


Ultra-Rare Variants Identify Biological Pathways and Candidate Genes in the Pathobiology of Non-Syndromic Cleft Palate Only

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
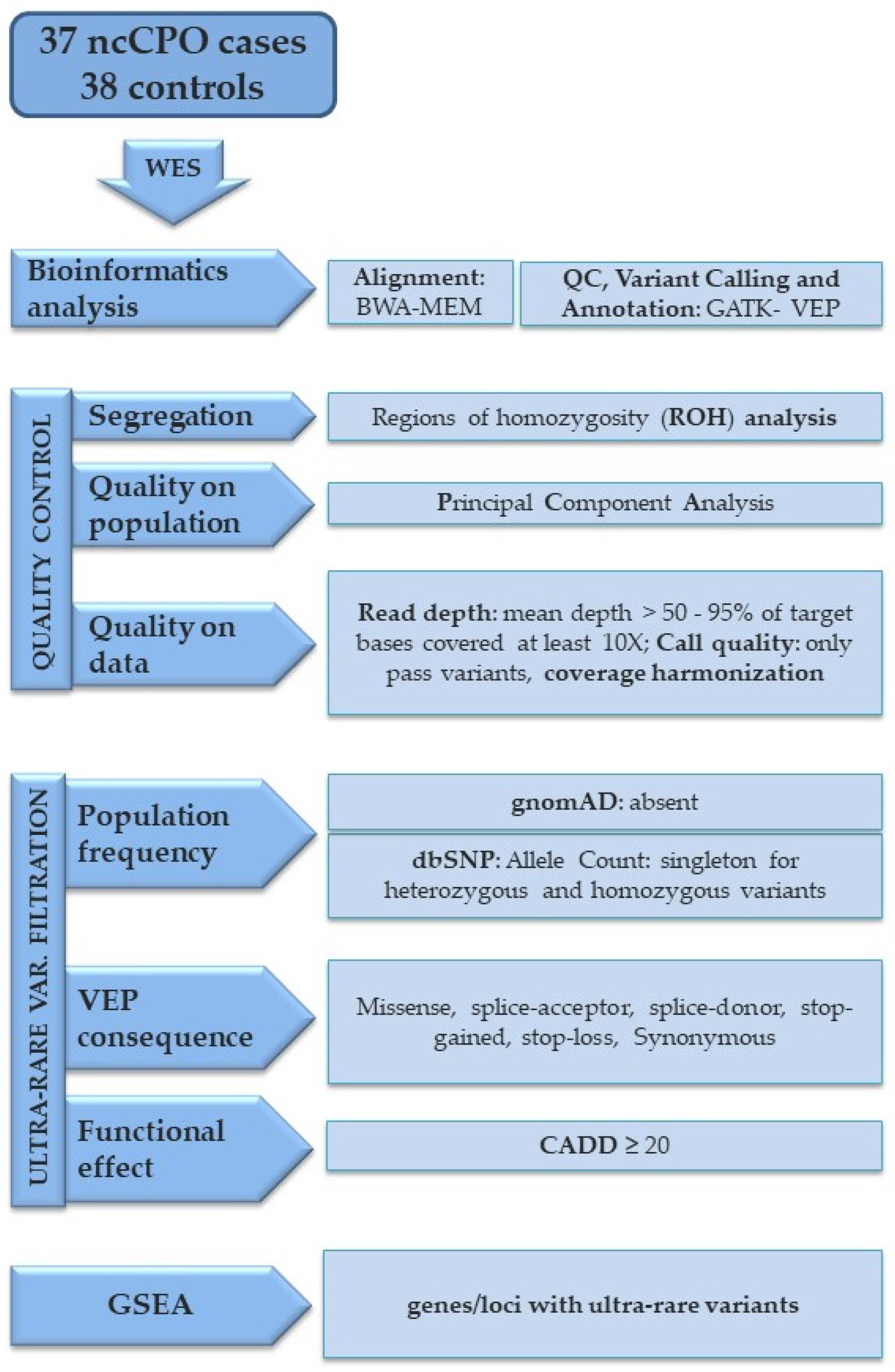
2. Materials and Methods
2.1. Sample Collection
2.2. Sequencing and Variant Detection
- Any gene should have <20% of bases covered <20× in each sample;
- Any gene should not differ by >10% in mean coverage between cases and controls.
