



Computational Insights into the Allosteric Modulation of a Phthalate-Degrading Hydrolase by Distal Mutations


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Alignment and Co-Evolution Analysis
2.2. Homology Modeling and Molecular Docking
2.3. Conventional Molecular Dynamics Simulation
2.4. Trajectory Analyses
2.5. Disulfide-Bond Design
3. Results
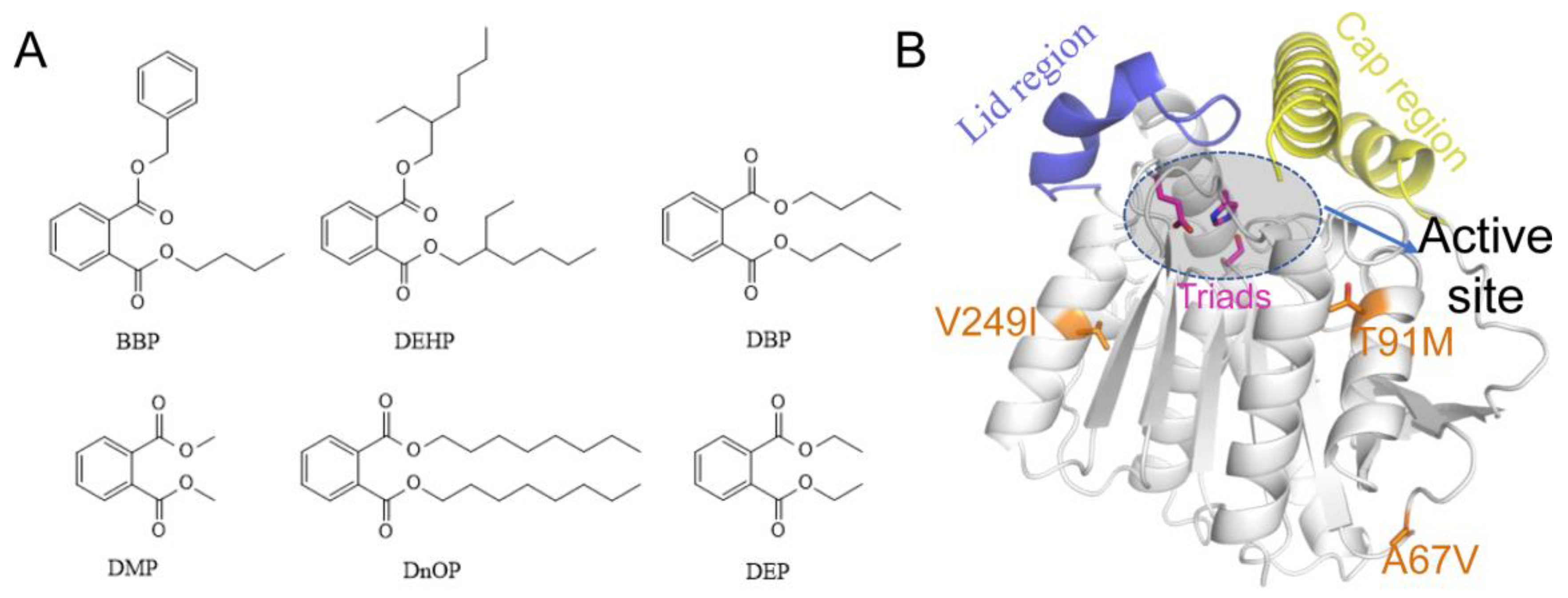
3.1. Sequential and Structural Analyses of EstJ6
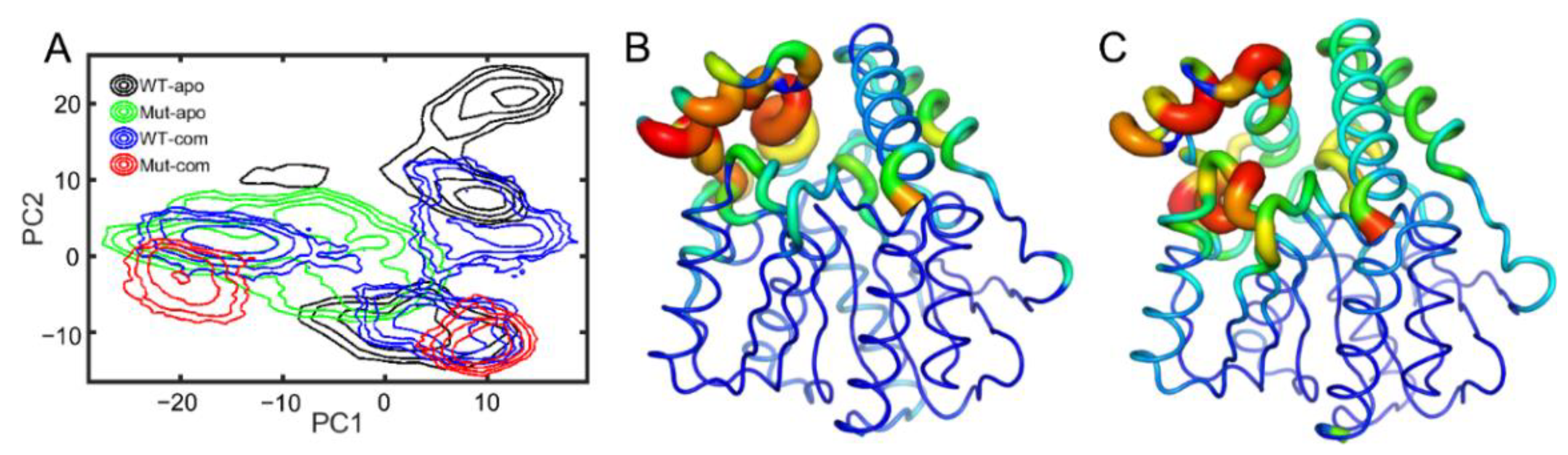
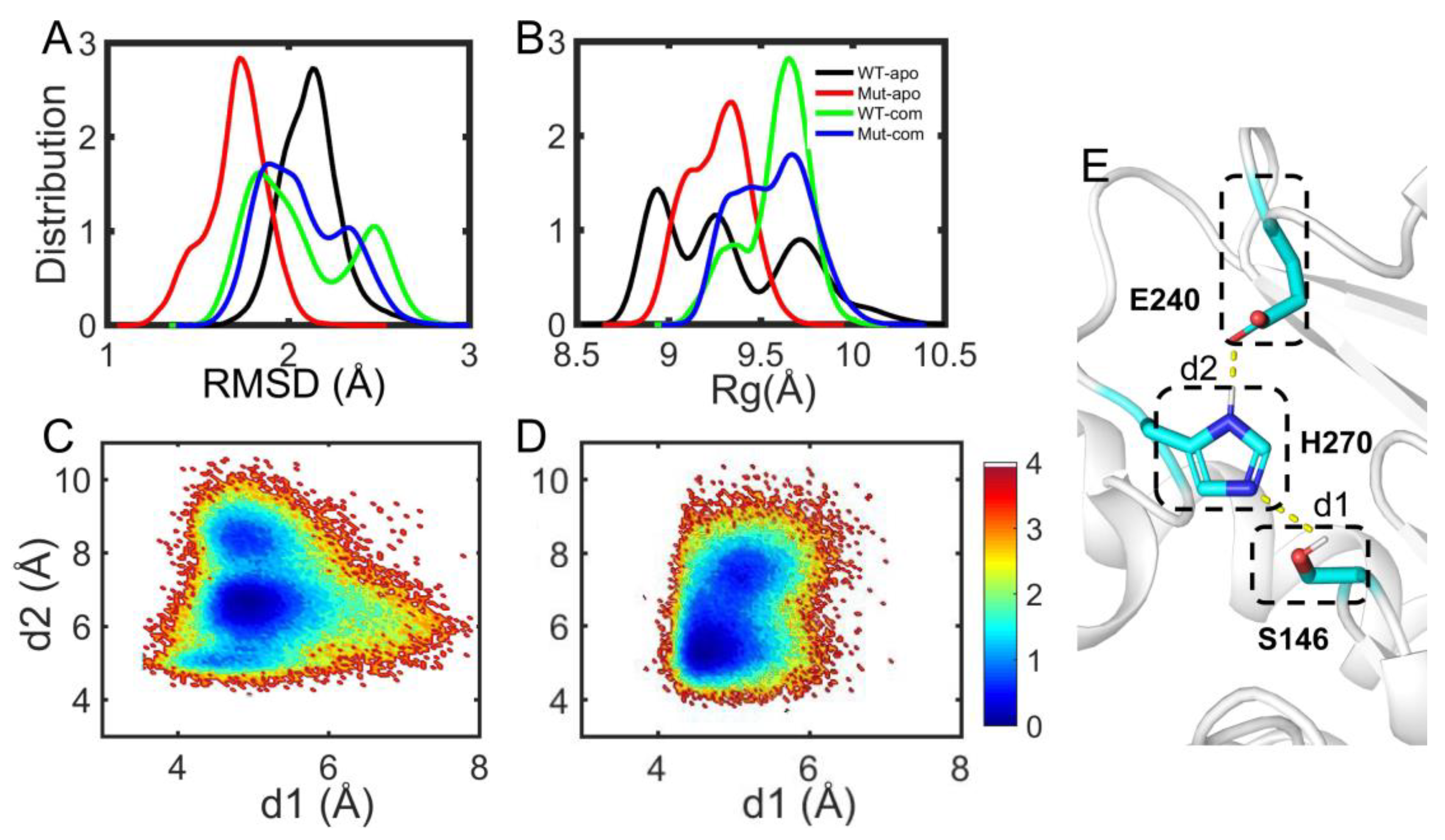
3.2. Global and Local Conformational Variations of EstJ6 Induced by Mutations and Substrate Binding
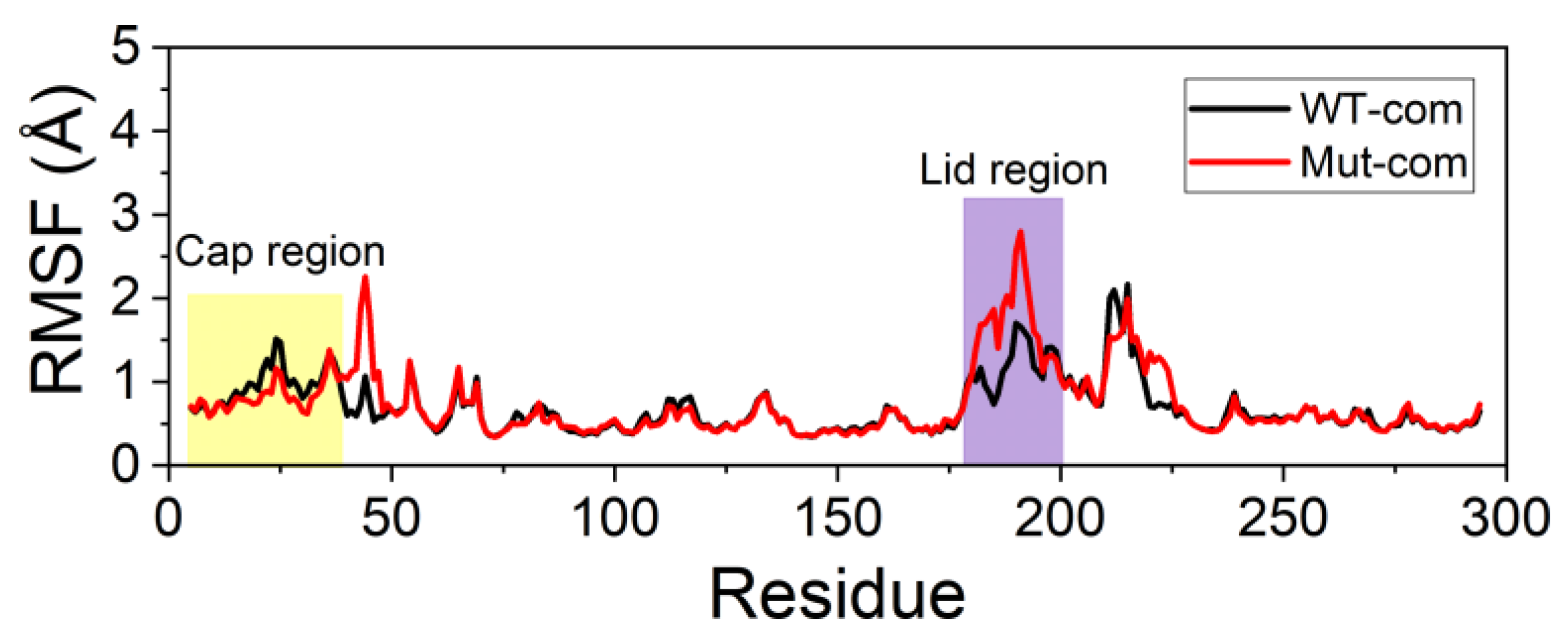
3.3. Mutations Increase the Flexibility of the Lid Region Resulting in a More Open Pocket
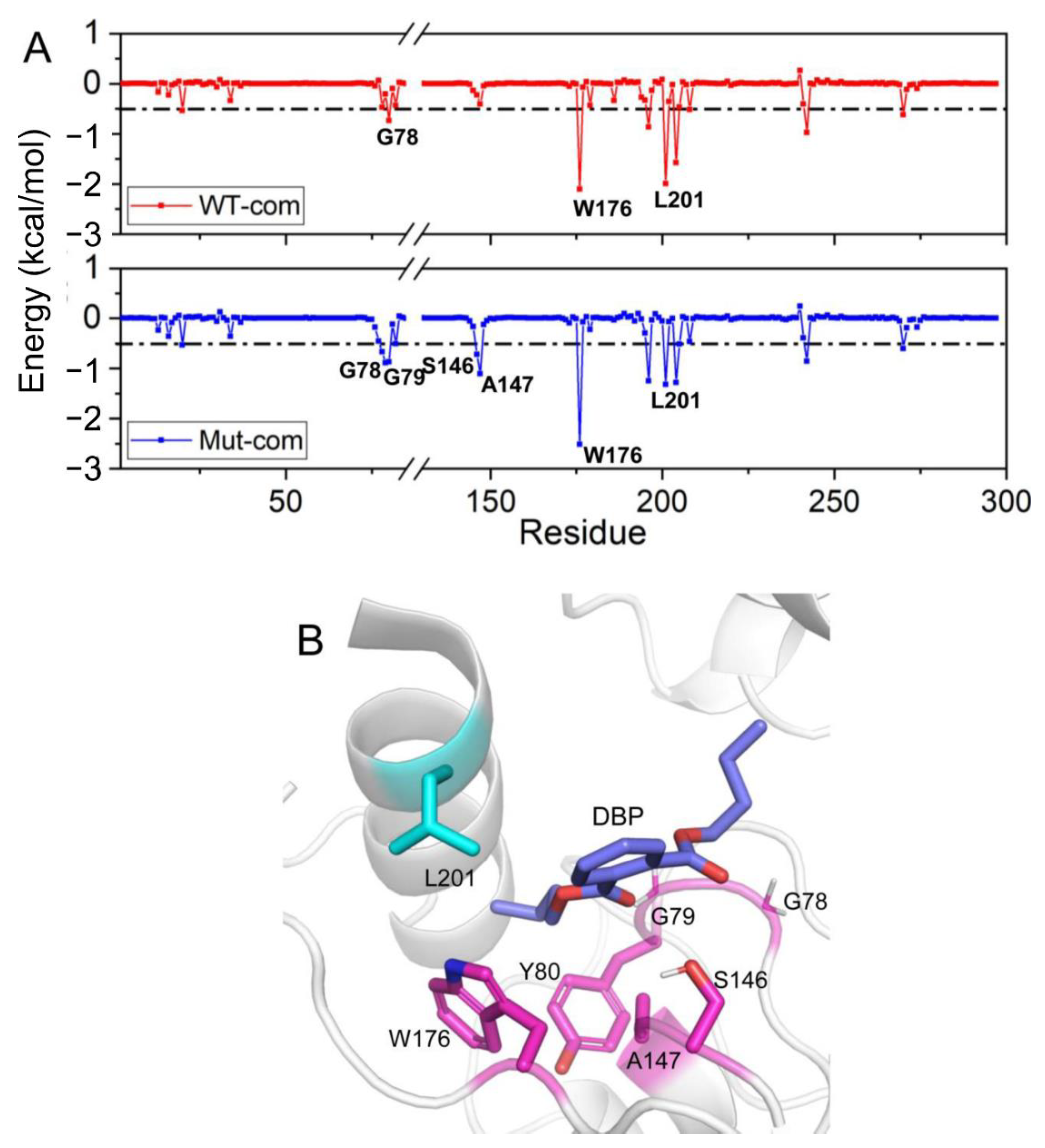
3.4. Effects of Distal Mutations on the Active Site
3.5. The Discrepancy of Residue-Residue Communications among All Systems
3.6. Distal Mutations Enhance the Binding of Substrate to EstJ6
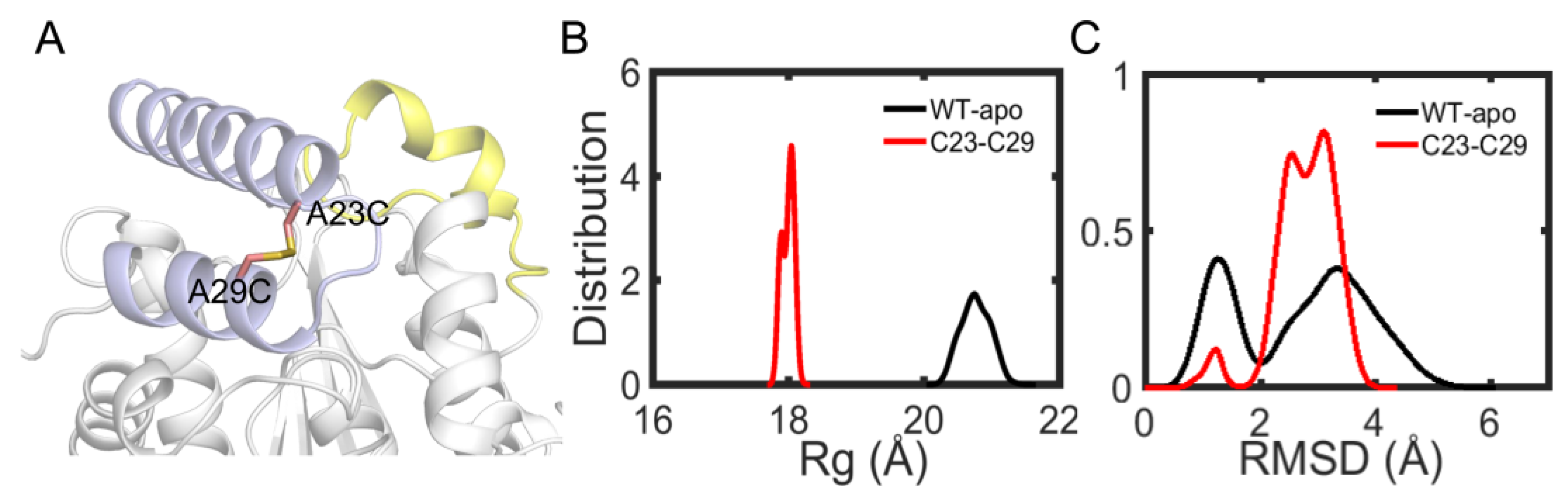
3.7. Rational Designed Disulfide Bonds Increase the Thermostability of EstJ6
4. Discussion
4.1. The Conformational Changes of the Lid Closely Related to the Activity of Esterases
4.2. The Potential and Challenge of Computation Biology in Rational Design of Proteins
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, X.; Sun, Z.; Chen, G.; Zhang, W.; Cai, Y.; Kong, R.; Wang, X.; Suo, Y.; You, J. Determination of phthalate esters in environmental water by magnetic Zeolitic Imidazolate Framework-8 solid-phase extraction coupled with high-performance liquid chromatography. J. Chromatogr. A 2015, 1409, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Liu, R.; Gao, H.; Tan, R.; Zeng, P.; Song, Y. Spatial distribution and ecological risk assessment of phthalic acid esters and phenols in surface sediment from urban rivers in Northeast China. Environ. Pollut. 