

The Application of Single-Cell RNA Sequencing in the Inflammatory Tumor Microenvironment


Abstract
:1. Introduction
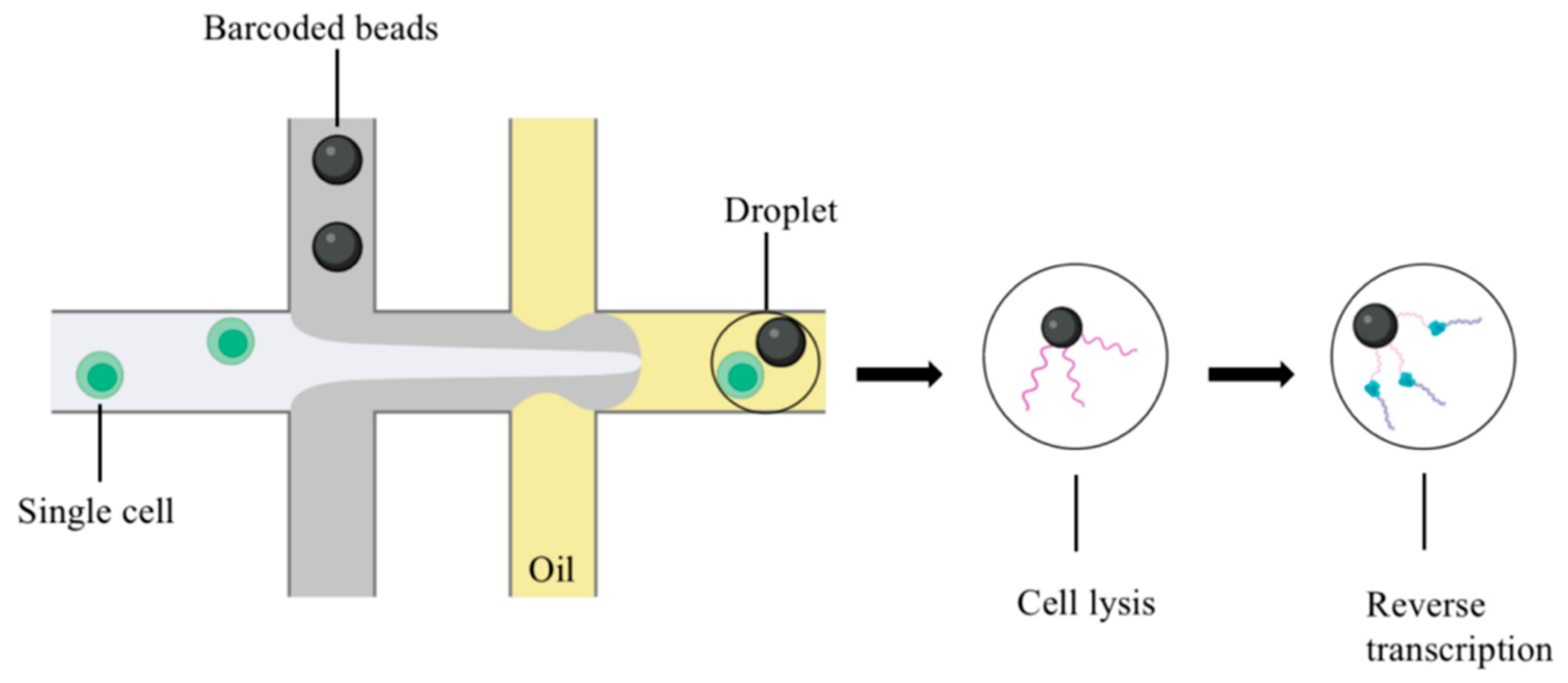
2. Overview of scRNA-seq
3. scRNA-seq in Immunology
3.1. Identification of Novel Immunocyte Subsets and Marker Genes
3.2. Revealing the Heterogeneity
3.3. Reconstructing the Trajectory of Immune Cell Development and Differentiation
3.4. Uncovering Immune Mechanisms
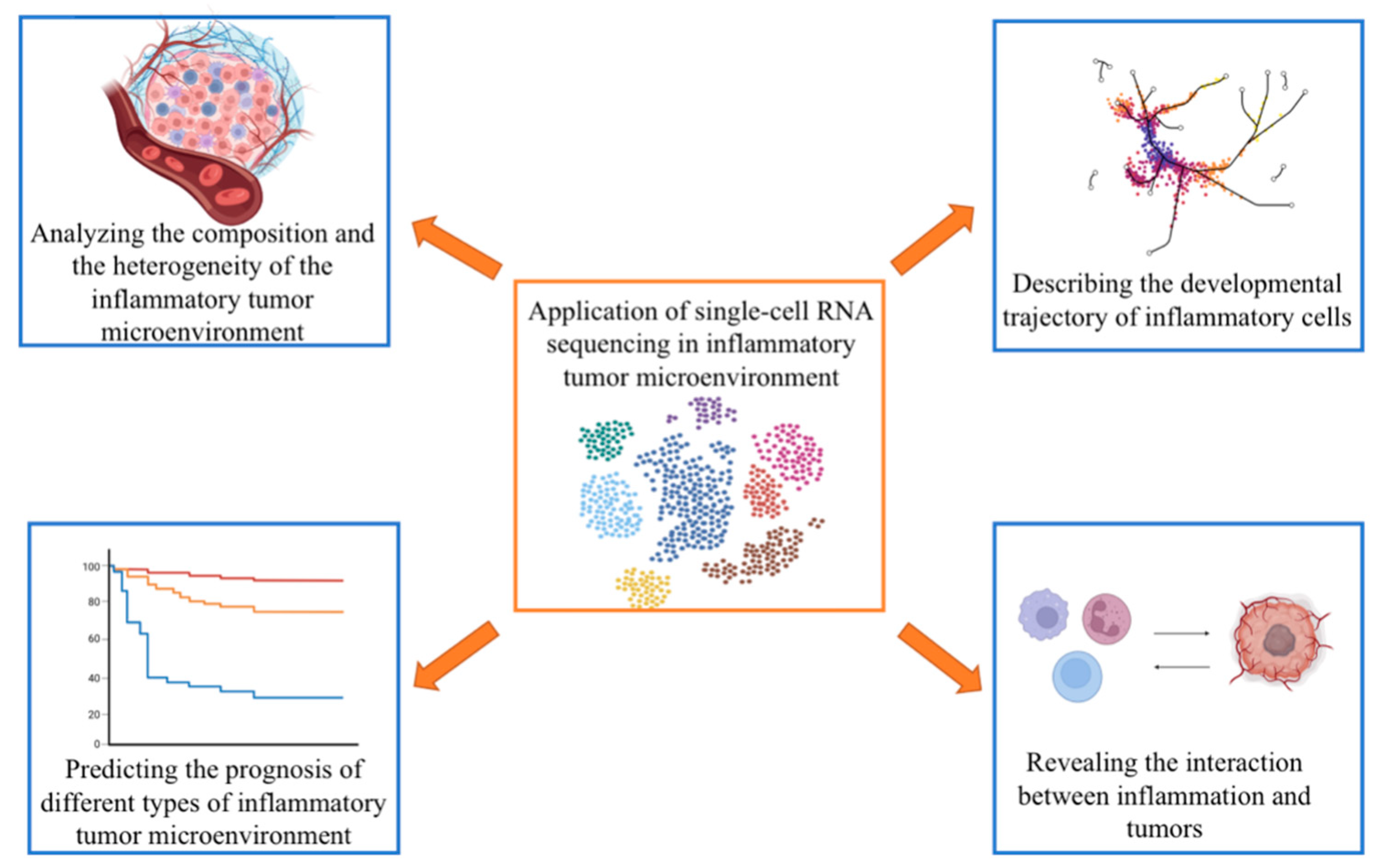
4. scRNA-seq in the Inflammatory TME
4.1. Analyzing the Composition and the Heterogeneity of the Inflammatory TME
4.2. Describing the Developmental Trajectory of Inflammatory Cells
4.3. Predicting the Prognosis of Different Types of Inflammatory TME
4.4. Revealing the Interaction between Inflammation and Tumors
5. Spatial Transcriptomics Combined with scRNA-seq in Tumor, Inflammation, and Immunity
5.1. Spatial Transcriptomics
5.2. Combined Applications in Tumor, Inflammation, and Immunity
6. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hagerling, C.; Casbon, A.J.; Werb, Z. Balancing the innate immune system in tumor development. Trends Cell Biol. 2015, 25, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Deng, Y.; Li, Z.; Chen, Y.; Zhu, X.; Tan, X.; Cao, G. Cancer Evo-Dev: A Theory of Inflammation-Induced Oncogenesis. Front. Immunol. 2021, 12, 768098. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Li, R.; Zhang, J.; An, L.; Zhou, G.; Lei, M.; Deng, J.; Yang, R.; Song, Z.; Zhong, W.; et al. Distinct immune and inflammatory response patterns contribute to the identification of poor prognosis and advanced clinical characters in bladder cancer patients. Front. Immunol. 2022, 13, 1008865. [Google Scholar] [CrossRef]
- Chen, K.; Wang, Q.; Li, M.; Guo, H.; Liu, W.; Wang, F.; Tian, X.; Yang, Y. Single-cell RNA-seq reveals dynamic change in tumor microenvironment during pancreatic ductal adenocarcinoma malignant progression. EBioMedicine 2021, 66, 103315. [Google Scholar] [CrossRef]
- Chen, H.; Ye, F.; Guo, G. Revolutionizing immunology with single-cell RNA sequencing. Cell Mol. Immunol. 2019, 16, 242–249. [Google Scholar] [CrossRef]
- Ding, S.; Chen, X.; Shen, K. Single-cell RNA sequencing in breast cancer: Understanding tumor heterogeneity and paving roads to individualized therapy. Cancer Commun. 2020, 40, 329–344. [Google Scholar] [CrossRef]
- Xie, J.; Sun, J.; Feng, J.; Yang, F.; Wang, J.; Wen, T.; Nie, Q. Kernel Differential Subgraph Analysis to Reveal the Key Period Affecting Glioblastoma. Biomolecules 2020, 10, 318. [Google Scholar] [CrossRef]
- Potter, S.S. Single-cell RNA sequencing for the study of development, physiology and disease. Nat. Rev. Nephrol. 2018, 14, 479–492. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, D.; Peng, M.; Tang, L.; Ouyang, J.; Xiong, F.; Guo, C.; Tang, Y.; Zhou, Y.; Liao, Q.; et al. Single-cell RNA sequencing in cancer research. J. Exp. Clin. Cancer Res. 2021, 40, 81. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Nordman, E.; Li, B.; Xu, N.; Bashkirov, V.I.; Lao, K.; Surani, M.A. RNA-Seq analysis to capture the transcriptome landscape of a single cell. Nat. Protoc. 