

Granzyme B in Autoimmune Skin Disease


{kind=link}
{kind=link}
Abstract
:1. Background
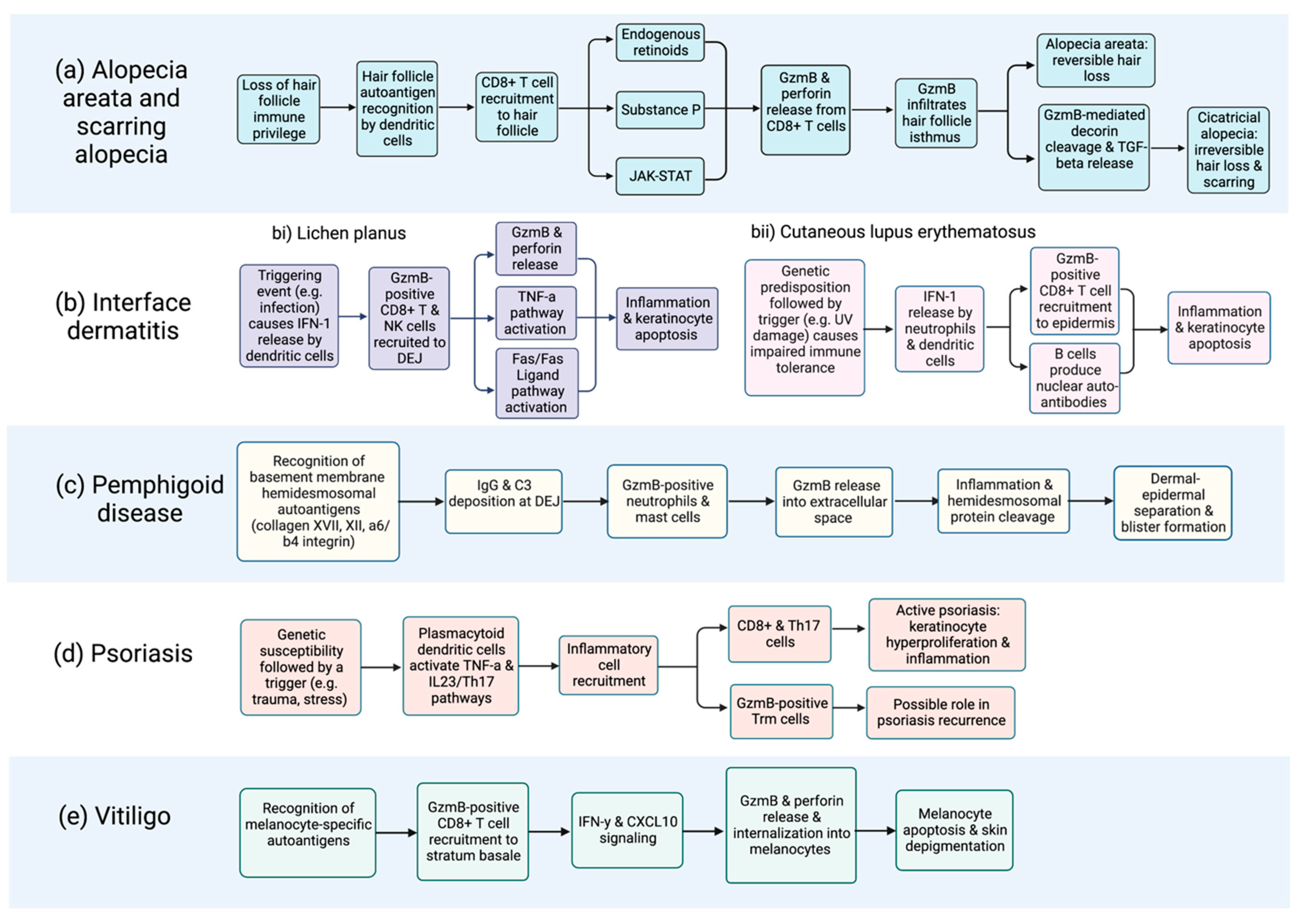
2. Alopecia Areata
3. Interface Dermatitis
4. Pemphigoid Diseases
5. Psoriasis
6. Systemic Sclerosis
7. Vitiligo
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mitratza, M.; Klijs, B.; Hak, A.E.; Kardaun, J.W.P.F.; Kunst, A.E. Systemic autoimmune disease as a cause of death: Mortality burden and comorbidities. Rheumatology 2020, 60, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.S.; Bynum, M.L.; Somers, E.C. Recent insights in the epidemiology of autoimmune diseases: Improved prevalence estimates and understanding of clustering of diseases. J. Autoimmun. 2009, 33, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Lerner, A.; Jeremias, P.; Matthias, T. The World Incidence and Prevalence of Autoimmune Diseases is Increasing. Int. J. Celiac Dis. 2015, 3, 151–155. [Google Scholar] [CrossRef] [Green Version]
- Dinse, G.E.; Parks, C.G.; Weinberg, C.R.; Co, C.A.; Wilkerson, J.; Zeldin, D.C.; Chan, E.K.L.; Miller, F.W. Increasing Prevalence of Antinuclear Antibodies in the United States. Arthritis Rheumatol. 2022, 74, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, M.D.; Remedios, K.A.; Abbas, A.K. Mechanisms of human autoimmunity. J. Clin. Investig. 2015, 125, 2228–2233. [Google Scholar] [CrossRef] [Green Version]
- Julian, M.K. Autoimmune disease. Nurs. Manag. 2014, 45, 24–29. [Google Scholar] [CrossRef]
- Lee, H.L.; Jang, J.W.; Lee, S.W.; Yoo, S.H.; Kwon, J.H.; Nam, S.W.; Bae, S.H.; Choi, J.Y.; Han, N.I.; Yoon, S.K. Inflammatory cytokines and change of Th1/Th2 balance as prognostic indicators for hepatocellular carcinoma in patients treated with transarterial chemoembolization. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Santamaria, P. Cytokines and Chemokines in Autoimmune Disease: An Overview. Adv. Exp. Med. Biol. 2003, 520, 1–7. [Google Scholar] [CrossRef]
- Rashtak, S.; Pittelkow, M.R. Skin Involvement in Systemic Autoimmune Diseases. Dermatol. Immun. 2008, 10, 344–358. [Google Scholar] [CrossRef]
- Grönhagen, C.; Nyberg, F. Cutaneous lupus erythematosus: An update. Indian Dermatol. Online J. 2014, 5, 7–13. [Google Scholar] [CrossRef]
- Hughes, M.; Herrick, A.L. Digital Ulcers in Systemic Sclerosis. Rheumatology 2017, 56, 14–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reunala, T.; Hervonen, K.; Salmi, T. Dermatitis Herpetiformis: An Update on Diagnosis and Management. Am. J. Clin. Dermatol. 2021, 22, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Whitton, M.E.; Pinart, M.; Batchelor, J.; Leonardi-Bee, J.; González, U.; Jiyad, Z.; Eleftheriadou, V.; Ezzedine, K. Interventions for Vitiligo. Cochrane Database Syst. Rev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hannon, C.W.; McCourt, C.; Lima, H.C.; Chen, S.; Bennett, C. Interventions for Cutaneous Disease in Systemic Lupus Ery-thematosus. Cochrane Database Syst. Rev. 2021. [Google Scholar] [CrossRef]
- Mehta, A.; Nadkarni, N.; Patil, S.; Godse, K.; Gautam, M.; Agarwal, S. Topical corticosteroids in dermatology. Indian J. Dermatol. Venereol. Leprol. 2016, 82, 371–378. [Google Scholar] [CrossRef]
- Jung, K.; Pawluk, M.A.; Lane, M.; Nabai, L.; Granville, D.J. Granzyme B in epithelial barrier dysfunction and related skin diseases. Am. J. Physiol. Physiol. 2022, 323, C170–C189. [Google Scholar] [CrossRef]
- Hiroyasu, S.; Hiroyasu, A.; Granville, D.J.; Tsuruta, D. Pathological functions of granzyme B in inflammatory skin diseases. J. Dermatol. Sci. 2021, 104, 76–82. [Google Scholar] [CrossRef]
- Turner, C.T.; Lim, D.; Granville, D.J. Granzyme B in skin inflammation and disease. Matrix Biol. 2019, 75–76, 126–140. [Google Scholar] [CrossRef]
- Lord, S.J.; Rajotte, R.V.; Korbutt, G.S.; Bleackley, R.C. Granzyme B: A natural born killer. Immunol. Rev. 2003, 193, 31–38. [Google Scholar] [CrossRef]