2.3. Gene-Set Enrichment Analysis (GSEA)
2.4. Gene-Set Burden and Variants Analysis
3. Results
3.1. Quality Checks
3.2. GSEA Results
3.3. Gene-Set Burden Test Results
3.4. Clinical Variants
4. Discussion
- GSEA highlighted genes with ultra-rare variants cluster into gene ontology sets that are highly relevant to nsCPO pathobiology (cytoskeleton rearrangement, cell-adhesion, ECM-cell interaction);
- GCA identified two top-ranking genes (COL2A1 and GLI3) significantly overlapping in the list of genes relevant to nsCPO pathobiology;
- Pathogenic and likely pathogenic variants in genes, accounting for autosomal dominant orofacial cleft syndromes (CDON, COL2A1, IRF6, SNRPB), have been identified in four cases (10.8%).
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferguson, M.W. Palate development. Development 1988, 103, 41–60. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Bian, Z.; Torensma, R.; Von den Hoff, J.W. Biological mechanisms in palatogenesis and cleft palate. J. Dent. Res. 2009, 88, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Grosen, D.; Chevrier, C.; Skytthe, A.; Bille, C.; Molsted, K.; Sivertsen, A.; Murray, J.C.; Christensen, K. A cohort study of recurrence patterns among more than 54,000 relatives of oral cleft cases in Denmark: Support for the multifactorial threshold model of inheritance. J. Med. Genet. 2010, 47, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, M.; Palmieri, A.; Carinci, F.; Scapoli, L. Non-syndromic Cleft Palate: An Overview on Human Genetic and Environmental Risk Factors. Front Cell Dev. Biol. 2020, 8, 592271. [Google Scholar] [CrossRef]
- Beaty, T.H.; Ruczinski, I.; Murray, J.C.; Marazita, M.L.; Munger, R.G.; Hetmanski, J.B.; Murray, T.; Redett, R.J.; Fallin, M.D.; Liang, K.Y.; et al. Evidence for gene-environment interaction in a genome wide study of nonsyndromic cleft palate. Genet Epidemiol. 2011, 35, 469–478. [Google Scholar] [CrossRef]
- Leslie, E.J.; Liu, H.; Carlson, J.C.; Shaffer, J.R.; Feingold, E.; Wehby, G.; Laurie, C.A.; Jain, D.; Laurie, C.C.; Doheny, K.F.; et al. A Genome-wide Association Study of Nonsyndromic Cleft Palate Identifies an Etiologic Missense Variant in GRHL3. Am. J. Hum. Genet. 2016, 98, 744–754. [Google Scholar] [CrossRef] [Green Version]
- Tam, V.; Patel, N.; Turcotte, M.; Bosse, Y.; Pare, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Gudmundsson, S.; Singer-Berk, M.; Watts, N.A.; Phu, W.; Goodrich, J.K.; Solomonson, M.; Genome Aggregation Database, C.; Rehm, H.L.; MacArthur, D.G.; O’Donnell-Luria, A. Variant interpretation using population databases: Lessons from gnomAD. Hum. Mutat. 2022, 43, 1012–1030. [Google Scholar] [CrossRef]
- Bonnefond, A.; Froguel, P. Rare and common genetic events in type 2 diabetes: What should biologists know? Cell Metab. 2015, 21, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Hoebel, A.K.; Drichel, D.; van de Vorst, M.; Bohmer, A.C.; Sivalingam, S.; Ishorst, N.; Klamt, J.; Golz, L.; Alblas, M.; Maaser, A.; et al. Candidate Genes for Nonsyndromic Cleft Palate Detected by Exome Sequencing. J. Dent. Res. 2017, 96, 1314–1321. [Google Scholar] [CrossRef]