2016, 219, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Theuerkorn, K.; Stelzer, N.; Gehre, M.; Thullner, M.; Richnow, H.H. Applicability of Stable Isotope Fractionation Analysis for the Characterization of Benzene Biodegradation in a BTEX-contaminated Aquifer. Environ. Sci. Technol. 2007, 41, 3689–3696. [Google Scholar] [CrossRef] [PubMed]
- Sayyad, G.; Price, G.; Sharifi, M.; Khosravi, K. Fate and transport modeling of phthalate esters from biosolid amended soil under corn cultivation. J. Hazard. Mater. 2017, 323, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Net, S.; Sempéré, R.; Delmont, A.; Paluselli, A.; Ouddane, B. Occurrence, Fate, Behavior and Ecotoxicological State of Phthalates in Different Environmental Matrices. Environ. Sci. Technol. 2015, 49, 4019–4035. [Google Scholar] [CrossRef]
- Xu, G.; Li, F.; Wang, Q. Occurrence and degradation characteristics of dibutyl phthalate (DBP) and di-(2-ethylhexyl) phthalate (DEHP) in typical agricultural soils of China. Sci. Total Environ. 2008, 393, 333–340. [Google Scholar] [CrossRef]
- Babu, B.; Wu, J.-T. Production of phthalate esters by nuisance freshwater algae and cyanobacteria. Sci. Total Environ. 2010, 408, 4969–4975. [Google Scholar] [CrossRef]
- Chen, C.Y. Biosynthesis of di-(2-ethylhexyl) phthalate (DEHP) and di-n-butyl phthalate (DBP) from red alga—Bangia atropurpurea. Water Res. 2004, 38, 1014–1018. [Google Scholar] [CrossRef]
- Meeker, J.D.; Sathyanarayana, S.; Swan, S. Phthalates and other additives in plastics: Human exposure and associated health outcomes. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 2097–2113. [Google Scholar] [CrossRef] [Green Version]
- Hauser, R.; Calafat, A. Phthalates and human health. Occup. Environ. Med. 2005, 62, 806–818. [Google Scholar] [CrossRef] [Green Version]