2010, 5, 516–535. [Google Scholar] [CrossRef] [PubMed]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. CEL-Seq: Single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012, 2, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Ramsköld, D.; Luo, S.; Wang, Y.C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C.; et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012, 30, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Kjällquist, U.; Moliner, A.; Zajac, P.; Fan, J.B.; Lönnerberg, P.; Linnarsson, S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 2011, 21, 1160–1167. [Google Scholar] [CrossRef]
- Salomon, R.; Kaczorowski, D.; Valdes-Mora, F.; Nordon, R.E.; Neild, A.; Farbehi, N.; Bartonicek, N.; Gallego-Ortega, D. Droplet-based single cell RNAseq tools: A practical guide. Lab. Chip 2019, 19, 1706–1727. [Google Scholar] [CrossRef] [PubMed]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Islam, S.; Zeisel, A.; Joost, S.; La Manno, G.; Zajac, P.; Kasper, M.; Lönnerberg, P.; Linnarsson, S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods 2014, 11, 163–166. [Google Scholar] [CrossRef]
- Ziegenhain, C.; Vieth, B.; Parekh, S.; Reinius, B.; Guillaumet-Adkins, A.; Smets, M.; Leonhardt, H.; Heyn, H.; Hellmann, I.; Enard, W. Comparative Analysis of Single-Cell RNA Sequencing Methods. Mol. Cell 2017, 65, 631–643.e4. [Google Scholar] [CrossRef]
- Kubo, M.; Nishiyama, T.; Tamada, Y.; Sano, R.; Ishikawa, M.; Murata, T.; Imai, A.; Lang, D.; Demura, T.; Reski, R.; et al. Single-cell transcriptome analysis of Physcomitrella leaf cells during reprogramming using microcapillary manipulation. Nucleic Acids Res. 2019, 47, 4539–4553. [Google Scholar] [CrossRef]
- Foley, J.W.; Zhu, C.; Jolivet, P.; Zhu, S.X.; Lu, P.; Meaney, M.J.; West, R.B. Gene expression profiling of single cells from archival tissue with laser-capture microdissection and Smart-3SEQ. Genome Res. 2019, 29, 1816–1825. [Google Scholar] [CrossRef] [Green Version]
- Hashimshony, T.; Senderovich, N.; Avital, G.; Klochendler, A.; de Leeuw, Y.; Anavy, L.; Gennert, D.; Li, S.; Livak, K.J.; Rozenblatt-Rosen, O.; et al. CEL-Seq2: Sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016, 17, 77. [Google Scholar] [CrossRef] [PubMed]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A.; et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 2014, 343, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Picelli S, Björklund Å K, Faridani OR, Sagasser S, Winberg G, Sandberg R: Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 2013, 10, 1096–1098. [CrossRef] [PubMed]
- Streets, A.M.; Zhang, X.; Cao, C.; Pang, Y.; Wu, X.; Xiong, L.; Yang, L.; Fu, Y.; Zhao, L.; Tang, F.; et al. Microfluidic single-cell whole-transcriptome sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, 7048–7053. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Venteicher, A.S.; Hebert, C.; Escalante, L.E.; Patel, A.P.; Yizhak, K.; Fisher, J.M.; Rodman, C.; Mount, C.; Filbin, M.G.; et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 2016, 539, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef]
