
Using ΔK280 TauRD Folding Reporter Cells to Screen TRKB Agonists as Alzheimer’s Disease Treatment Strategy

 , , and
, , and 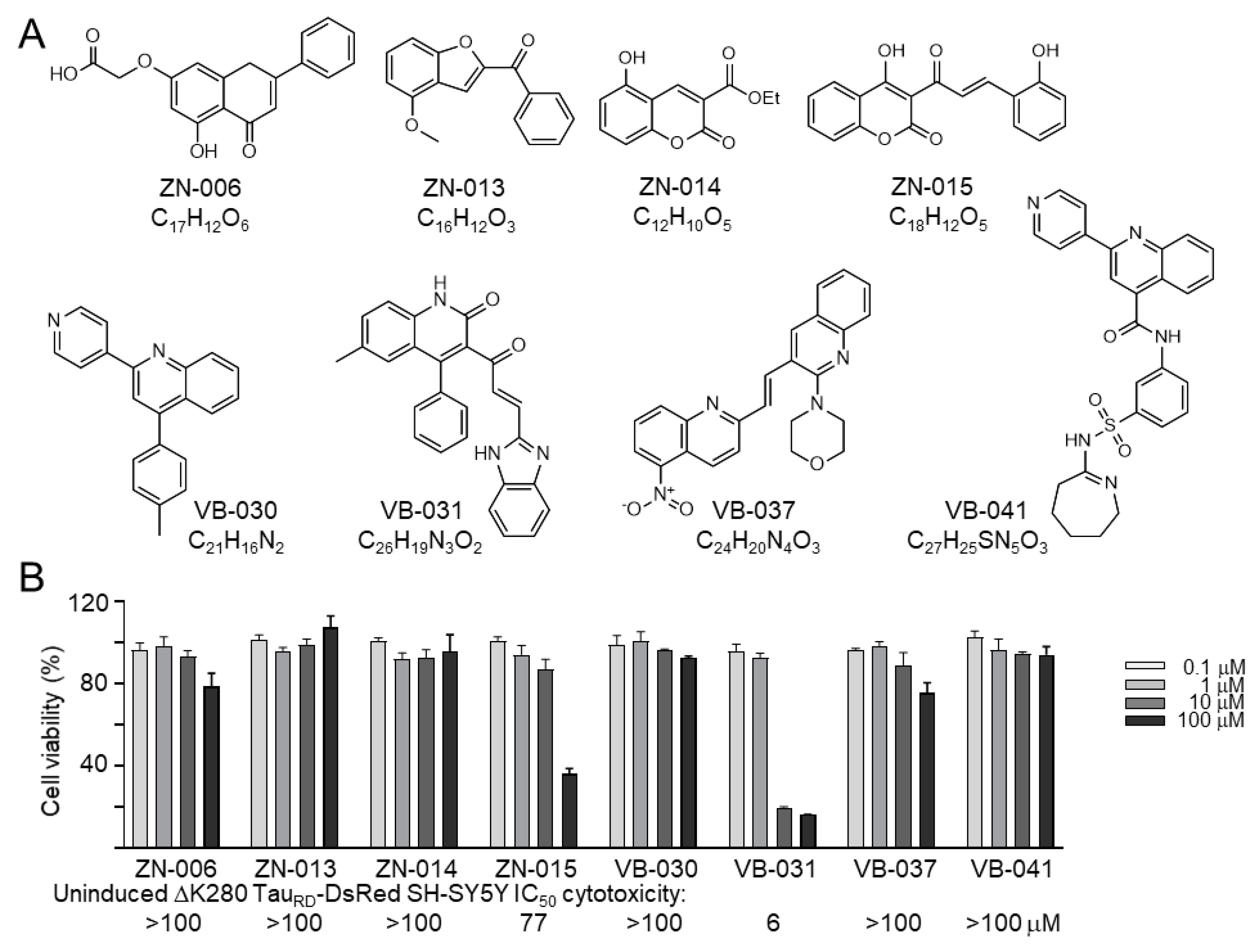{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Compounds
2.2. Cell Culture and Cell Viability Assay
2.3. Antioxidant Assays
2.4. Biochemical Fluorescence-Based TauRD Aggregation Assay
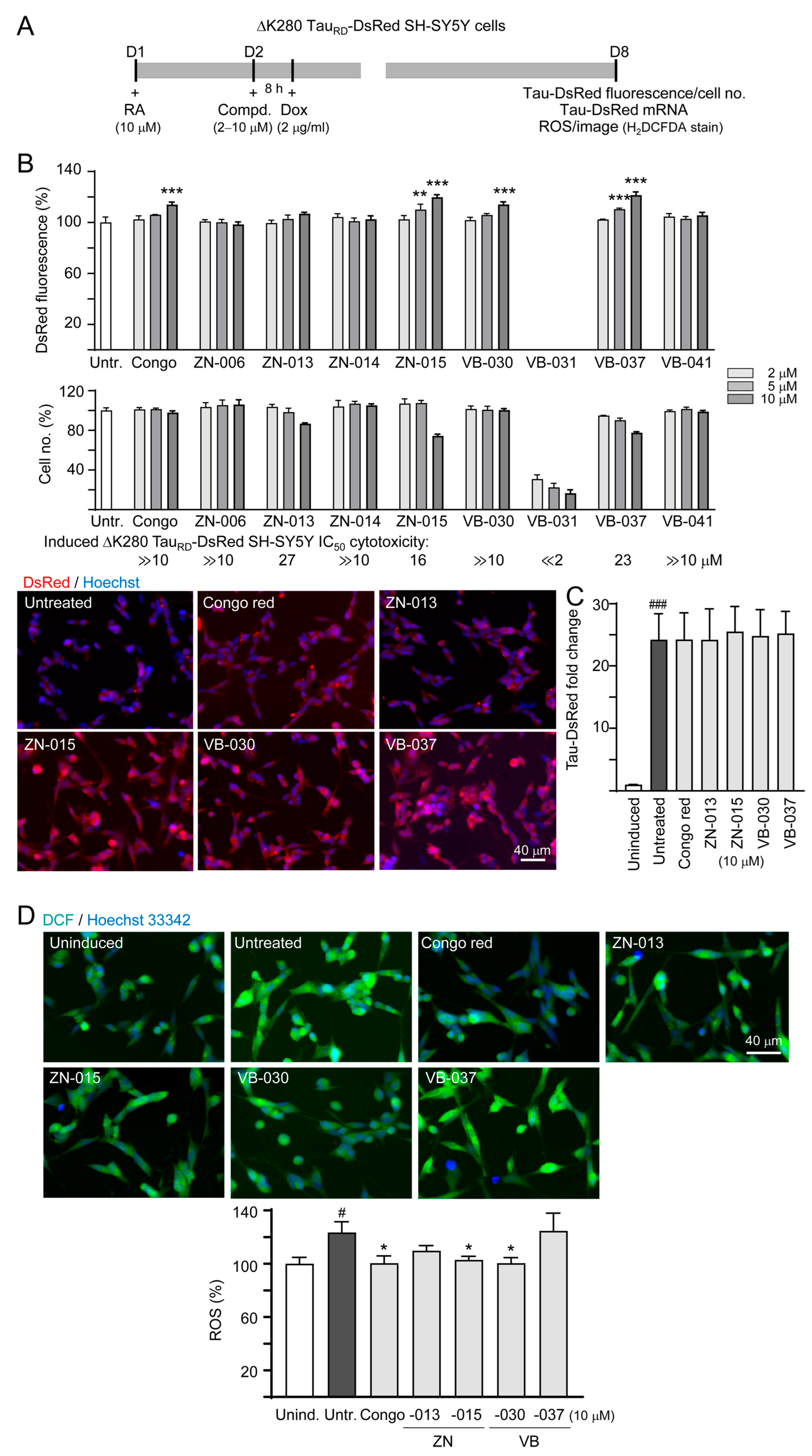
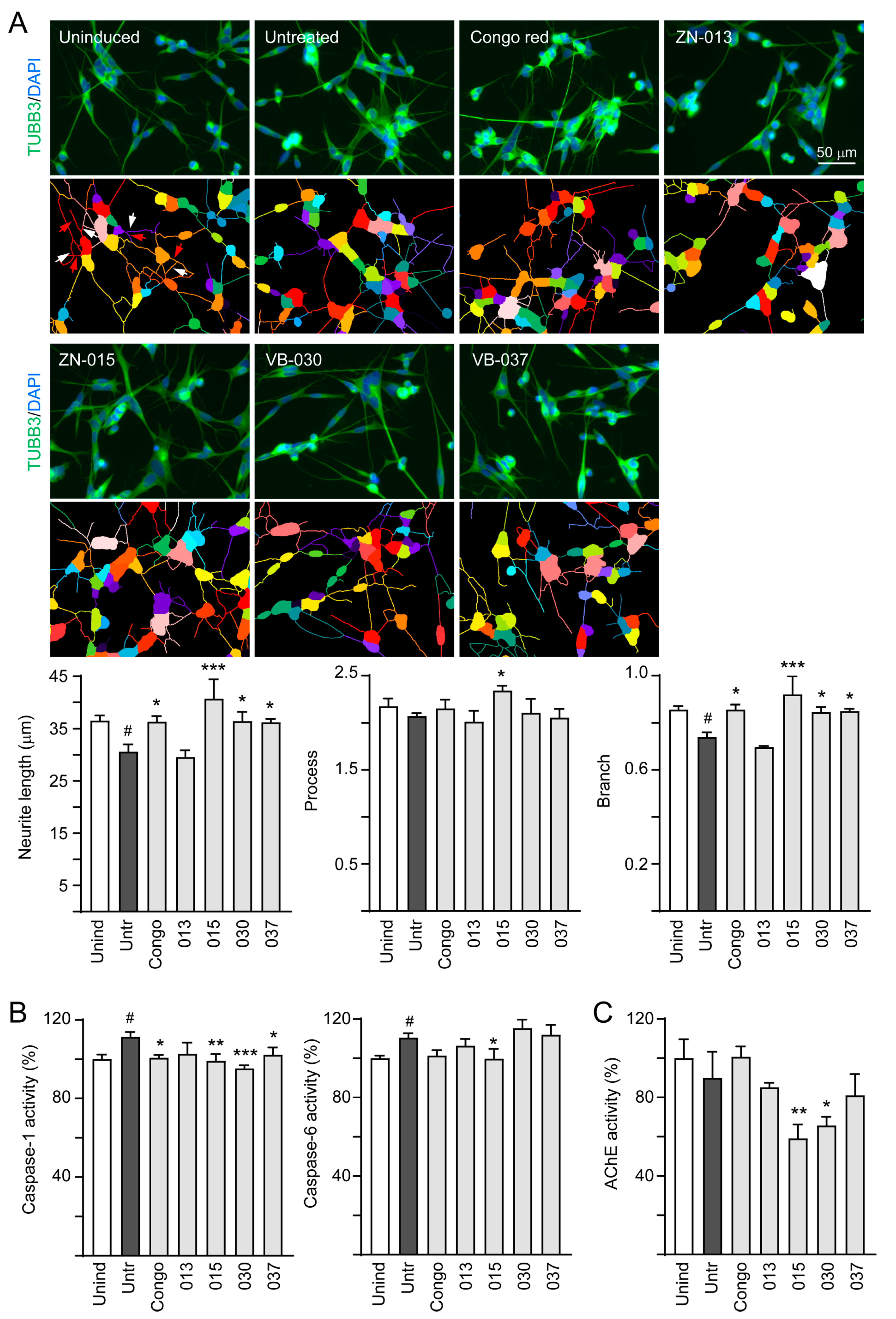
2.5. High-Content Analysis of Cellular ΔK280 TauRD-DsRed Aggregation, Reactive Oxygen Species (ROS) and Neurite Outgrowth
2.6. Real-Time PCR Analysis
2.7. Caspase-1/6 and Acetylcholinesterase (AChE) Activity Assays
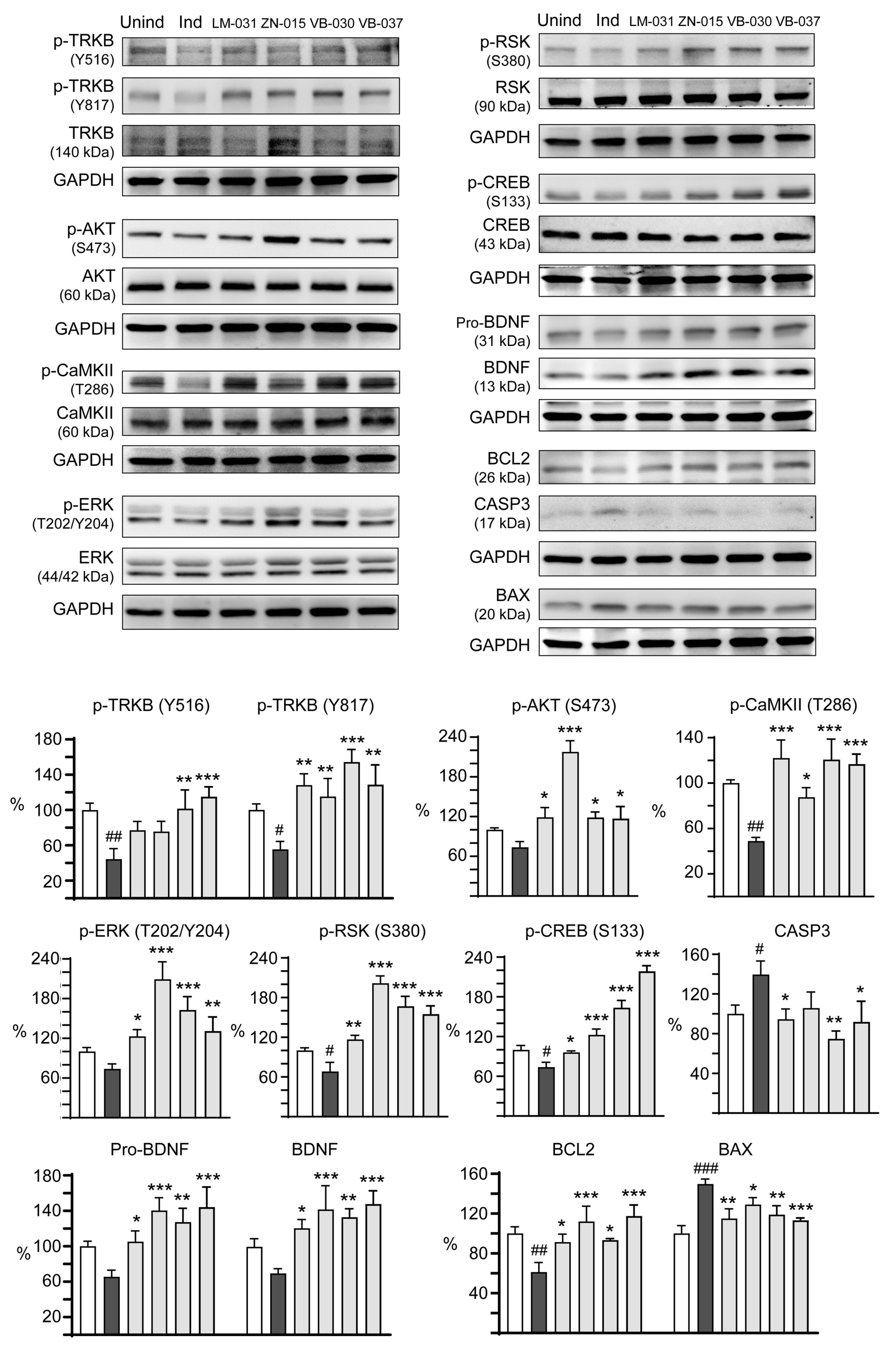
2.8. Western Blot Analysis
2.9. RNA Interference
2.10. Tryptophan Fluorescence Quenching Assay
2.11. Statistical Analysis
3. Results
3.1. Cytotoxicity of Heterocyclic ZN/VB Compounds
3.2. Screening Compounds Promoting ΔK280 TauRD-DsRed Folding and Reducing Cellular Oxidative Stress
3.3. Chemical Chaperone and Antioxidant Activities of ZN/VB Compounds
3.4. Neuroprotective Effects of ZN/VB Compounds
3.5. Targets of ZN/VB Compounds on TRKB Pathway
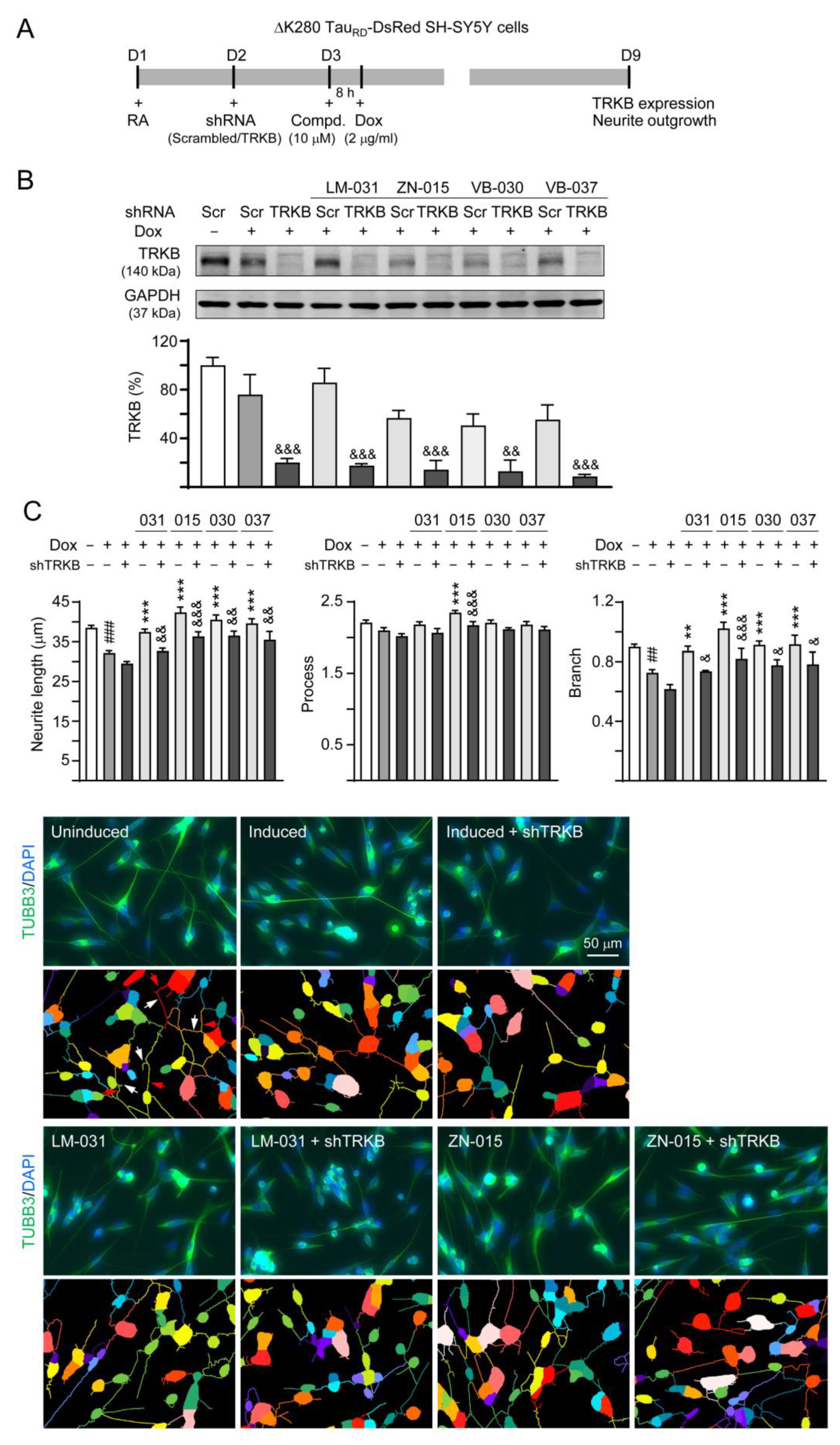
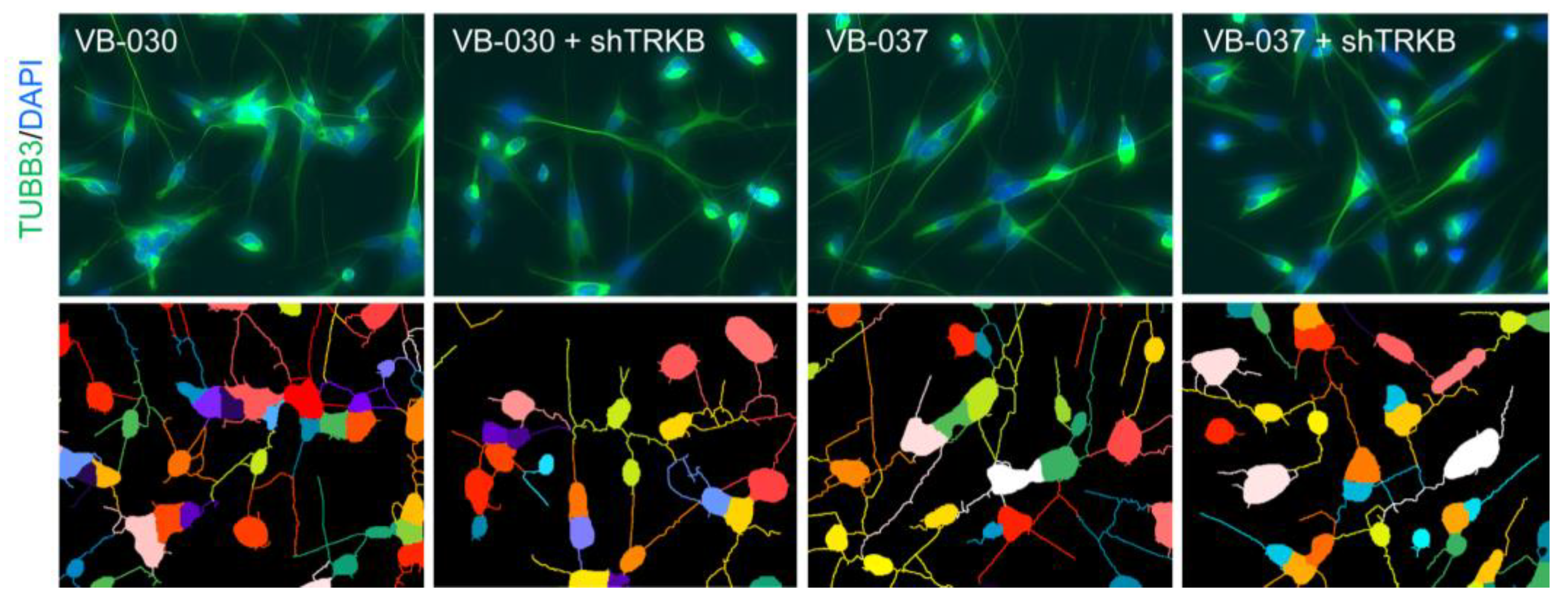
3.6. Effects of TRKB Knockdown on Neurite Outgrowth
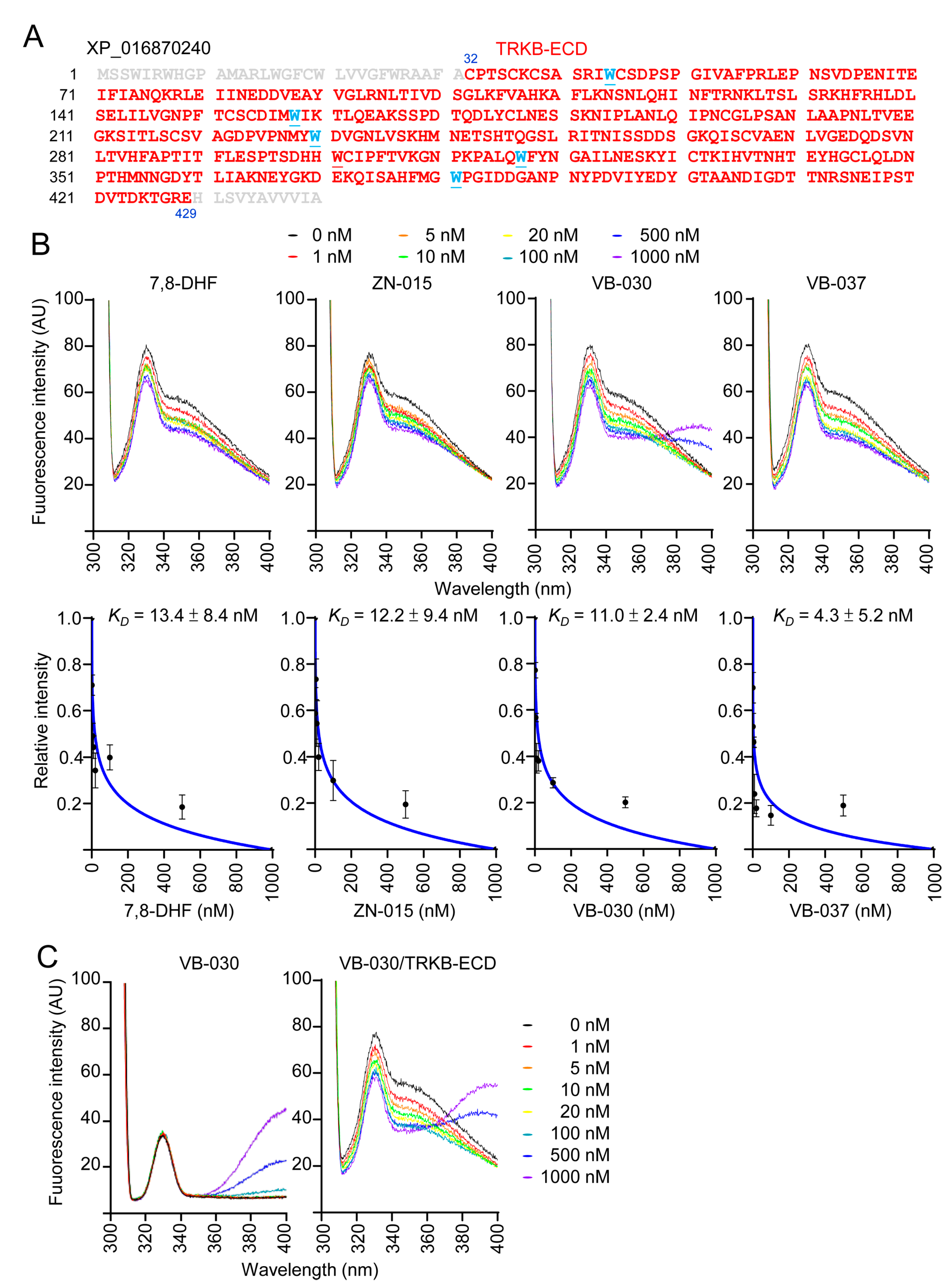
3.7. Binding Affinity of ZN-015, VB-030 and VB-037 with TRKB-ECD
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iqbal, K.; Alonso Adel, C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Drubin, D.G.; Kirschner, M.W. Tau protein function in living cells. J. Cell Biol. 1986, 103, 2739–2746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terwel, D.; Dewachter, I.; Van Leuven, F. Axonal transport, tau protein, and neurodegeneration in Alzheimer’s disease. Neuromol. Med. 2002, 2, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza, I.; Poorkaj, P.; Hong, M.; Nochlin, D.; Lee, V.M.; Bird, T.D.; Schellenberg, G.D. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc. Natl. Acad. Sci. USA 1999, 96, 5598–5603. [Google Scholar] [CrossRef] [Green Version]