- Liu, H.; Busch, T.; Eliason, S.; Anand, D.; Bullard, S.; Gowans, L.J.J.; Nidey, N.; Petrin, A.; Augustine-Akpan, E.A.; Saadi, I.; et al. Exome sequencing provides additional evidence for the involvement of ARHGAP29 in Mendelian orofacial clefting and extends the phenotypic spectrum to isolated cleft palate. Birth Defects Res. 2017, 109, 27–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magini, P.; Pippucci, T.; Tsai, I.C.; Coppola, S.; Stellacci, E.; Bartoletti-Stella, A.; Turchetti, D.; Graziano, C.; Cenacchi, G.; Neri, I.; et al. A mutation in PAK3 with a dual molecular effect deregulates the RAS/MAPK pathway and drives an X-linked syndromic phenotype. Hum. Mol. Genet. 2014, 23, 3607–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pippucci, T.; Maresca, A.; Magini, P.; Cenacchi, G.; Donadio, V.; Palombo, F.; Papa, V.; Incensi, A.; Gasparre, G.; Valentino, M.L.; et al. Homozygous NOTCH3 null mutation and impaired NOTCH3 signaling in recessive early-onset arteriopathy and cavitating leukoencephalopathy. EMBO Mol. Med. 2015, 7, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Pippucci, T.; Licchetta, L.; Baldassari, S.; Marconi, C.; De Luise, M.; Myers, C.; Nardi, E.; Provini, F.; Cameli, C.; Minardi, R.; et al. Contribution of ultrarare variants in mTOR pathway genes to sporadic focal epilepsies. Ann. Clin. Transl. Neurol. 2019, 6, 475–485. [Google Scholar] [CrossRef] [Green Version]
- Kendig, K.I.; Baheti, S.; Bockol, M.A.; Drucker, T.M.; Hart, S.N.; Heldenbrand, J.R.; Hernaez, M.; Hudson, M.E.; Kalmbach, M.T.; Klee, E.W.; et al. Sentieon DNASeq Variant Calling Workflow Demonstrates Strong Computational Performance and Accuracy. Front Genet. 2019, 10, 736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, C.; Mou, C.; Dong, Y.; Tu, Y. dbNSFP v4: A comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 2020, 12, 103. [Google Scholar] [CrossRef]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef]
- Magi, A.; Giangregorio, T.; Semeraro, R.; Carangelo, G.; Palombo, F.; Romeo, G.; Seri, M.; Pippucci, T. AUDACITY: A comprehensive approach for the detection and classification of Runs of Homozygosity in medical and population genomics. Comput Struct Biotechnol. J. 2020, 18, 1956–1967. [Google Scholar] [CrossRef]
- Chen, J.; Xu, H.; Aronow, B.J.; Jegga, A.G. Improved human disease candidate gene prioritization using mouse phenotype. BMC Bioinform. 2007, 8, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, M.R.; Diaz Perez, K.K.; Sun, M.; Ho, S.; Chopra, P.; Mukhopadhyay, N.; Hetmanski, J.B.; Taub, M.A.; Moreno-Uribe, L.M.; Valencia-Ramirez, L.C.; et al. Genome-wide Enrichment of De Novo Coding Mutations in Orofacial Cleft Trios. Am. J. Hum. Genet. 