- Kotliar, D.; Lin, A.E.; Logue, J.; Hughes, T.K.; Khoury, N.M.; Raju, S.S.; Wadsworth, M.H., 2nd; Chen, H.; Kurtz, J.R.; Dighero-Kemp, B.; et al. Single-Cell Profiling of Ebola Virus Disease In Vivo Reveals Viral and Host Dynamics. Cell 2020, 183, 1383–1401.e19. [Google Scholar] [CrossRef]
- Shen, H.; Yang, M.; Li, S.; Zhang, J.; Peng, B.; Wang, C.; Chang, Z.; Ong, J.; Du, P. Mouse totipotent stem cells captured and maintained through spliceosomal repression. Cell 2021, 184, 2843–2859.e20. [Google Scholar] [CrossRef]
- Ku, C.; Sebé-Pedrós, A. Using single-cell transcriptomics to understand functional states and interactions in microbial eukaryotes. Philos. Trans. R Soc. Lond. B. Biol. Sci. 2019, 374, 20190098. [Google Scholar] [CrossRef]
- de Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef]
- Kislev, N.; Izgilov, R.; Adler, R.; Benayahu, D. Exploring the Cell Stemness and the Complexity of the Adipose Tissue Niche. Biomolecules 2021, 11, 1906. [Google Scholar] [CrossRef] [PubMed]
- Stubbington, M.J.T.; Rozenblatt-Rosen, O.; Regev, A.; Teichmann, S.A. Single-cell transcriptomics to explore the immune system in health and disease. Science 2017, 358, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Ao, T.; Kikuta, J.; Ishii, M. The Effects of Vitamin D on Immune System and Inflammatory Diseases. Biomolecules 2021, 11, 1624. [Google Scholar] [CrossRef]
- Zhang, S.; Xie, L.; Cui, Y.; Carone, B.R.; Chen, Y. Detecting Fear-Memory-Related Genes from Neuronal scRNA-seq Data by Diverse Distributions and Bhattacharyya Distance. Biomolecules 2022, 12, 1130. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, S.; Liu, C.; Yang, J.; Lin, Q.; Zheng, S.; Chen, C.; Zhou, Q.; Chen, R. Single-cell RNA sequencing reveals spatiotemporal heterogeneity and malignant progression in pancreatic neuroendocrine tumor. Int. J. Biol. Sci. 2021, 17, 3760–3775. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, A.; Muñoz-Manchado, A.B.; Codeluppi, S.; Lönnerberg, P.; La Manno, G.; Juréus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef]
- Pollen, A.A.; Nowakowski, T.J.; Shuga, J.; Wang, X.; Leyrat, A.A.; Lui, J.H.; Li, N.; Szpankowski, L.; Fowler, B.; Chen, P.; et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat. Biotechnol. 2014, 32, 1053–1058. [Google Scholar] [CrossRef]
- Wills, Q.F.; Livak, K.J.; Tipping, A.J.; Enver, T.; Goldson, A.J.; Sexton, D.W.; Holmes, C. Single-cell gene expression analysis reveals genetic associations masked in whole-tissue experiments. Nat. Biotechnol. 2013, 31, 748–752. [Google Scholar] [CrossRef]
- Shalek, A.K.; Satija, R.; Adiconis, X.; Gertner, R.S.; Gaublomme, J.T.; Raychowdhury, R.; Schwartz, S.; Yosef, N.; Malboeuf, C.; Lu, D.; et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 2013, 498, 236–240. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, S.; Min, M.; Ni, Y.; Lu, Z.; Sun, X.; Wu, J.; Liu, B.; Ying, X.; Liu, Y. Dissecting transcriptional heterogeneity in primary gastric adenocarcinoma by single cell RNA sequencing. Gut 2021, 70, 464–475. [Google Scholar] [CrossRef]