- Momeni, P.; Pittman, A.; Lashley, T.; Vandrovcova, J.; Malzer, E.; Luk, C.; Hulette, C.; Lees, A.; Revesz, T.; Hardy, J.; et al. Clinical and pathological features of an Alzheimer’s disease patient with the MAPT ∆K280 mutation. Neurobiol. Aging 2009, 30, 388–393. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.H.; Chen, I.C.; Lin, H.Y.; Chen, H.C.; Lin, C.H.; Lin, T.H.; Weng, Y.-T.; Chao, C.-Y.; Wu, Y.-R.; Chen, C.; et al. The aqueous extract of Glycyrrhiza inflata can upregulate unfolded protein response-mediated chaperones to reduce tau misfolding in cell models of Alzheimer’s disease. Drug Des. Devel. Ther. 2016, 10, 885–896. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.H.; Chang, K.H.; Chiu, Y.J.; Weng, Z.K.; Sun, Y.C.; Lin, W.; Lee-Chen, G.-J.; Chen, C.-M. Neuroprotective action of coumarin derivatives through activation of TRKB-CREB-BDNF pathway and reduction of caspase activity in neuronal cells expressing pro-aggregated Tau protein. Int. J. Mol. Sci. 2022, 23, 12734. [Google Scholar] [CrossRef]
- Rosa, E.; Mahendram, S.; Ke, Y.D.; Ittner, L.M.; Ginsberg, S.D.; Fahnestock, M. Tau downregulates BDNF expression in animal and cellular models of Alzheimer’s disease. Neurobiol. Aging 2016, 48, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Nagappan, G.; Guan, X.; Nathan, P.J.; Wren, P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 401–416. [Google Scholar] [CrossRef]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Hains, J.M.; Armanini, M.; Laramee, G.R.; Johnson, S.A.; Winslow, J.W. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 1991, 7, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuruta, F.; Masuyama, N.; Gotoh, Y. The phosphatidylinositol 3-kinase (PI3K)-Akt pathway suppresses Bax translocation to mitochondria. J. Biol. Chem. 2002, 277, 14040–14047. [Google Scholar] [CrossRef] [Green Version]