- Voskoboinik, I.; Whisstock, J.C.; Trapani, J.A. Perforin and granzymes: Function, dysfunction and human pathology. Nat. Rev. Immunol. 2015, 15, 388–400. [Google Scholar] [CrossRef]
- Isaaz, S.; Baetz, K.; Olsen, K.; Podack, E.; Griffiths, G.M. Serial killing by cytotoxic T lymphocytes: T cell receptor triggers degranulation, re-filling of the lytic granules and secretion of lytic proteins via a non-granule pathway. Eur. J. Immunol. 1995, 25, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Huang, Y.; He, J.; Zhong, L.; Zhao, Y. Dual roles of granzyme B. Scand. J. Immunol. 2021, 94, e13086. [Google Scholar] [CrossRef]
- Boivin, W.A.; Cooper, D.M.; Hiebert, P.R.; Granville, D.J. Intracellular versus extracellular granzyme B in immunity and disease: Challenging the dogma. Lab. Investig. 2009, 89, 1195–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkinson, L.G.; Toro, A.; Zhao, H.; Brown, K.; Tebbutt, S.J.; Granville, D.J. Granzyme B mediates both direct and indirect cleavage of extracellular matrix in skin after chronic low-dose ultraviolet light irradiation. Aging Cell 2014, 14, 67–77. [Google Scholar] [CrossRef]
- Shah, H.; Eisenbarth, S.; Tormey, C.A.; Siddon, A.J. Behind the scenes with basophils: An emerging therapeutic target. Immunother. Adv. 2021, 1, ltab008. [Google Scholar] [CrossRef]
- Martin, A.; Seignez, C.; Racoeur, C.; Isambert, N.; Mabrouk, N.; Scagliarini, A.; Reveneau, S.; Arnould, L.; Bettaieb, A.; Jeannin, J.-F.; et al. Tumor-derived granzyme B-expressing neutrophils acquire antitumor potential after lipid A treatment. Oncotarget 2018, 9, 28364–28378. [Google Scholar] [CrossRef] [Green Version]
- Abebe, E.C.; Dejenie, T.A.; Ayele, T.M.; Baye, N.D.; Teshome, A.A.; Muche, Z.T. The Role of Regulatory B Cells in Health and Diseases: A Systemic Review. J. Inflamm. Res. 2021, 14, 75–84. [Google Scholar] [CrossRef]
- Karrich, J.J.; Jachimowski, L.C.M.; Nagasawa, M.; Kamp, A.; Balzarolo, M.; Wolkers, M.; Uittenbogaart, C.H.; Van Ham, S.M.; Blom, B. IL-21–stimulated human plasmacytoid dendritic cells secrete granzyme B, which impairs their capacity to induce T-cell proliferation. Blood 2013, 121, 3103–3111. [Google Scholar] [CrossRef] [Green Version]
- Choy, J.C.; McDonald, P.C.; Suarez, A.C.; Hung, V.H.Y.; Wilson, J.E.; McManus, B.M.; Granville, D.J. Granzyme B in Atherosclerosis and Transplant Vascular Disease: Association with Cell Death and Atherosclerotic Disease Severity. Mod. Pathol. 2003, 16, 460–470. [Google Scholar] [CrossRef]
- Park, S.; Anderson, N.L.; Canaria, D.A.; Olson, M.R. Granzyme-Producing CD4 T Cells in Cancer and Autoimmune Disease. Immunohorizons 2021, 5, 909–917. [Google Scholar] [CrossRef]
- Grover, P.; Goel, P.N.; Greene, M.I. Regulatory T Cells: Regulation of Identity and Function. Front. Immunol. 2021, 12, 750542. [Google Scholar] [CrossRef]
- Hernandez-Pigeon, H.; Jean, C.; Charruyer, A.; Haure, M.-J.; Titeux, M.; Tonasso, L.; Quillet-Mary, A.; Baudouin, C.; Charveron, M.; Laurent, G. Human Keratinocytes Acquire Cellular Cytotoxicity under UV-B Irradiation: Implication of Granzyme B and Perforin. J. Biol. Chem. 2006, 281, 13525–13532. [Google Scholar] [CrossRef] [Green Version]
- Kaiserman, D.; Bird, P.I. Control of granzymes by serpins. Cell Death Differ. 2009, 17, 586–595. [Google Scholar] [CrossRef] [Green Version]
- Hiroyasu, S.; Zeglinski, M.R.; Zhao, H.; Pawluk, M.A.; Turner, C.T.; Kasprick, A.; Tateishi, C.; Nishie, W.; Burleigh, A.; Lennox, P.A.; et al. Granzyme B inhibition reduces disease severity in autoimmune blistering diseases. Nat. Commun. 2021, 12, 302. [Google Scholar] [CrossRef]