- Grün, D.; van Oudenaarden, A. Design and Analysis of Single-Cell Sequencing Experiments. Cell 2015, 163, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fang, Y.; Bao, L.; Wu, F.; Wang, S.; Hao, S. Intercellular Communication Reveals Therapeutic Potential of Epithelial-Mesenchymal Transition in Triple-Negative Breast Cancer. Biomolecules 2022, 12, 1478. [Google Scholar] [CrossRef] [PubMed]
- Corker, A.; Neff, L.S.; Broughton, P.; Bradshaw, A.D.; DeLeon-Pennell, K.Y. Organized Chaos: Deciphering Immune Cell Heterogeneity’s Role in Inflammation in the Heart. Biomolecules 2021, 12, 11. [Google Scholar] [CrossRef]
- Liang, L.; Yu, J.; Li, J.; Li, N.; Liu, J.; Xiu, L.; Zeng, J.; Wang, T.; Wu, L. Integration of scRNA-Seq and Bulk RNA-Seq to Analyse the Heterogeneity of Ovarian Cancer Immune Cells and Establish a Molecular Risk Model. Front. Oncol. 2021, 11, 711020. [Google Scholar] [CrossRef]
- Pan, Y.; Lu, F.; Fei, Q.; Yu, X.; Xiong, P.; Yu, X.; Dang, Y.; Hou, Z.; Lin, W.; Lin, X.; et al. Single-cell RNA sequencing reveals compartmental remodeling of tumor-infiltrating immune cells induced by anti-CD47 targeting in pancreatic cancer. J. Hematol. Oncol. 2019, 12, 124. [Google Scholar] [CrossRef]
- Metzger, T.C.; Khan, I.S.; Gardner, J.M.; Mouchess, M.L.; Johannes, K.P.; Krawisz, A.K.; Skrzypczynska, K.M.; Anderson, M.S. Lineage tracing and cell ablation identify a post-Aire-expressing thymic epithelial cell population. Cell Rep. 2013, 5, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Paul, F.; Arkin, Y.; Giladi, A.; Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Winter, D.; Lara-Astiaso, D.; Gury, M.; Weiner, A.; et al. Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell 2015, 163, 1663–1677. [Google Scholar] [CrossRef] [PubMed]
- Schlitzer, A.; Sivakamasundari, V.; Chen, J.; Sumatoh, H.R.; Schreuder, J.; Lum, J.; Malleret, B.; Zhang, S.; Larbi, A.; Zolezzi, F.; et al. Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat. Immunol. 2015, 16, 718–728. [Google Scholar] [CrossRef]
- Ishizuka, I.E.; Chea, S.; Gudjonson, H.; Constantinides, M.G.; Dinner, A.R.; Bendelac, A.; Golub, R. Single-cell analysis defines the divergence between the innate lymphoid cell lineage and lymphoid tissue-inducer cell lineage. Nat. Immunol. 2016, 17, 269–276. [Google Scholar] [CrossRef]
- DeLaughter, D.M.; Bick, A.G.; Wakimoto, H.; McKean, D.; Gorham, J.M.; Kathiriya, I.S.; Hinson, J.T.; Homsy, J.; Gray, J.; Pu, W.; et al. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev. Cell 2016, 39, 480–490. [Google Scholar] [CrossRef] [Green Version]