2020, 107, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Shi, G. On rare variants in principal component analysis of population stratification. BMC Genet. 2020, 21, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghavan, N.S.; Brickman, A.M.; Andrews, H.; Manly, J.J.; Schupf, N.; Lantigua, R.; Wolock, C.J.; Kamalakaran, S.; Petrovski, S.; Tosto, G.; et al. Whole-exome sequencing in 20,197 persons for rare variants in Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2018, 5, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Lessard, J.L.; Wee, E.L.; Zimmerman, E.F. Presence of contractile proteins in mouse fetal palate prior to shelf elevation. Teratology 1974, 9, 113–125. [Google Scholar] [CrossRef]
- Kim, S.; Lewis, A.E.; Singh, V.; Ma, X.; Adelstein, R.; Bush, J.O. Convergence and extrusion are required for normal fusion of the mammalian secondary palate. PLoS Biol. 2015, 13, e1002122. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, M.; Di Stazio, M.; Scapoli, L.; Marchesini, J.; Di Bari, F.; Pezzetti, F.; Carinci, F.; Palmieri, A.; Carinci, P.; Savoia, A. Cleft lip with or without cleft palate: Implication of the heavy chain of non-muscle myosin IIA. J. Med. Genet. 2007, 44, 387–392. [Google Scholar] [CrossRef] [Green Version]
- Birnbaum, S.; Reutter, H.; Mende, M.; de Assis, N.A.; Diaz-Lacava, A.; Herms, S.; Scheer, M.; Lauster, C.; Braumann, B.; Schmidt, G.; et al. Further evidence for the involvement of MYH9 in the etiology of non-syndromic cleft lip with or without cleft palate. Eur. J. Oral. Sci. 2009, 117, 200–203. [Google Scholar] [CrossRef]
- Chiquet, B.T.; Hashmi, S.S.; Henry, R.; Burt, A.; Mulliken, J.B.; Stal, S.; Bray, M.; Blanton, S.H.; Hecht, J.T. Genomic screening identifies novel linkages and provides further evidence for a role of MYH9 in nonsyndromic cleft lip and palate. Eur. J. Hum. Genet. 2009, 17, 195–204. [Google Scholar] [CrossRef]
- Funato, N.; Yanagisawa, H. Deletion of the T-box transcription factor gene, Tbx1, in mice induces differential expression of genes associated with cleft palate in humans. Arch. Oral. Biol. 2018, 95, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Li, M.X.; Li, Z.; Zhang, R.; Yu, Y.; Wang, L.S.; Wang, Q.; Ding, Z.; Zhang, J.P.; Zhang, M.R.; Xu, L.C. Effects of small interfering RNA-mediated silencing of susceptibility genes of non-syndromic cleft lip with or without cleft palate on cell proliferation and migration. Int. J. Pediatr. Otorhinolaryngol. 2020, 138, 110382. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Lu, J.; Lian, G.; Ferland, R.J.; Dettenhofer, M.; Sheen, V.L. Formin 1 and filamin B physically interact to coordinate chondrocyte proliferation and differentiation in the growth plate. Hum. Mol. Genet. 2014, 23, 4663–4673. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S. FLNB Disorders. In GeneReviews((R)); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Li, B.C.; Hogue, J.; Eilers, M.; Mehrotra, P.; Hyland, J.; Holm, T.; Prosen, T.; Slavotinek, A.M. Clinical report: Two patients with atelosteogenesis type I caused by missense mutations affecting the same FLNB residue. Am. J. Med. Genet A 2013, 161A, 619–625. [Google Scholar] [CrossRef]