- Klein, J.L.; Shows, T.B.; Dupont, B.; Trapani, J.A. Genomic organization and chromosomal assignment for a serine protease gene (CSPB) expressed by human cytotoxic lymphocytes. Genomics 1989, 5, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, D.; Lieberman, J. Death by a Thousand Cuts: Granzyme Pathways of Programmed Cell Death. Annu. Rev. Immunol. 2008, 26, 389–420. [Google Scholar] [CrossRef] [Green Version]
- Waugh, S.M.; Craik, C.S.; Harris, J.L.; Fletterick, R. The structure of the pro-apoptotic protease granzyme B reveals the molecular determinants of its specificity. Nat. Struct. Biol. 2000, 7, 762–765. [Google Scholar] [CrossRef]
- Estebanez-Perpina, E.; Fuentes-Prior, P.; Belorgey, D.; Braun, M.; Kiefersauer, R.; Maskos, K.; Huber, R.; Rubin, H.; Bode, W. Crystal Structure of the Caspase Activator Human Granzyme B, a Proteinase Highly Specific for an Asp-P1 Residue. Biol. Chem. 2000, 381, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Bird, C.H.; Sutton, V.; McDonald, L.; Coughlin, P.B.; De Jong, T.A.; Trapani, J.; Bird, P. A Cytosolic Granzyme B Inhibitor Related to the Viral Apoptotic Regulator Cytokine Response Modifier A Is Present in Cytotoxic Lymphocytes. J. Biol. Chem. 1996, 271, 27802–27809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, V.; Klein, T.; Lim, D.J.; Solis, N.; Machado, Y.; Hiroyasu, S.; Nabai, L.; Shen, Y.; Zeglinski, M.R.; Zhao, H.; et al. Granzyme B is elevated in autoimmune blistering diseases and cleaves key anchoring proteins of the dermal-epidermal junction. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willoughby, C.A.; Bull, H.G.; Garcia-Calvo, M.; Jiang, J.; Chapman, K.T.; Thornberry, N.A. Discovery of potent, selective human granzyme B inhibitors that inhibit CTL mediated apoptosis. Bioorganic Med. Chem. Lett. 2002, 12, 2197–2200. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Rano, T.A.; Peterson, E.P.; Rasper, D.M.; Timkey, T.; Garcia-Calvo, M.; Houtzager, V.M.; Nordstrom, P.A.; Roy, S.; Vaillancourt, J.P.; et al. A Combinatorial Approach Defines Specificities of Members of the Caspase Family and Granzyme B. J. Biol. Chem. 1997, 272, 17907–17911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, J.L.; Peterson, E.P.; Hudig, D.; Thornberry, N.A.; Craik, C.S. Definition and Redesign of the Extended Substrate Specificity of Granzyme B. J. Biol. Chem. 1998, 273, 27364–27373. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Zeglinski, M.; Turner, C.; Raithatha, S.A.; Wu, Z.; Russo, V.; Oram, C.; Hiroyasu, S.; Nabai, L.; Zhao, H.; et al. Topical small molecule granzyme B inhibitor improves remodeling in a murine model of impaired burn wound healing. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.T.; Zeglinski, M.R.; Richardson, K.C.; Santacruz, S.; Hiroyasu, S.; Wang, C.; Zhao, H.; Shen, Y.; Sehmi, R.; Lima, H.; et al. Granzyme B Contributes to Barrier Dysfunction in Oxazolone-Induced Skin Inflammation through E-Cadherin and FLG Cleavage. J. Investig. Dermatol. 2020, 141, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Wijngaard, R.V.D.; Wankowicz-Kalinska, A.; Le Poole, C.; Tigges, B.; Westerhof, W.; Das, P. Local Immune Response in Skin of Generalized Vitiligo Patients. Lab. Investig. 2000, 80, 1299–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunger, R.E.; Brönnimann, M.; Kappeler, A.; Mueller, C.; Braathen, L.R.; Yawalkar, N. Detection of perforin and granzyme B mRNA expressing cells in lichen sclerosus. Exp. Dermatol. 2007, 16, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Lage, D.; Pimentel, V.N.; Soares, T.C.B.; Souza, E.M.; Metze, K.; Cintra, M.L. Perforin and granzyme B expression in oral and cutaneous lichen planus—A comparative study. J. Cutan. Pathol. 2011, 38, 973–978. [Google Scholar] [CrossRef]
- Hussein, M.R.; Ali, F.M.N.; Omar, A.-E.M.M. Immunohistological analysis of immune cells in blistering skin lesions. J. Clin. Pathol. 2007, 60, 62–71. [Google Scholar] [CrossRef] [Green Version]