- Shalek, A.K.; Satija, R.; Shuga, J.; Trombetta, J.J.; Gennert, D.; Lu, D.; Chen, P.; Gertner, R.S.; Gaublomme, J.T.; Yosef, N.; et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 2014, 510, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Zheng, Y.; Zhang, G.; Hu, Y.; Fan, X.; Hou, Y.; Wen, L.; Li, L.; Xu, Y.; Wang, Y.; et al. Single-cell RNA-seq analysis reveals the progression of human osteoarthritis. Ann. Rheum. Dis. 2019, 78, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef]
- Chen, Z.; Zhou, L.; Liu, L.; Hou, Y.; Xiong, M.; Yang, Y.; Hu, J.; Chen, K. Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma. Nat. Commun. 2020, 11, 5077. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K. Chronic Inflammation as an Immunological Abnormality and Effectiveness of Exercise. Biomolecules 2019, 9, 223. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, H.C.; Zhao, J.; Wu, M.H.; Shih, T.C. Immunosuppressive Roles of Galectin-1 in the Tumor Microenvironment. Biomolecules 2021, 11, 1398. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Mo, S.; Li, X.; He, Y.; Yang, J. Single-cell RNA-seq reveals the immune escape and drug resistance mechanisms of mantle cell lymphoma. Cancer Biol. Med. 2020, 17, 726–739. [Google Scholar] [CrossRef]
- Liu, W.; Hu, H.; Shao, Z.; Lv, X.; Zhang, Z.; Deng, X.; Song, Q.; Han, Y.; Guo, T.; Xiong, L.; et al. Characterizing the tumor microenvironment at the single-cell level reveals a novel immune evasion mechanism in osteosarcoma. Bone Res. 2023, 11, 4. [Google Scholar] [CrossRef]
- Anand, P.; Guillaumet-Adkins, A.; Dimitrova, V.; Yun, H.; Drier, Y.; Sotudeh, N.; Rogers, A.; Ouseph, M.M.; Nair, M.; Potdar, S.; et al. Single-cell RNA-seq reveals developmental plasticity with coexisting oncogenic states and immune evasion programs in ETP-ALL. Blood 2021, 137, 2463–2480. [Google Scholar] [CrossRef]
- Xie, S.; Cai, Y.; Chen, D.; Xiang, Y.; Cai, W.; Mao, J.; Ye, J. Single-cell transcriptome analysis reveals heterogeneity and convergence of the tumor microenvironment in colorectal cancer. Front. Immunol. 2022, 13, 1003419. [Google Scholar] [CrossRef]
- Zhu, L.; Zhu, X.; Wu, Y. Effects of Glucose Metabolism, Lipid Metabolism, and Glutamine Metabolism on Tumor Microenvironment and Clinical Implications. Biomolecules 2022, 12, 580. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer Cell 2017, 31, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Alves, J.M.; Posada, D. Sensitivity to sequencing depth in single-cell cancer genomics. Genome Med. 2018, 10, 29. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Navin, N.E. Delineating cancer evolution with single-cell sequencing. Sci. Transl. Med. 2015, 7, 296fs229. [Google Scholar] [CrossRef]
- Li, X.; Sun, Z.; Peng, G.; Xiao, Y.; Guo, J.; Wu, B.; Li, X.; Zhou, W.; Li, J.; Li, Z.; et al. Single-cell RNA sequencing reveals a pro-invasive cancer-associated fibroblast subgroup associated with poor clinical outcomes in patients with gastric cancer. Theranostics 2022, 12, 620–638. [Google Scholar] [CrossRef]
- de Jong, M.M.E.; Kellermayer, Z.; Papazian, N.; Tahri, S.; Hofste Op Bruinink, D.; Hoogenboezem, R.; Sanders, M.A.; van de Woestijne, P.C.; Bos, P.K.; Khandanpour, C.; et al. The multiple myeloma microenvironment is defined by an inflammatory stromal cell landscape. Nat. Immunol. 2021, 22, 769–780. [Google Scholar] [CrossRef]
