Biomolecules 13, no. 2: 195. https://doi.org/10.3390/biom13020195
APA StyleLiu, Z., Chen, Y., & Shen, T. (2023). Evidence Based on an Integrative Analysis of Multi-Omics Data on METTL7A as a Molecular Marker in Pan-Cancer. Biomolecules, 13(2), 195. https://doi.org/10.3390/biom13020195