- Blanco, P.; Pitard, V.; Taupin, J.-L.; Pellegrin, J.-L. Increase in activated CD8+ T lymphocytes expressing perforin and granzyme B correlates with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2005, 52, 201–211. [Google Scholar] [CrossRef]
- Kok, H.M.; Hoogen, L.L.V.D.; Van Roon, J.A.G.; Adriaansen, E.J.M.; Fritsch-Stork, R.D.E.; Nguyen, T.Q.; Goldschmeding, R.; Radstake, T.R.D.J.; Bovenschen, N. Systemic and local granzyme B levels are associated with disease activity, kidney damage and interferon signature in systemic lupus erythematosus. Rheumatology 2017, 56, 2129–2134. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.; Kiran, R.; Wanchu, A.; Bhatnagar, A. Soluble granzyme B and cytotoxic T lymphocyte activity in the pathogenesis of systemic lupus erythematosus. Cell. Immunol. 2011, 269, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Boniface, K.; Vergier, B.; Mossalayi, D.; Taieb, A.; Ezzedine, K.; Seneschal, J. Type I interferon signature in the initiation of the immune response in vitiligo. Pigment. Cell Melanoma Res. 2014, 27, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.Y.; Chiu, Y.-C.; Chan, Y.-P.; Lin, Y.-J.; Chung, P.-H.; Chung, W.-H.; Ku, C.-L. Skin Interstitial Fluid and Plasma Multiplex Cytokine Analysis Reveals IFN-γ Signatures and Granzyme B as Useful Biomarker for Activity, Severity and Prognosis Assessment in Vitiligo. Front. Immunol. 2022, 13, 872458. [Google Scholar] [CrossRef] [PubMed]
- Koguchi-Yoshioka, H.; Watanabe, R.; Matsumura, Y.; Okiyama, N.; Ishitsuka, Y.; Nakamura, Y.; Fujisawa, Y.; Fujimoto, M. The Possible Linkage of Granzyme B-Producing Skin T Cells with the Disease Prognosis of Alopecia Areata. J. Investig. Dermatol. 2020, 141, 427–429.e10. [Google Scholar] [CrossRef] [PubMed]
- Ghoreishi, M.; Martinka, M.; Dutz, J. Type 1 interferon signature in the scalp lesions of alopecia areata. Br. J. Dermatol. 2010, 163, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Del Duca, E.; Ruiz, J.R.; Pavel, A.; Sanyal, R.D.; Song, T.; Gay-Mimbrera, J.; Zhang, N.; Estrada, Y.; Peng, X.; Renert-Yuval, Y.; et al. Frontal fibrosing alopecia shows robust T helper 1 and Janus kinase 3 skewing. Br. J. Dermatol. 2020, 183, 1083–1093. [Google Scholar] [CrossRef]
- Pratt, C.H.; King, L.E.; Messenger, A.G.; Christiano, A.M.; Sundberg, J.P. Alopecia Areata. Nat. Rev. Dis. Primers 2017, 3, 17011. [Google Scholar] [CrossRef] [Green Version]
- Xing, L.; Dai, Z.; Jabbari, A.; Cerise, J.E.; Higgins, C.A.; Gong, W.; de Jong, A.; Harel, S.; DeStefano, G.M.; Rothman, L.; et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat. Med. 2014, 20, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Phan, K.; Sebaratnam, D.F. JAK inhibitors for alopecia areata: A systematic review and meta-analysis. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 850–856. [Google Scholar] [CrossRef]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. JAK–STAT Signaling as a Target for Inflammatory and Auto-immune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef]
- Olayinka, J.T.; Richmond, J.M. Immunopathogenesis of alopecia areata. Curr. Res. Immunol. 2021, 2, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Duncan, F.J.; Silva, K.A.; Johnson, C.J.; King, B.L.; Szatkiewicz, J.P.; Kamdar, S.P.; Ong, D.E.; Napoli, J.L.; Wang, J.; King, L.E.; et al. Endogenous Retinoids in the Pathogenesis of Alopecia Areata. J. Investig. Dermatol. 2013, 133, 334–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duriancik, D.M.; Lackey, D.E.; Hoag, K.A. Vitamin A as a Regulator of Antigen Presenting Cells. J. Nutr. 2010, 140, 1395–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebenhaar, F.; Sharov, A.A.; Peters, E.M.; Sharova, T.Y.; Syska, W.; Mardaryev, A.N.; Freyschmidt-Paul, P.; Sundberg, J.P.; Maurer, M.; Botchkarev, V.A. Substance P as an Immunomodulatory Neuropeptide in a Mouse Model for Autoimmune Hair Loss (Alopecia Areata). J. Investig. Dermatol. 2007, 127, 1489–1497. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, T.M.; O’Connell, J.; O’Brien, D.I.; Goode, T.; Bredin, C.P.; Shanahan, F. The role of substance P in inflammatory disease. J. Cell. Physiol. 2004, 201, 167–180. [Google Scholar] [CrossRef]