- He, D.; Wang, D.; Lu, P.; Yang, N.; Xue, Z.; Zhu, X.; Zhang, P.; Fan, G. Single-cell RNA sequencing reveals heterogeneous tumor and immune cell populations in early-stage lung adenocarcinomas harboring EGFR mutations. Oncogene 2021, 40, 355–368. [Google Scholar] [CrossRef]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Yeaton, A.; Cayanan, G.; Loghavi, S.; Dolgalev, I.; Leddin, E.M.; Loo, C.E.; Torabifard, H.; Nicolet, D.; Wang, J.; Corrigan, K.; et al. The Impact of Inflammation-Induced Tumor Plasticity during Myeloid Transformation. Cancer Discov. 2022, 12, 2392–2413. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zheng, L.; Yoo, J.K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356.e16. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wu, L.; Zhong, Y.; Zhou, K.; Hou, Y.; Wang, Z.; Zhang, Z.; Xie, J.; Wang, C.; Chen, D.; et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell 2021, 184, 404–421.e16. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, C.; Chen, J.; Chen, L.; Ren, X.; Hou, M.; Cui, X.; Jiang, Y.; Liu, E.; Zong, Y.; et al. Single-cell dissection of remodeled inflammatory ecosystem in primary and metastatic gallbladder carcinoma. Cell Discov. 2022, 8, 101. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.; Lin, Y.; Navin, N. Advancing Cancer Research and Medicine with Single-Cell Genomics. Cancer Cell 2020, 37, 456–470. [Google Scholar] [CrossRef]
- Zhang, P.; Yang, M.; Zhang, Y.; Xiao, S.; Lai, X.; Tan, A.; Du, S.; Li, S. Dissecting the Single-Cell Transcriptome Network Underlying Gastric Premalignant Lesions and Early Gastric Cancer. Cell Rep. 2019, 27, 1934–1947.e5. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Cheng, J.; Fan, X.M. Role of cyclooxygenase-2 in gastric cancer development and progression. World J. Gastroenterol. 2013, 19, 7361–7368. [Google Scholar] [CrossRef]
- Vishwakarma, M.; Piddini, E. Outcompeting cancer. Nat. Rev. Cancer 2020, 20, 187–198. [Google Scholar] [CrossRef]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Zhang, P.; Wang, J.; Lin, C.; Liu, H.; Li, H.; He, H.; Li, R.; Zhang, H.; Zhang, W. Somatic ARID1A mutation stratifies patients with gastric cancer to PD-1 blockade and adjuvant chemotherapy. Cancer Immunol. Immunother. 2022. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Crosetto N, Bienko M, van Oudenaarden A: Spatially resolved transcriptomics and beyond. Nat. Rev. Genet. 2015, 16, 57–66. [CrossRef] [PubMed]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Bergenstråhle, L.; Stenbeck, L.; Abid, A.; Andersson, A.; Borg, Å.; Maaskola, J.; Lundeberg, J.; Zou, J. Integrating spatial gene expression and breast tumour morphology via deep learning. Nat. Biomed Eng. 2020, 4, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Berglund, E.; Maaskola, J.; Schultz, N.; Friedrich, S.; Marklund, M.; Bergenstråhle, J.; Tarish, F.; Tanoglidi, A.; Vickovic, S.; Larsson, L.; et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat. Commun. 2018, 9, 2419. [Google Scholar] [CrossRef]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstråhle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 497–514.e22. [Google Scholar] [CrossRef]