- Julinova, M.; Slavík, R. Removal of phthalates from aqueous solution by different adsorbents: A short review. J. Environ. Manag. 2012, 94, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Hu, R.-W.; Zhao, H.-M.; Huang, H.-B.; Xiang, L.; Liu, B.-L.; Feng, N.-X.; Li, H.; Li, Y.-W.; Cai, Q.-Y.; et al. Mechanistic insight into esterase-catalyzed hydrolysis of phthalate esters (PAEs) based on integrated multi-spectroscopic analyses and docking simulation. J. Hazard. Mater. 2020, 408, 124901. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.K.; Kundapur, R.; Shouche, Y.S.; Karegoudar, T.B. Degradation of Plasticizer Di-n-butylphthalate by Delftia sp. TBKNP-05. Curr. Microbiol. 2006, 52, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Dong, S.; Chen, D.; Rui, Q.; Guo, J.; Wang, D.; Jiang, J. Potential of esterase DmtH in transforming plastic additive dimethyl terephthalate to less toxic mono-methyl terephthalate. Ecotoxicol. Environ. Saf. 2019, 187, 109848. [Google Scholar] [CrossRef] [PubMed]
- Wrighton, K.H. Discovering antibiotics through soil metagenomics. Nat. Rev. Drug Discov. 2018, 17, 241. [Google Scholar] [CrossRef] [PubMed]
- Jansson, J.K.; Hofmockel, K.S. The soil microbiome—from metagenomics to metaphenomics. Curr. Opin. Microbiol. 2018, 43, 162–168. [Google Scholar] [CrossRef]
- Nahurira, R.; Ren, L.; Song, J.; Jia, Y.; Wang, J.; Fan, S.; Wang, H.; Yan, Y. Degradation of Di(2-Ethylhexyl) Phthalate by a Novel Gordonia alkanivorans Strain YC-RL2. Curr. Microbiol. 2017, 74, 309–319. [Google Scholar] [CrossRef]
- Qiu, J.; Zhang, Y.; Shi, Y.; Jiang, J.; Wu, S.; Li, L.; Shao, Y.; Xin, Z. Identification and characterization of a novel phthalate-degrading hydrolase from a soil metagenomic library. Ecotoxicol. Environ. Saf. 2020, 190, 110148. [Google Scholar] [CrossRef]