- Jugessur, A.; Shi, M.; Gjessing, H.K.; Lie, R.T.; Wilcox, A.J.; Weinberg, C.R.; Christensen, K.; Boyles, A.L.; Daack-Hirsch, S.; Nguyen, T.T.; et al. Maternal genes and facial clefts in offspring: A comprehensive search for genetic associations in two population-based cleft studies from Scandinavia. PLoS ONE 2010, 5, e11493. [Google Scholar] [CrossRef] [Green Version]
- Haaland, O.A.; Romanowska, J.; Gjerdevik, M.; Lie, R.T.; Gjessing, H.K.; Jugessur, A. A genome-wide scan of cleft lip triads identifies parent-of-origin interaction effects between ANK3 and maternal smoking, and between ARHGEF10 and alcohol consumption. F1000Res 2019, 8, 960. [Google Scholar] [CrossRef]
- Oh, W.J.; Westmoreland, J.J.; Summers, R.; Condie, B.G. Cleft palate is caused by CNS dysfunction in Gad1 and Viaat knockout mice. PLoS ONE 2010, 5, e9758. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Kakizaki, T.; Fujihara, K.; Miyata, S.; Zhang, Y.; Suto, T.; Kato, D.; Saito, S.; Shibasaki, K.; Ishizaki, Y.; et al. Impact of GAD65 and/or GAD67 deficiency on perinatal development in rats. FASEB J. 2022, 36, e22123. [Google Scholar] [CrossRef]
- Suzuki, A.; Abdallah, N.; Gajera, M.; Jun, G.; Jia, P.; Zhao, Z.; Iwata, J. Genes and microRNAs associated with mouse cleft palate: A systematic review and bioinformatics analysis. Mech. Dev. 2018, 150, 21–27. [Google Scholar] [CrossRef]
- Chiquet, B.T.; Yuan, Q.; Swindell, E.C.; Maili, L.; Plant, R.; Dyke, J.; Boyer, R.; Teichgraeber, J.F.; Greives, M.R.; Mulliken, J.B.; et al. Knockdown of Crispld2 in zebrafish identifies a novel network for nonsyndromic cleft lip with or without cleft palate candidate genes. Eur. J. Hum. Genet. 2018, 26, 1441–1450. [Google Scholar] [CrossRef]
- Figueiredo, J.C.; Ly, S.; Raimondi, H.; Magee, K.; Baurley, J.W.; Sanchez-Lara, P.A.; Ihenacho, U.; Yao, C.; Edlund, C.K.; van den Berg, D.; et al. Genetic risk factors for orofacial clefts in Central Africans and Southeast Asians. Am. J. Med. Genet. A 2014, 164A, 2572–2580. [Google Scholar] [CrossRef]
- Ray, D.; Venkataraghavan, S.; Zhang, W.; Leslie, E.J.; Hetmanski, J.B.; Weinberg, S.M.; Murray, J.C.; Marazita, M.L.; Ruczinski, I.; Taub, M.A.; et al. Pleiotropy method reveals genetic overlap between orofacial clefts at multiple novel loci from GWAS of multi-ethnic trios. PLoS Genet. 2021, 17, e1009584. [Google Scholar] [CrossRef] [PubMed]