- Guo, H.; Cheng, Y.; Shapiro, J.; McElwee, K. The role of lymphocytes in the development and treatment of alopecia areata. Expert Rev. Clin. Immunol. 2015, 11, 1335–1351. [Google Scholar] [CrossRef]
- McPhee, C.G.; Duncan, F.J.; Silva, K.A.; King, L.E.; HogenEsch, H.; Roopenian, D.C.; Everts, H.B.; Sundberg, J.P. Increased Expression of Cxcr3 and Its Ligands, Cxcl9 and Cxcl10, during the Development of Alopecia Areata in the Mouse. J. Investig. Dermatol. 2012, 132, 1736–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, K.; Yamada, Y.; Sekiguchi, K.; Matsuda, S.; Mori, S.; Matsumoto, T. Induction of alopecia areata in C3H/HeJ mice using cryopreserved lymphocytes. J. Dermatol. Sci. 2021, 102, 177–183. [Google Scholar] [CrossRef]
- Villasante Fricke, A.C.; Miteva, M. Epidemiology and Burden of Alopecia Areata: A Systematic Review. Clin. Cosmet. Investig. Dermatol. 2015, 8, 397–403. [Google Scholar]
- Fukumoto, T.; Fukumoto, R.; Magno, E.; Oka, M.; Nishigori, C.; Horita, N. Treatments for alopecia areata: A systematic review and network meta-analysis. Dermatol. Ther. 2021, 34, e14916. [Google Scholar] [CrossRef] [PubMed]
- Lai, V.W.Y.; Chen, G.; Gin, D.; Sinclair, R. Systemic treatments for alopecia areata: A systematic review. Australas. J. Dermatol. 2018, 60, e1–e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanti, V.; Röwert-Huber, J.; Vogt, A.; Blume-Peytavi, U. Cicatricial alopecia. JDDG J. Dtsch. Dermatol. Ges. 2018, 16, 435–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoulis, A.C.; Christodoulou, C.; Liakou, A.I.; Kouris, A.; Korkoliakou, P.; Kaloudi, E.; Kanelleas, A.; Papageorgiou, C.; Rigopoulos, D. Quality of life and psychosocial impact of scarring and non-scarring alopecia in women. JDDG J. Dtsch. Dermatol. Ges. 2015, 13, 137–141. [Google Scholar] [CrossRef]
- Boivin, W.A.; Shackleford, M.; Hoek, A.V.; Zhao, H.; Hackett, T.L.; Knight, D.A.; Granville, D.J. Granzyme B Cleaves Decorin, Biglycan and Soluble Betaglycan, Releasing Active Transforming Growth Factor-β1. PLoS ONE 2012, 7, e33163. [Google Scholar] [CrossRef]
- Honardoust, D.; Varkey, M.; Hori, K.; Ding, J.; Shankowsky, H.A.; Tredget, E.E. Small leucine-rich proteoglycans, decorin and fibromodulin, are reduced in postburn hypertrophic scar. Wound Repair Regen. 2011, 19, 368–378. [Google Scholar] [CrossRef]
- Meenakshi, J.; Vidyameenakshi, S.; Ananthram, D.; Ramakrishnan, K.M.; Jayaraman, V.; Babu, M. Low decorin expression along with inherent activation of ERK1,2 in ear lobe keloids. Burns 2009, 35, 519–526. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Wong, M.Y.; Chan, S.Y.; Do, D.V.; Khoo, A.; Ong, C.T.; Cheong, H.H.; Lim, I.J.; Phan, T.T. Syndecan-2 and Decorin: Proteoglycans with a Difference—Implications in Keloid Pathogenesis. J. Trauma: Inj. Infect. Crit. Care 2010, 68, 999–1008. [Google Scholar] [CrossRef]
- Ang, L.S.; Boivin, W.A.; Williams, S.J.; Zhao, H.; Abraham, T.; Carmine-Simmen, K.; McManus, B.M.; Bleackley, R.C.; Granville, D.J. Serpina3n attenuates granzyme B-mediated decorin cleavage and rupture in a murine model of aortic aneurysm. Cell Death Dis. 2011, 2, e209. [Google Scholar] [CrossRef] [Green Version]
- Moretti, S.; Amato, L.; Massi, D.; Bianchi, B.; Gallerani, I.; Fabbri, P. Evaluation of inflammatory infiltrate and fibrogenic cytokines in pseudopelade of Brocq suggests the involvement of T-helper 2 and 3 cytokines. Br. J. Dermatol. 2004, 151, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Aguh, C.; Dina, Y.; Talbot, C.C.; Garza, L. Fibroproliferative genes are preferentially expressed in central centrifugal cicatricial alopecia. J. Am. Acad. Dermatol. 2018, 79, 904–912.e1. [Google Scholar] [CrossRef] [PubMed]