- Qiu, J.; Yang, H.; Shao, Y.; Li, L.; Sun, S.; Wang, L.; Tan, Y.; Xin, Z. Enhancing the activity and thermal stability of a phthalate-degrading hydrolase by random mutagenesis. Ecotoxicol. Environ. Saf. 2020, 209, 111795. [Google Scholar] [CrossRef]
- Maria-Solano, M.A.; Serrano-Hervás, E.; Romero-Rivera, A.; Iglesias-Fernández, J.; Osuna, S. Role of conformational dynamics in the evolution of novel enzyme function. Chem. Commun. 2018, 54, 6622–6634. [Google Scholar] [CrossRef] [Green Version]
- Osuna, S. The challenge of predicting distal active site mutations in computational enzyme design. WIREs Comput. Mol. Sci. 2020, 11, e1502. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, H.; Becker, A.; Palm, G.J.; Berndt, L.; Badenhorst, C.P.S.; Godehard, S.P.; Reisky, L.; Lammers, M.; Bornscheuer, U.T. Sequence-Based Prediction of Promiscuous Acyltransferase Activity in Hydrolases. Angew. Chem. Int. Ed. 2020, 59, 11607–11612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, L.; Aller, S.G. Tools and Procedures for Visualization of Proteins and Other Biomolecules. Curr. Protoc. Mol. Biol. 2015, 110, 19.12.1–19.12.47. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Ye, L.; Wang, X.; Wang, X.; Liu, H.; Qian, X.; Zhu, Y.; Yu, H. Molecular docking, molecular dynamics simulation, and structure-based 3D-QSAR studies on estrogenic activity of hydroxylated polychlorinated biphenyls. Sci. Total Environ. 2012, 441, 230–238. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Harvey, M.; De Fabritiis, G. An Implementation of the Smooth Particle Mesh Ewald Method on GPU Hardware. J. Chem. Theory Comput. 2009, 5, 2371–2377. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Sethi, A.; Eargle, J.; Black, A.A.; Luthey-Schulten, Z. Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. USA 2009, 106, 6620–6625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardini, M.; Dijkstra, B.W. α/β Hydrolase fold enzymes: The family keeps growing. Curr. Opin. Struct. Biol. 1999, 9, 732–737. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Torres, A.D.C.L.; de Lima, L.N.; Tardioli, P.W.; Júnior, R.D.S. Mathematical modeling of enzymatic syntheses of biosurfactants catalyzed by immobilized lipases. React. Kinet. Catal. Lett. 2020, 130, 699–712. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Mohamed, R.; Salleh, A.B.; Leow, T.C.; Yahaya, N.M.; Rahman, M.B.A. Site-directed mutagenesis: Role of lid region for T1 lipase specificity. Protein Eng. Des. Sel. 2018, 31, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Dehury, B.; Patra, M.C.; Maharana, J.; Sahu, J.; Sen, P.; Modi, M.K.; Choudhury, M.D.; Barooah, M. Structure-Based Computational Study of Two Disease Resistance Gene Homologues (Hm1 and Hm2) in Maize (Zea mays L.) with Implications in Plant-Pathogen Interactions. PLoS ONE 2014, 9, e97852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Guo, J.; Li, L.; Liu, X.; Yao, X.; Liu, H. The solvent at antigen-binding site regulated C3d–CR2 interactions through the C-terminal tail of C3d at different ion strengths: Insights from molecular dynamics simulation. Biochim. et Biophys. Acta (BBA)-Gen. Subj. 2016, 1860, 2220–2231. [Google Scholar] [CrossRef]
- Guo, J.; Zhou, H.-X. Allosteric activation of SENP1 by SUMO1 β-grasp domain involves a dock-and-coalesce mechanism. eLife 2016, 5, e18249. [Google Scholar] [CrossRef]
- Li, M.; Xu, Y.; Guo, J. Insights into the negative regulation of EGFR upon the binding of an allosteric inhibitor. Chem. Biol. Drug Des. 2022, 99, 650–661. [Google Scholar] [CrossRef]