- Slavec, L.; Karas Kuzelicki, N.; Locatelli, I.; Gersak, K. Genetic markers for non-syndromic orofacial clefts in populations of European ancestry: A meta-analysis. Sci. Rep. 2022, 12, 1214. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Huang, X.; Yuan, L. Molecular genetics of the COL2A1-related disorders. Mutat Res. Rev. Mutat Res. 2016, 768, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nikopensius, T.; Jagomagi, T.; Krjutskov, K.; Tammekivi, V.; Saag, M.; Prane, I.; Piekuse, L.; Akota, I.; Barkane, B.; Krumina, A.; et al. Genetic variants in COL2A1, COL11A2, and IRF6 contribute risk to nonsyndromic cleft palate. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Goudy, S.L.; Ketova, T.; Litingtung, Y.; Chiang, C. Gli3-deficient mice exhibit cleft palate associated with abnormal tongue development. Dev. Dyn. 2008, 237, 3079–3087. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, Y.; Huang, Y.; Pan, Y.; Shi, B.; Ma, J.; Ma, L.; Lan, F.; Zhou, Y.; Shi, J.; et al. The association study of nonsyndromic cleft lip with or without cleft palate identified risk variants of the GLI3 gene in a Chinese population. J. Genet. 2017, 96, 687–693. [Google Scholar] [CrossRef]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.; Hua, Y.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R.; et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347, 1254806. [Google Scholar] [CrossRef] [Green Version]
- Hoornaert, K.P.; Vereecke, I.; Dewinter, C.; Rosenberg, T.; Beemer, F.A.; Leroy, J.G.; Bendix, L.; Bjorck, E.; Bonduelle, M.; Boute, O.; et al. Stickler syndrome caused by COL2A1 mutations: Genotype-phenotype correlation in a series of 100 patients. Eur. J. Hum. Genet. 2010, 18, 872–880. [Google Scholar] [CrossRef] [Green Version]
- Lansdon, L.A.; Dickinson, A.; Arlis, S.; Liu, H.; Hlas, A.; Hahn, A.; Bonde, G.; Long, A.; Standley, J.; Tyryshkina, A.; et al. Genome-wide analysis of copy number variation in humans with cleft lip and/or cleft palate identifies COBLL1, RIC1, and ARHGEF38 as clefting genes. Am. J. Hum. Genet. 2022, 110, 71–91. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Category | Term | Nominal p * | Adjusted p * | Genes |
|---|---|---|---|---|
| GO: MF | cytoskeletal protein binding (GO:0008092) | 1.43 × 10−7 | 1.57 × 10−4 | RAB11B, XIRP1, ABL2, MAP1A, OBSCN, PARVB, ADGRV1, TUBGCP5, FMNL2, TTN, AP1G1, FSCN2, TAOK1, KIF17, UTRN, FKBP4, FLII, FLNB, VPS18, ANK3, CLIP2, APC, PPARG, CLSTN1, CLIP3, SETD2, PARVA, VPS41, MAST1, MYH15, MYBPC3, MYH2, MYH3, MYH4, TTLL9, EML2, PANX1, ACE, FRMD4B, IQGAP1, MAPK8IP3, TTBK2, CYFIP1, BAIAP2L1, NAV3, KCNQ2, MYO18B, GNB3, TBCD, KPNB1, NRCAM, SYNM, SYNE1, NUMA1, CLIP4, ARL4C, FARP1, MACF1, KIRREL1, DAAM2, RFLNA, MYO15A, TTLL13 |
| GO: MF | microtubule plus-end binding (GO:0051010) | 1.92 × 10−5 | 1.06 × 10−2 | CLIP2, APC, CLIP3, TTBK2, NUMA1, CLIP4 |
| GO: MF | cell adhesion molecule binding (GO:0050839) | 3.97 × 10−5 | 1.24 × 10−2 | RAB11B, ABCF3, CDHR3, TRPC4, OBSCN, FMNL2, TENM2, TAGLN2, UTRN, FLNB, ANK3, ITGA10, APC, NTNG1, AHSA1, TENM3, TNN, PARVA, TES, ADAM15, ITGB7, IQGAP1, PTPRB, PTPRH, BAIAP2L1, PTPRZ1, NRCAM, DOCK9, LAMA3, LAMB2, GRIN2A, GRIN2B, MACF1, KIRREL1, CDH7 |