- Hauge, C.; Frödin, M. RSK and MSK in MAP kinase signaling. J. Cell Sci. 2006, 119, 3021–3023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabuchi, A.; Sakaya, H.; Kisukeda, T.; Fushiki, H.; Tsuda, M. Involvement of an upstream stimulatory factor as well as cAMP-responsive element-binding protein in the activation of brain-derived neurotrophic factor gene promoter I. J. Biol. Chem. 2002, 277, 35920–35931. [Google Scholar] [CrossRef] [Green Version]
- Riccio, A.; Ahn, S.; Davenport, C.M.; Blendy, J.A.; Ginty, D.D. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science 1999, 286, 2358–2361. [Google Scholar] [CrossRef]
- Sharma, V.K.; Singh, T.G. CREB: A multifaceted target for Alzheimer’s disease. Curr. Alzheimer Res. 2020, 17, 1280–1293. [Google Scholar] [CrossRef]
- Tao, X.; Finkbeiner, S.; Arnold, D.B.; Shaywitz, A.J.; Greenberg, M.E. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 1998, 20, 709–726. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.J.; Lin, T.H.; Chen, C.M.; Lin, C.H.; Teng, Y.S.; Lin, C.Y.; Sun, Y.-C.; Hsieh-Li, H.M.; Su, M.-T.; Lee-Chen, G.-J.; et al. Novel synthetic coumarin-chalcone derivative (E)-3-(3-(4-(dimethylamino)phenyl)acryloyl)-4-hydroxy-2H-chromen-2-one activates CREB-mediated neuroprotection in Aβ and tau cell models of Alzheimer’s disease. Oxid. Med. Cell. Longev. 2021, 2021, 3058861. [Google Scholar] [CrossRef]
- Hsiao, Y.H.; Hung, H.C.; Chen, S.H.; Gean, P.W. Social interaction rescues memory deficit in an animal model of Alzheimer’s disease by increasing BDNF-dependent hippocampal neurogenesis. J. Neurosci. 2014, 34, 16207–16219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagahara, A.H.; Merrill, D.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bubols, G.B.; da Rocha Vianna, D.; Medina-Remon, A.; von Poser, G.; Lamuela-Raventos, R.M.; Eifler-Lima, V.L.; Garcia, S. The antioxidant activity of coumarins and flavonoids. Mini Rev. Med. Chem. 2013, 13, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Dawood, K.M. An update on benzofuran inhibitors: A patent review. Expert Opin. Ther. Pat. 2019, 29, 841–870. [Google Scholar] [CrossRef]