- Hünenberger, P.; Mark, A.; Van Gunsteren, W. Fluctuation and Cross-correlation Analysis of Protein Motions Observed in Nanosecond Molecular Dynamics Simulations. J. Mol. Biol. 1995, 252, 492–503. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yu, X.; Xu, Y. Improvement of catalytic activity of Aspergillus terreus lipase by site-directed mutagenesis. Sheng Wu Gong Cheng Xue Bao = Chin. J. Biotechnol. 2018, 34, 1091–1105. [Google Scholar]
- Eargle, J.; Luthey-Schulten, Z. NetworkView: 3D display and analysis of protein{middle dot}RNA interaction networks. Bioinformatics 2012, 28, 3000–3001. [Google Scholar] [CrossRef] [Green Version]
- Ning, L.; Guo, J.; Jin, N.; Liu, H.; Yao, X. The role of Cys179–Cys214 disulfide bond in the stability and folding of prion protein: Insights from molecular dynamics simulations. J. Mol. Model. 2014, 20, 1–8. [Google Scholar] [CrossRef]
- Secundo, F.; Carrea, G.; Tarabiono, C.; Gatti-Lafranconi, P.; Brocca, S.; Lotti, M.; Jaeger, K.-E.; Puls, M.; Eggert, T. The lid is a structural and functional determinant of lipase activity and selectivity. J. Mol. Catal. B Enzym. 2006, 39, 166–170. [Google Scholar] [CrossRef]
- Barbe, S.; Lafaquière, V.; Guieysse, D.; Monsan, P.; Remaud-Siméon, M.; André, I. Insights into lid movements of Burkholderia cepacia lipase inferred from molecular dynamics simulations. Proteins: Struct. Funct. Bioinform. 2009, 77, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Pleiss, J.; Fischer, M.; Schmid, R.D. Anatomy of lipase binding sites: The scissile fatty acid binding site. Chem. Phys. Lipids 1998, 93, 67–80. [Google Scholar] [CrossRef]
- Karkhane, A.A.; Yakhchali, B.; Jazii, F.R.; Bambai, B. The effect of substitution of Phe181 and Phe182 with Ala on activity, substrate specificity and stabilization of substrate at the active site of Bacillus thermocatenulatus lipase. J. Mol. Catal. B Enzym. 2009, 61, 162–167. [Google Scholar] [CrossRef]