| GO: MF | protein-containing complex binding (GO:0044877) | 4.52 × 10−5 | 1.24 × 10−2 | XIRP1, NOG, ABL2, MAP1A, PDK2, TSHR, ARID1A, FMNL2, TTN, HIRA, ARID1B, AP1G1, FCGR2A, OTOF, AEBP1, MIF, FSCN2, PLCB2, CNGB1, UTRN, NCOA2, FLII, ANK3, ABI3BP, FOS, ANXA11, ITGA10, APC, CPD, CDC42BPB, PEX26, MTOR, TNN, PARVA, MYH15, CTSK, GAD1, MYH2, MYH3, MYH4, SPHK2, ADAM15, VPS33A, PANX1, ITGA2B, ITGB7, IQGAP1, ITPR1, MED24, CYFIP1, SACS, PTPRZ1, GLI3, GNB3, KPNB1, DOCK2, HNF1B, CARMIL2, SYNM, SYNE1, NUMA1, LAMA3, GRB2, LAMB2, LAMB3, GRIN2A, GRIN2B, LIN28A, RELL2, H1-0, H1-5, LRP2, MACF1, MYO15A, CPEB1 |
| Pathway | ECM-receptor interaction (M7098) | 7.03 × 10−6 | 1.69 × 10−2 | COL2A1, COL4A2, COL5A2, ITGA10, TNN, LAMA1, ITGA2B, ITGB7, LAMA3, LAMA4, LAMB2, LAMB3 |
| Pathway | Integrin signaling pathway (P00034) | 1.07 × 10−5 | 1.28 × 10−2 | PARVB, MAP3K4, COL2A1, COL4A2, COL5A2, FLNB, COL7A1, ITGA10, PARVA, MAP2K3, LAMA1, ITGA2B, ITGB7, LAMA3, LAMA4, LAMB2, LAMB3 |
| Pathway | Ensemble of genes encoding core extracellular matrix including ECM glycoproteins, collagens and proteoglycans (M5884) | 2.77 × 10−5 | 1.86 × 10−2 | CRIM1, AEBP1, COL2A1, COL4A2, COL5A2, COL7A1, ABI3BP, COL28A1, EFEMP2, NTNG1, ZP1, SNED1, TNN, LAMA1, TINAGL1, IGSF10, PXDN, VWA5B1, LAMA3, LAMA4, LAMB2, LAMB3 |
| Pathway | Axon guidance (1270303) | 5.21 × 10−5 | 2.42 × 10−2 | EPHB3, SCN11A, ERBB3, ABL2, RPS6KA2, TRPC4, SCN10A, FGF6, ALCAM, PLXNB1, RASAL2, COL4A2, ANK3, RASA3, CNTNAP1, ITGA10, RAPGEF2, PSMA5, LAMA1, FRS2, ITGA2B, IQGAP1, KSR1, KCNQ2, CACNB1, TRPC7, NRCAM, CAMK2G, KSR2, GRB2, SRGAP2, GRIN2A, GRIN2B, ROBO1 |
| Cases | Controls | ||||
|---|---|---|---|---|---|
| GENES | Variants | No_Variants | Variants | No_Variants | p-Value |
| ZFYVE26 | 7 | 28 | 0 | 38 | 0.0041 |
| THSD7B | 6 | 29 | 0 | 38 | 0.0095 |
| SPATC1 | 6 | 29 | 0 | 38 | 0.0095 |
| IGHG1 | 6 | 29 | 0 | 38 | 0.0095 |
| EXO1 | 0 | 35 | 7 | 31 | 0.0119 |
| JAKMIP3 | 9 | 26 | 2 | 36 | 0.0210 |
| AHNAK | 9 | 26 | 2 | 36 | 0.0210 |
| PLEKHN1 | 5 | 30 | 0 | 38 | 0.0216 |
| NBEAL2 | 5 | 30 | 0 | 38 | 0.0216 |
| USP42 | 5 | 30 | 0 | 38 | 0.0216 |
| OR4K1 | 5 | 30 | 0 | 38 | 0.0216 |
| STARD9 | 5 | 30 | 0 | 38 | 0.0216 |
| UNC13A | 5 | 30 | 0 | 38 | 0.0216 |
| OLFML2B | 4 | 31 | 0 | 38 | 0.0481 |
| MROH9 | 4 | 31 | 0 | 38 | 0.0481 |
| SLC4A5 | 4 | 31 | 0 | 38 | 0.0481 |
| VWA3B | 4 | 31 | 0 | 38 | 0.0481 |
| CAND2 | 4 | 31 | 0 | 38 | 0.0481 |
| USP4 | 4 | 31 | 0 | 38 | 0.0481 |
| LRP2BP | 4 | 31 | 0 | 38 | 0.0481 |
| ERAP2 | 4 | 31 | 0 | 38 | 0.0481 |
| DOPEY1 | 4 | 31 | 0 | 38 | 0.0481 |
| GLI3 | 4 | 31 | 0 | 38 | 0.0481 |
| TRBV6-7 | 4 | 31 | 0 | 38 | 0.0481 |
| LOXL2 | 4 | 31 | 0 | 38 | 0.0481 |
| APOBEC1 | 4 | 31 | 0 | 38 | 0.0481 |
| COL2A1 | 4 | 31 | 0 | 38 | 0.0481 |
| LIMA1 | 4 | 31 | 0 | 38 | 0.0481 |
| HAL | 4 | 31 | 0 | 38 | 0.0481 |
| RAI1 | 4 | 31 | 0 | 38 | 0.0481 |
| MPP3 | 4 | 31 | 0 | 38 | 0.0481 |
| RSAD1 | 4 | 31 | 0 | 38 | 0.0481 |
| CILP2 | 4 | 31 | 0 | 38 | 0.0481 |
| ZNF600 | 4 | 31 | 0 | 38 | 0.0481 |
| LILRA4 | 4 | 31 | 0 | 38 | 0.0481 |
| TUBB1 | 4 | 31 | 0 | 38 | 0.0481 |
| UTRN | 6 | 29 | 1 | 37 | 0.0497 |
| NLRC3 | 6 | 29 | 1 | 37 | 0.0497 |