- Saiselet, M.; Rodrigues-Vitória, J.; Tourneur, A.; Craciun, L.; Spinette, A.; Larsimont, D.; Andry, G.; Lundeberg, J.; Maenhaut, C.; Detours, V. Transcriptional output, cell-type densities, and normalization in spatial transcriptomics. J. Mol. Cell Biol. 2020, 12, 906–908. [Google Scholar] [CrossRef]
- Longo, S.K.; Guo, M.G.; Ji, A.L.; Khavari, P.A. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat. Rev. Genet. 2021, 22, 627–644. [Google Scholar] [CrossRef]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef]
- Vickovic, S.; Eraslan, G.; Salmén, F.; Klughammer, J.; Stenbeck, L.; Schapiro, D.; Äijö, T.; Bonneau, R.; Bergenstråhle, L.; Navarro, J.F.; et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 2019, 16, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.S.; Xi, J.; Si, Y.; Park, S.R.; Hsu, J.E.; Kim, M.; Jun, G.; Kang, H.M.; Lee, J.H. Microscopic examination of spatial transcriptome using Seq-Scope. Cell 2021, 184, 3559–3572.e22. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Liao, S.; Cheng, M.; Ma, K.; Wu, L.; Lai, Y.; Qiu, X.; Yang, J.; Xu, J.; Hao, S.; et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 2022, 185, 1777–1792.e21. [Google Scholar] [CrossRef] [PubMed]
- Ke, R.; Mignardi, M.; Pacureanu, A.; Svedlund, J.; Botling, J.; Wählby, C.; Nilsson, M. In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods 2013, 10, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Moor, A.E.; Itzkovitz, S. Spatial transcriptomics: Paving the way for tissue-level systems biology. Curr. Opin. Biotechnol. 2017, 46, 126–133. [Google Scholar] [CrossRef]
- Guo, W.; Zhou, B.; Yang, Z.; Liu, X.; Huai, Q.; Guo, L.; Xue, X.; Tan, F.; Li, Y.; Xue, Q.; et al. Integrating microarray-based spatial transcriptomics and single-cell RNA-sequencing reveals tissue architecture in esophageal squamous cell carcinoma. EBioMedicine 2022, 84, 104281. [Google Scholar] [CrossRef]
- Moncada, R.; Barkley, D.; Wagner, F.; Chiodin, M.; Devlin, J.C.; Baron, M.; Hajdu, C.H.; Simeone, D.M.; Yanai, I. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol. 2020, 38, 333–342. [Google Scholar] [CrossRef]
- Qi, J.; Sun, H.; Zhang, Y.; Wang, Z.; Xun, Z.; Li, Z.; Ding, X.; Bao, R.; Hong, L.; Jia, W.; et al. Single-cell and spatial analysis reveal interaction of FAP(+) fibroblasts and SPP1(+) macrophages in colorectal cancer. Nat. Commun. 2022, 13, 1742. [Google Scholar] [CrossRef]
- Kumar, M.P.; Du, J.; Lagoudas, G.; Jiao, Y.; Sawyer, A.; Drummond, D.C.; Lauffenburger, D.A.; Raue, A. Analysis of Single-Cell RNA-Seq Identifies Cell-Cell Communication Associated with Tumor Characteristics. Cell Rep. 2018, 25, 1458–1468.e4. [Google Scholar] [CrossRef]