- Harries, M.J.; Paus, R. The Pathogenesis of Primary Cicatricial Alopecias. Am. J. Pathol. 2010, 177, 2152–2162. [Google Scholar] [CrossRef]
- Joshi, R. Interface Dermatitis. Indian J. Dermatol. Venereol. Leprol. 2013, 79, 349. [Google Scholar] [CrossRef] [PubMed]
- Grassi, M.; Capello, F.; Bertolino, L.; Seia, Z.; Pippione, M. Identification of granzyme B-expressing CD-8-positive T cells in lymphocytic inflammatory infiltrate in cutaneous lupus erythematosus and in dermatomyositis. Clin. Exp. Dermatol. 2009, 34, 910–914. [Google Scholar] [CrossRef]
- Sontheimer, R.D. Lichenoid Tissue Reaction/Interface Dermatitis: Clinical and Histological Perspectives. J. Investig. Dermatol. 2009, 129, 1088–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okiyama, N.; Fujimoto, M. Clinical perspectives and murine models of lichenoid tissue reaction/interface dermatitis. J. Dermatol. Sci. 2015, 78, 167–172. [Google Scholar] [CrossRef]
- Boch, K.; Langan, E.A.; Kridin, K.; Zillikens, D.; Ludwig, R.J.; Bieber, K. Lichen Planus. Front. Med. 2021, 8, 737813. [Google Scholar] [CrossRef]
- Wenzel, J.; Scheler, M.; Proelss, J.; Bieber, T.; Tüting, T. Type I interferon-associated cytotoxic inflammation in lichen planus. J. Cutan. Pathol. 2006, 33, 672–678. [Google Scholar] [CrossRef]
- Shimizu, M.; Higaki, Y.; Higaki, M.; Kawashima, M. The role of granzyme B-expressing CD8-positive T cells in apoptosis of keratinocytes in lichen planus. Arch. Dermatol. Res. 1997, 289, 527–532. [Google Scholar] [CrossRef]
- Ammar, M.; Mokni, M.; Boubaker, S.; El Gaied, A.; Ben Osman, A.; Louzir, H. Involvement of granzyme B and granulysin in the cytotoxic response in lichen planus. J. Cutan. Pathol. 2008, 35, 630–634. [Google Scholar] [CrossRef]
- Pimentel, V.N.; De Matos, L.S.; Soares, T.C.B.; Adam, R.; Metze, K.; Correa, M.E.P.; De Souza, C.A.; Cintra, M.L. Perforin and granzyme B involvement in oral lesions of lichen planus and chronic GVHD. J. Oral Pathol. Med. 2010, 39, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Blake, S.C.; Daniel, B.S. Cutaneous lupus erythematosus: A review of the literature. Int. J. Women’s Dermatol. 2019, 5, 320–329. [Google Scholar] [CrossRef]
- Tsokos, G.C.; Lo, M.S.; Reis, P.C.; Sullivan, K.E. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2016, 12, 716–730. [Google Scholar] [CrossRef] [PubMed]
- Garelli, C.J.; Refat, M.A.; Nanaware, P.P.; Ramirez-Ortiz, Z.G.; Rashighi, M.; Richmond, J.M. Current Insights in Cutaneous Lupus Erythematosus Immunopathogenesis. Front. Immunol. 2020, 11, 1353. [Google Scholar] [CrossRef]
- Casciola-Rosen, L.; Andrade, F.; Ulanet, D.; Wong, W.B.; Rosen, A. Cleavage by Granzyme B Is Strongly Predictive of Autoantigen Status. J. Exp. Med. 1999, 190, 815–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdou, A.G.; Shoeib, M.; Bakry, O.A.; El-Bality, H. Immunohistochemical Expression of Granzyme B and Perforin in Discoid Lupus Erythematosus. Ultrastruct. Pathol. 2013, 37, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, J.; Uerlich, M.; Worrenkamper, E.; Freutel, S.; Bieber, T.; Tuting, T. Scarring skin lesions of discoid lupus erythematosus are characterized by high numbers of skin-homing cytotoxic lymphocytes associated with strong expression of the type I interferon-induced protein MxA. Br. J. Dermatol. 2005, 153, 1011–1015. [Google Scholar] [CrossRef]
- DeWane, M.E.; Waldman, R.; Lu, J. Dermatomyositis: Clinical features and pathogenesis. J. Am. Acad. Dermatol. 2020, 82, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Torchia, D.; Caproni, M.; Volpi, W.; Barletta, E.; Fabbri, P. The Fas/Fas ligand system, rather than granzyme B, may represent the main mediator of epidermal apoptosis in dermatomyositis. Clin. Exp. Dermatol. 2010, 35, 669–670. [Google Scholar] [CrossRef]