| URB1 | 6 | 29 | 1 | 37 | 0.0497 |
| Gene | Genomic Position * | Variant Type | Variant Class | CADD Score | Protein Consequence | Population Frequency | Population |
|---|---|---|---|---|---|---|---|
| COL2A1 | 12-48377883-G-T | SNV | missense | 28 | p.Pro643His | 7.16 × 10−6 | Iranian |
| COL2A1 | 12-48369250-C-T | SNV | missense | 9 | p.Gly1246Ser | 0.000598 | Italian |
| COL2A1 | 12-48373812-G-A | SNV | Stop gain | 46 | p.Arg887Ter | 0 | Italian |
| COL2A1 | 12-48376305-C-T | SNV | missense | 19 | p.Ala761Thr | 8.77 × 10−6 | Italian |
| GLI3 | 7-42007446-C-T | SNV | missense | 27.5 | p.Gly727Arg | 0.007131 | Italian |
| GLI3 | 7-42063171-C-G | SNV | missense | 23.5 | p.Gly465Arg | 0.003811 | Iranian |
| GLI3 | 7-42187851-C-T | SNV | missense | 28.4 | p.Arg114Lys | 0.002434 | Italian |
| GLI3 | 7-42187885-G-A | SNV | missense | 24.7 | p.Pro103Ser | 4.03 × 10−6 | Italian |
| Genes | Genomic Position * | Protein Consequence | HGVS Nomenclature | ACMG Classif. | OMIM Disease and Inheritance | ClinVar Accession | Genotype |
|---|---|---|---|---|---|---|---|
| CDON | 11-125875899-G-A | p.Gln536Ter | NM_001243597.2: c.1606C>T | LP | Holoprosencephaly 11; AD | Het | |
| COL2A1 | 12-48373812-G-A | p.Arg887Ter | NM_001844.5: c.2659C>T | P | Stickler syndrome, type I; AD | VCV000817513.4 | Het |
| IRF6 | 1-209974677-A-C | p.Trp28Gly | NM_006147.4: c.82T>G | LP | {Orofacial cleft 6}; AD | VCV000464464.1 | Het |
| SNRPB | 20-2442575-T-A | p.Ter232Cysext *?§ | NM_198216.2: c.686-136A>T | LP | Cerebrocostomandibular syndrome; AD | Hom |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iovino, E.; Scapoli, L.; Palmieri, A.; Sgarzani, R.; Nouri, N.; Pellati, A.; Carinci, F.; Seri, M.; Pippucci, T.; Martinelli, M. Ultra-Rare Variants Identify Biological Pathways and Candidate Genes in the Pathobiology of Non-Syndromic Cleft Palate Only. Biomolecules 2023, 13, 236. https://doi.org/10.3390/biom13020236
Iovino E, Scapoli L, Palmieri A, Sgarzani R, Nouri N, Pellati A, Carinci F, Seri M, Pippucci T, Martinelli M. Ultra-Rare Variants Identify Biological Pathways and Candidate Genes in the Pathobiology of Non-Syndromic Cleft Palate Only. Biomolecules. 2023; 13(2):236. https://doi.org/10.3390/biom13020236
Chicago/Turabian StyleIovino, Emanuela, Luca Scapoli, Annalisa Palmieri, Rossella Sgarzani, Nayereh Nouri, Agnese Pellati, Francesco Carinci, Marco Seri, Tommaso Pippucci, and Marcella Martinelli. 2023. "Ultra-Rare Variants Identify Biological Pathways and Candidate Genes in the Pathobiology of Non-Syndromic Cleft Palate Only" Biomolecules 13, no. 2: 236. https://doi.org/10.3390/biom13020236
APA StyleIovino, E., Scapoli, L., Palmieri, A., Sgarzani, R., Nouri, N., Pellati, A., Carinci, F., Seri, M., Pippucci, T., & Martinelli, M. (2023). Ultra-Rare Variants Identify Biological Pathways and Candidate Genes in the Pathobiology of Non-Syndromic Cleft Palate Only. Biomolecules, 13(2), 236. https://doi.org/10.3390/biom13020236