- Matada, B.S.; Pattanashettar, R.; Yernale, N.G. A comprehensive review on the biological interest of quinoline and its derivatives. Bioorg. Med. Chem. 2021, 32, 115973. [Google Scholar] [CrossRef]
- Gao, L.; Tian, M.; Zhao, H.Y.; Xu, Q.Q.; Huang, Y.M.; Si, Q.C.; Tian, Q.; Wu, Q.-M.; Hu, X.-M.; Sun, L.-B.; et al. TrkB activation by 7, 8-dihydroxyflavone increases synapse AMPA subunits and ameliorates spatial memory deficits in a mouse model of Alzheimer’s disease. J. Neurochem. 2016, 136, 620–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Liu, X.; Schroeder, J.P.; Chan, C.B.; Song, M.; Yu, S.P.; Weinshenker, D.; Ye, K. 7,8-dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2014, 39, 638–650. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.C.; Chang, K.H.; Chiu, Y.J.; Chen, Y.R.; Lung, T.H.; Hsieh-Li, H.M.; Su, M.-T.; Sun, Y.-C.; Chen, C.-M.; Lin, W.; et al. Multi-target effects of novel synthetic coumarin derivatives protecting Aβ-GFP SH-SY5Y cells against Aβ toxicity. Cells 2021, 10, 3095. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Hsieh, Y.S.; Sun, Y.C.; Huang, W.H.; Chen, S.L.; Weng, Z.K.; Lin, T.-H.; Wu, Y.-R.; Chang, K.-H.; Huang, H.-J.; et al. Virtual screening and testing of GSK-3 inhibitors using human SH-SY5Y cells expressing Tau folding reporter and mouse hippocampal primary culture under Tau cytotoxicity. Biomol. Ther. 2022. [CrossRef]
- Lee, S.Y.; Chiu, Y.J.; Yang, S.M.; Chen, C.M.; Huang, C.C.; Lee-Chen, G.J.; Lin, W.; Chang, K. Novel synthetic chalcone-coumarin hybrid for Aβ aggregation reduction, antioxidation, and neuroprotection. CNS Neurosci. Ther. 2018, 24, 1286–1298. [Google Scholar] [CrossRef]