- Goebels, N.; Michaelis, D.; Engelhardt, M.; Huber, S.; Bender, A.; Pongratz, D.; Johnson, M.A.; Wekerle, H.; Tschopp, J.; Jenne, D.; et al. Differential expression of perforin in muscle-infiltrating T cells in polymyositis and dermatomyositis. J. Clin. Investig. 1996, 97, 2905–2910. [Google Scholar] [CrossRef] [Green Version]
- Mammen, A.L. Dermatomyositis and Polymyositis: Clinical Presentation, Autoantibodies, and Pathogenesis. Ann. N. Y. Acad. Sci. 2010, 1184, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Daniel, B.S.; Murrell, D.F. Review of Autoimmune Blistering Diseases: The Pemphigoid Diseases. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Kridin, K.; Ludwig, R. The Growing Incidence of Bullous Pemphigoid: Overview and Potential Explanations. Front. Med. 2018, 5, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido, P.M.; Queirós, C.S.; Travassos, A.R.; Borges-Costa, J.; Filipe, P. Emerging treatments for bullous pemphigoid. J. Dermatol. Treat. 2021, 33, 649–661. [Google Scholar] [CrossRef]
- Kirtschig, G.; Middleton, P.; Bennett, C.; Murrell, D.F.; Wojnarowska, F.; Khumalo, N.P. Interventions for Bullous Pemphigoid. Cochrane Database Syst. Rev. 2010. [Google Scholar] [CrossRef]
- Hiroyasu, S.; Turner, C.T.; Richardson, K.; Granville, D.J. Proteases in Pemphigoid Diseases. Front. Immunol. 2019, 10, 1454. [Google Scholar] [CrossRef]
- Nishie, W. Collagen XVII Processing and Blistering Skin Diseases. Acta Derm. Venereol. 2020, 100, 102–107. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, A.W.; Read, C. Pathophysiology, Clinical Presentation, and Treatment of Psoriasis: A Review. JAMA—J. Am. Med. Assoc. 2020, 323, 1945–1960. [Google Scholar] [CrossRef]
- Rendon, A.; Schäkel, K. Psoriasis Pathogenesis and Treatment. Int. J. Mol. Sci. 2019, 20, 1475. [Google Scholar] [CrossRef] [Green Version]
- Rønholt, K.; Iversen, L. Old and New Biological Therapies for Psoriasis. Int. J. Mol. Sci. 2017, 18, 2297. [Google Scholar] [CrossRef] [Green Version]
- Yawalkar, N.; Schmid, S.; Braathen, L.; Pichler, W. Perforin and granzyme B may contribute to skin inflammation in atopic dermatitis and psoriasis. Br. J. Dermatol. 2001, 144, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Kamata, Y.; Kimura, U.; Matsuda, H.; Tengara, S.; Kamo, A.; Umehara, Y.; Iizumi, K.; Kawasaki, H.; Suga, Y.; Ogawa, H.; et al. Relationships among plasma granzyme B level, pruritus and dermatitis in patients with atopic dermatitis. J. Dermatol. Sci. 2016, 84, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Fenix, K.; Wijesundara, D.K.; Cowin, A.J.; Grubor-Bauk, B.; Kopecki, Z. Immunological Memory in Imiquimod-Induced Murine Model of Psoriasiform Dermatitis. Int. J. Mol. Sci. 2020, 21, 7228. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Lian, N.; Liu, L.; Chen, M. Tapering and discontinuation of systemic medications in psoriasis patients with low disease activity. Dermatol. Ther. 2020, 33, e13599. [Google Scholar] [CrossRef]
- Piaserico, S.; Gisondi, P.; Simone, C.; Marinello, E.; Conti, A.; Amerio, P.; Peserico, A. Down-titration of Adalimumab and Etanercept in Psoriatic Patients: A Multicentre Observational Study. Acta Derm. Venereol. 2016, 96, 251–252. [Google Scholar] [CrossRef] [Green Version]
- Denton, C.; Khanna, D. Systemic Sclerosis. Lancet 2017, 390, 1685–1699. [Google Scholar] [CrossRef]
- Henriques, A.; Santiago, M.; Martinho, A.; Trindade, H.; Silva-Santos, B.; Henriques, M.J.; Da Silva, J.A.P.; Paiva, A.; Silva, C. Subset-specific alterations in frequencies and functional signatures of γδ T cells in systemic sclerosis patients. Inflamm. Res. 2016, 65, 985–994. [Google Scholar] [CrossRef]