- Panizza, P.; Cesarini, S.; Diaz, P.; Giordano, S.R. Saturation mutagenesis in selected amino acids to shift Pseudomonas sp. acidic lipase Lip I.3 substrate specificity and activity. Chem. Commun. 2015, 51, 1330–1333. [Google Scholar] [PubMed]
- Ren, L.-Q.; Chang, T.-T.; Ren, D.-P.; Zhou, Y.; Ye, B.-C. Rational design to improve activity of the Est3563 esterase from Acinetobacter sp. LMB-5. Enzym. Microb. Technol. 2019, 131, 109331. [Google Scholar] [CrossRef]
- Murphy, P.M.; Bolduc, J.M.; Gallaher, J.L.; Stoddard, B.L.; Baker, D. Alteration of enzyme specificity by computational loop remodeling and design. Proc. Natl. Acad. Sci. USA 2009, 106, 9215–9220. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Wijma, H.J.; Song, L.; Cui, Y.; Otzen, M.; Tian, Y.; Du, J.; Li, T.; Niu, D.; Chen, Y.; et al. Computational redesign of enzymes for regio- and enantioselective hydroamination. Nat. Chem. Biol. 2018, 14, 664–670. [Google Scholar] [CrossRef]
- Kuhlman, B.; Dantas, G.; Ireton, G.C.; Varani, G.; Stoddard, B.L.; Baker, D. Design of a Novel Globular Protein Fold with Atomic-Level Accuracy. Science 2003, 302, 1364–1368. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Althoff, E.A.; Clemente, F.R.; Doyle, L.; Röthlisberger, D.; Zanghellini, A.; Gallaher, J.L.; Betker, J.L.; Tanaka, F.; Barbas, C.F.; et al. De Novo Computational Design of Retro-Aldol Enzymes. Science 2008, 319, 1387–1391. [Google Scholar] [CrossRef] [Green Version]
- Röthlisberger, D.; Khersonsky, O.; Wollacott, A.M.; Jiang, L.; DeChancie, J.; Betker, J.; Gallaher, J.L.; Althoff, E.A.; Zanghellini, A.; Dym, O.; et al. Kemp elimination catalysts by computational enzyme design. Nature 2008, 453, 