- Sharma, O.P.; and Bhat, T.K. DPPH antioxidant assay revisited. Food Chem. 2009, 113, 1202–1205. [Google Scholar] [CrossRef]
- Ou, B.; Hampsch-Woodill, M.; Prior, R.L. Development and validation of an improved oxygen radical absorbance capacity assay using fluorescein as the fluorescent probe. J. Agric. Food Chem. 2001, 49, 4619–4926. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.H.; Chiu, Y.J.; Lin, C.H.; Lin, C.Y.; Chao, C.Y.; Chen, Y.C.; Yang, S.; Lin, W.; Hsieh-Li, H.M.; Wu, Y.; et al. Exploration of multi-target effects of 3-benzoyl-5-hydroxychromen-2-one in Alzheimer’s disease cell and mouse models. Aging Cell 2020, 19, e13169. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.N.; Lin, T.H.; Teng, Y.S.; Sun, Y.C.; Chang, K.H.; Lin, C.Y.; Hsieh-Li, H.M.; Su, M.-T.; Chen, C.-M.; Lee-Chen, G.-J. Flavones 7,8-DHF, quercetin, and apigenin against Tau toxicity via activation of TRKB Signaling in ΔK280 TauRD-DsRed SH-SY5Y Cells. Front. Aging Neurosci. 2021, 13, 758895. [Google Scholar] [CrossRef]
- Liu, X.; Obianyo, O.; Chan, C.B.; Huang, J.; Xue, S.; Yang, J.J.; Zeng, F.; Goodman, M.; Ye, K. Biochemical and biophysical investigation of the brain-derived neurotrophic factor mimetic 7,8-dihydroxyflavone in the binding and activation of the TrkB receptor. J. Biol. Chem. 2014, 289, 27571–27584. [Google Scholar] [CrossRef] [Green Version]
- Gendron, T.F.; Petrucelli, L. The role of tau in neurodegeneration. Mol. Neurodegener. 2009, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Sivananthan, S.N.; Lee, A.W.; Goodyer, C.G.; LeBlanc, A.C. Familial amyloid precursor protein mutants cause caspase-6-dependent but amyloid beta-peptide independent neuronal degeneration in primary human neuron cultures. Cell Death Dis. 2010, 1, e100. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Pétrin, D.; Zhang, Z.; Bergeron, C.; Goodyer, C.G.; LeBlanc, A.C. Caspase-1 activation of caspase-6 in human apoptotic neurons. Cell Death Differ. 