- Ulanet, D.B.; Flavahan, N.A.; Casciola-Rosen, L.; Rosen, A. Selective cleavage of nucleolar autoantigen B23 by granzyme B in differentiated vascular smooth muscle cells: Insights into the association of specific autoantibodies with distinct disease phenotypes. Arthritis Rheum. 2004, 50, 233–241. [Google Scholar] [CrossRef]
- Mulligan-Kehoe, M.J.; Drinane, M.C.; Casciola-Rosen, L.; Hummers, L.K.; Hall, A.E.; Rosen, A.; Wigley, F.M.; Simons, M. Granzyme B-dependent matrix degradation generates anti-angiostatic activity in scleroderma patients. Vasc. Pharmacol. 2006, 45, e147. [Google Scholar] [CrossRef]
- Schachna, L.; Wigley, F.M.; Morris, S.; Gelber, A.C.; Rosen, A.; Casciola-Rosen, L. Recognition of Granzyme B-generated autoantigen fragments in scleroderma patients with ischemic digital loss. Arthritis Rheum. 2002, 46, 1873–1884. [Google Scholar] [CrossRef]
- Darrah, E.; Rosen, A. Granzyme B cleavage of autoantigens in autoimmunity. Cell Death Differ. 2010, 17, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Bergqvist, C.; Ezzedine, K. Vitiligo: A Review. Dermatology 2020, 236, 571–592. [Google Scholar] [CrossRef]
- Zhang, Y.; Cai, Y.; Shi, M.; Jiang, S.; Cui, S.; Wu, Y.; Gao, X.-H.; Chen, H.-D. The Prevalence of Vitiligo: A Meta-Analysis. PLoS ONE 2016, 11, e0163806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashighi, M.; Harris, J.E. Vitiligo Pathogenesis and Emerging Treatments. Dermatol. Clin. 2017, 35, 257–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krüger, C.; Schallreuter, K.U. A review of the worldwide prevalence of vitiligo in children/adolescents and adults. Int. J. Dermatol. 2012, 51, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, T.M.; Jin, Y.; Gowan, K.; Fain, P.R.; Spritz, R.A. Risk of Generalized Vitiligo Is Associated with the Common 55R-94A-247H Variant Haplotype of GZMB (Encoding Granzyme B). J. Investig. Dermatol. 2013, 133, 1677–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, K.-H.; Kim, S.K.; Seo, J.-K.; Shin, M.K.; Lee, M.-H. Association of GZMB polymorphisms and susceptibility to non-segmental vitiligo in a Korean population. Sci. Rep. 2021, 11, 397. [Google Scholar] [CrossRef]
- Xu, M.; Liu, Y.; Liu, Y.; Li, X.; Chen, G.; Dong, W.; Xiao, S. Genetic polymorphisms of GZMB and vitiligo: A genetic association study based on Chinese Han population. Sci. Rep. 2018, 8, 13001. [Google Scholar] [CrossRef] [Green Version]
- Tulic, M.K.; Cavazza, E.; Cheli, Y.; Jacquel, A.; Luci, C.; Cardot-Leccia, N.; Hadhiri-Bzioueche, H.; Abbe, P.; Gesson, M.; Sormani, L.; et al. Innate lymphocyte-induced CXCR3B-mediated melanocyte apoptosis is a potential initiator of T-cell autoreactivity in vitiligo. Nat. Commun. 2019, 10, 2178. [Google Scholar] [CrossRef] [Green Version]
- Boniface, K.; Jacquemin, C.; Darrigade, A.-S.; Dessarthe, B.; Martins, C.; Boukhedouni, N.; Vernisse, C.; Grasseau, A.; Thiolat, D.; Rambert, J.; et al. Vitiligo Skin Is Imprinted with Resident Memory CD8 T Cells Expressing CXCR3. J. Investig. Dermatol. 2017, 138, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wei, Y.; Sun, Y.; Shi, W.; Yang, J.; Zhu, L.; Li, M. Interferon-gamma Inhibits Melanogenesis and Induces Apoptosis in Melanocytes: A Pivotal Role of CD8+ Cytotoxic T Lymphocytes in Vitiligo. Acta Dermato-Venereologica 2015, 95, 664–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gleave, A.; Granville, D.J. Granzyme B in Autoimmune Skin Disease. Biomolecules 2023, 13, 388. https://doi.org/10.3390/biom13020388
Gleave A, Granville DJ. Granzyme B in Autoimmune Skin Disease. Biomolecules. 2023; 13(2):388. https://doi.org/10.3390/biom13020388
Chicago/Turabian StyleGleave, Anna, and David J. Granville. 2023. "Granzyme B in Autoimmune Skin Disease" Biomolecules 13, no. 2: 388. https://doi.org/10.3390/biom13020388
APA StyleGleave, A., & Granville, D. J. (2023). Granzyme B in Autoimmune Skin Disease. Biomolecules, 13(2), 388. https://doi.org/10.3390/biom13020388