- Schelker, M.; Feau, S.; Du, J.; Ranu, N.; Klipp, E.; MacBeath, G.; Schoeberl, B.; Raue, A. Estimation of immune cell content in tumour tissue using single-cell RNA-seq data. Nat. Commun. 2017, 8, 2032. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Protocol | Single-Cell Capture | mRNA Reverse Transcription | cDNA Amplification | Library Construction | Reference |
|---|---|---|---|---|---|
| Tang method | Micromanipulation | poly(A) tailing + second-strand synthesis | PCR | Full-length | [11] |
| CEL-seq | LCM/Flow cytometry | second-strand synthesis | IVT | 3′-Only | [12] |
| SMART-seq | Micromanipulation/LCM/Flow cytometry | template-switching method | PCR | Full-length | [13] |
| STRT-seq | LCM | template-switching method | PCR | 5′-Only or 3′-Only | [14] |
| Drop-seq | Microfluidics | template-switching method | PCR | 3′-Only | [15] |
| Cancer | Sample | Application | Combined with ST | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Composition 1 | Heterogeneity 2 | Trajectory 3 | Prognosis 4 | Interaction 5 | ||||
| Bladder Carcinoma (BC) | tumor samples & para tumor samples | Y | Y | Y | Y | Y | [54] | |
| Gastric Cancer (GC) | tumor samples & adjacent mucosal samples | Y | Y | Y | Y | Y | [67] | |
| Gastric Cancer (GC) | tumour samples & non-tumour samples | Y | Y | Y | Y | [76] | ||
| Multiple Myeloma (MM) | tumour samples & non-tumour samples | Y | Y | Y | [68] | |||
| Lung Adenocarcinoma (LUAD) | stage-I/II LUAD samples harboring EGFR mutations samples & tumor-adjacent Lung tissues | Y | Y | Y | Y | [69] | ||
| Acute Myeloid Leukemia (AML) | a novel animal model carrying a recurrent TET2 missense mutation | Y | Y | Y | [71] | |||
| Hepatocellular Carcinoma (HCC) | tumor samples & adjacent normal samples | Y | Y | Y | Y | [72] | ||
| Hepatocellular Carcinoma (HCC) | samples from primary or early-relapse HCC patients | Y | Y | Y | Y | Y | [73] | |
| Gallbladder carcinoma (GBC) | chronic cholecystitis samples & treatment-naive GBCs samples & metastases samples | Y | Y | Y | Y | [74] | ||
| Esophageal Squamous Cell Carcinoma (ESCC) | tumor samples | Y | Y | Y | Y | Y | Y | [96] |
| Pancreatic Ductal Adenocarcinomas (PDAC) | tumor samples | Y | Y | Y | Y | [97] | ||
| Colorectal Cancer (CRC) | tumor samples & adjacent normal samples | Y | Y | Y | Y | Y | Y | [98] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Shi, Y.; Cao, G. The Application of Single-Cell RNA Sequencing in the Inflammatory Tumor Microenvironment. Biomolecules 2023, 13, 344. https://doi.org/10.3390/biom13020344
Zhao J, Shi Y, Cao G. The Application of Single-Cell RNA Sequencing in the Inflammatory Tumor Microenvironment. Biomolecules. 2023; 13(2):344. https://doi.org/10.3390/biom13020344
Chicago/Turabian StyleZhao, Jiayi, Yiwei Shi, and Guangwen Cao. 2023. "The Application of Single-Cell RNA Sequencing in the Inflammatory Tumor Microenvironment" Biomolecules 13, no. 2: 344. https://doi.org/10.3390/biom13020344
APA StyleZhao, J., Shi, Y., & Cao, G. (2023). The Application of Single-Cell RNA Sequencing in the Inflammatory Tumor Microenvironment. Biomolecules, 13(2), 344. https://doi.org/10.3390/biom13020344