2006, 13, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Mathew, A.; Balaji E, V.; Pai, S.R.K.; Kishore, A.; Pai, V.; Pemmireddy, R.; Chandrashekar, K.S. Current drug targets in Alzheimer’s associated memory impairment: A comprehensive review. CNS Neurol. Disord. Drug Targets 2023, 22, 255–275. [Google Scholar] [CrossRef]
- Jang, S.W.; Liu, X.; Yepes, M.; Shepherd, K.R.; Miller, G.W.; Liu, Y.; Wilson, W.D.; Xiao, G.; Blanchi, B.; Sun, Y.E.; et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. USA 2010, 107, 2687–2692. [Google Scholar] [CrossRef]
- Qian, W.; Liu, F. Regulation of alternative splicing of tau exon 10. Neurosci. Bull. 2014, 30, 367–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.M.; Yen, C.Y.; Chen, W.L.; Lin, C.H.; Wu, Y.R.; Chang, K.H.; Lee-Chen, G.-J. Pathomechanism characterization and potential therapeutics identification for Parkinson’s disease targeting neuroinflammation. Int. J. Mol. Sci. 2021, 22, 1062. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Renis, M.; Calderone, A.; Russo, A.; Barcellona, M.L.; Rizza, V. Stress proteins and SH-groups in oxidant-induced cell damage after acute ethanol administration in rat. Free Radic. Biol. Med. 1996, 20, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Cornelius, C.; Dinkova-Kostova, A.T.; Calabrese, E.J.; Mattson, M.P. Cellular stress responses, the hormesis paradigm, and vitagenes: Novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid. Redox Signal. 2010, 13, 1763–1811. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, E.J.; Kozumbo, W.J. The hormetic dose-response mechanism: Nrf2 activation. Pharmacol. Res. 2021, 167, 105526. [Google Scholar] [CrossRef]
- Devi, L.; Ohno, M. TrkB reduction exacerbates Alzheimer’s disease-like signaling aberrations and memory deficits without affecting β-amyloidosis in 5XFAD mice. Transl. Psychiatry 2015, 5, e562. [Google Scholar] [CrossRef] [Green Version]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Razani, E.; Pourbagheri-Sigaroodi, A.; Safaroghli-Azar, A.; Zoghi, A.; Shanaki-Bavarsad, M.; Bashash, D. The PI3K/Akt signaling axis in Alzheimer’s disease: A valuable target to stimulate or suppress? Cell Stress Chaperones 2021, 26, 871–887. [Google Scholar] [CrossRef]
- Sun, X.Y.; Tuo, Q.Z.; Liuyang, Z.Y.; Xie, A.J.; Feng, X.L.; Yan, X.; Qiu, M.; Li, S.; Wang, X.-L.; Cao, F.-Y.; et al. Extrasynaptic NMDA receptor-induced tau overexpression mediates neuronal death through suppressing survival signaling ERK phosphorylation. Cell Death Dis. 2016, 7, e2449. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, Y.J. Effect of obesity on cognitive impairment in vascular dementia rat model via BDNF-ERK-CREB pathway. Biol. Res. Nurs. 2021, 23, 248–257. [Google Scholar] [CrossRef]
- Yu, X.W.; Oh, M.M.; Disterhoft, J.F. CREB, cellular excitability, and cognition: Implications for aging. Behav. Brain Res. 2017, 322, 206–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amidfar, M.; de Oliveira, J.; Kucharska, E.; Budni, J.; Kim, Y.K. The role of CREB and BDNF in neurobiology and treatment of Alzheimer’s disease. Life Sci. 2020, 257, 118020. [Google Scholar] [CrossRef]
- Miller, S.; Yasuda, M.; Coats, J.K.; Jones, Y.; Martone, M.E.; Mayford, M. Disruption of dendritic translation of CaMKIIα impairs stabilization of synaptic plasticity and memory consolidation. Neuron 2002, 36, 507–519. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Liu, J.; Ye, Z.; Huang, J.; He, F.; Xiao, W.; Hu, X.; Luo, Z. CaMKII-mediated CREB phosphorylation is involved in Ca2+-induced BDNF mRNA transcription and neurite outgrowth promoted by electrical stimulation. PLoS ONE 2016, 11, e0162784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.; Jung, I.H.; Yi, J.H.; Kim, J.H.; Park, J.H.; Lee, S.; Jung, J.W.; Lee, Y.C.; Ryu, J.H.; Kim, D.H. The seed of Zizyphus jujuba var. spinosa attenuates Alzheimer’s disease-associated hippocampal synaptic deficits through BDNF/TrkB signaling. Biol. Pharm. Bull. 2017, 40, 2096–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Ahn, E.H.; Liu, X.; Wang, Z.H.; Luo, S.; Liao, J.; Ye, K. Optimized TrkB agonist ameliorates Alzheimer’s disease pathologies and improves cognitive functions via inhibiting δ-secretase. ACS Chem. Neurosci. 2021, 12, 2448–2461. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yao, H.; Xu, Y.; Hao, R.; Zhang, W.; Liu, H.; Huang, Y.; Guo, W.; Lu, B. Therapeutic potential of a TrkB agonistic antibody for Alzheimer’s disease. Theranostics 2020, 10, 6854–6874. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weng, Z.-K.; Lin, T.-H.; Chang, K.-H.; Chiu, Y.-J.; Lin, C.-H.; Tseng, P.-H.; Sun, Y.-C.; Lin, W.; Lee-Chen, G.-J.; Chen, C.-M. Using ΔK280 TauRD Folding Reporter Cells to Screen TRKB Agonists as Alzheimer’s Disease Treatment Strategy. Biomolecules 2023, 13, 219. https://doi.org/10.3390/biom13020219
Weng Z-K, Lin T-H, Chang K-H, Chiu Y-J, Lin C-H, Tseng P-H, Sun Y-C, Lin W, Lee-Chen G-J, Chen C-M. Using ΔK280 TauRD Folding Reporter Cells to Screen TRKB Agonists as Alzheimer’s Disease Treatment Strategy. Biomolecules. 2023; 13(2):219. https://doi.org/10.3390/biom13020219
Chicago/Turabian StyleWeng, Zheng-Kui, Te-Hsien Lin, Kuo-Hsuan Chang, Ya-Jen Chiu, Chih-Hsin Lin, Pei-Hsuan Tseng, Ying-Chieh Sun, Wenwei Lin, Guey-Jen Lee-Chen, and Chiung-Mei Chen. 2023. "Using ΔK280 TauRD Folding Reporter Cells to Screen TRKB Agonists as Alzheimer’s Disease Treatment Strategy" Biomolecules 13, no. 2: 219. https://doi.org/10.3390/biom13020219
APA StyleWeng, Z. -K., Lin, T. -H., Chang, K. -H., Chiu, Y. -J., Lin, C. -H., Tseng, P. -H., Sun, Y. -C., Lin, W., Lee-Chen, G. -J., & Chen, C. -M. (2023). Using ΔK280 TauRD Folding Reporter Cells to Screen TRKB Agonists as Alzheimer’s Disease Treatment Strategy. Biomolecules, 13(2), 219. https://doi.org/10.3390/biom13020219