190–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyukhtenko, S.; Rajarshi, G.; Karageorgos, I.; Zvonok, N.; Gallagher, E.S.; Huang, H.; Vemuri, K.; Hudgens, J.W.; Ma, X.; Nasr, M.L.; et al. Effects of Distal Mutations on the Structure, Dynamics and Catalysis of Human Monoacylglycerol Lipase. Sci. Rep. 2018, 8, 1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehr, D.D.; Schnell, J.R.; McElheny, D.; Bae, S.-H.; Duggan, B.; Benkovic, S.J.; Dyson, H.J.; Wright, P.E. A Distal Mutation Perturbs Dynamic Amino Acid Networks in Dihydrofolate Reductase. Biochemistry 2013, 52, 4605–4619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragland, D.A.; Nalivaika, E.A.; Nalam, M.N.L.; Prachanronarong, K.L.; Cao, H.; Bandaranayake, R.M.; Cai, Y.; Kurt-Yilmaz, N.; Schiffer, C.A. Drug Resistance Conferred by Mutations Outside the Active Site through Alterations in the Dynamic and Structural Ensemble of HIV-1 Protease. J. Am. Chem. Soc. 2014, 136, 11956–11963. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, F.A.; Withers, S.G. Teaching old enzymes new tricks: Engineering and evolution of glycosidases and glycosyl transferases for improved glycoside synthesisThis paper is one of a selection of papers published in this Special Issue, entitled CSBMCB—Systems and Chemical Biology, and has undergone the Journal’s usual peer review process. Biochem. Cell Biol. 2008, 86, 169–177. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contributions | WT-com (kcal/mol) | Mut-com (kcal/mol) |
|---|---|---|
| ΔEvdw | −41.34 ± 4.24 | −44.50 ± 0.55 |
| ΔEele | −7.37 ± 6.98 | −12.07 ± 10.13 |
| ΔGpol,sol | 19.85 ± 6.35 | 23.02 ± 5.83 |
| ΔGnpol,sol | −5.83 ± 0.43 | −6.24 ± 0.09 |
| ΔEMM | −48.71 ± 9.14 | −56.57 ± 10.19 |
| ΔGsol | 14.02 ± 6.26 | 16.78 ± 5.92 |
| ΔGtotal | −34.68 ± 4.47 | −39.79 ± 4.70 |
| Docking score | −5.5 ± 2.12 | −6.45 ± 0.92 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, R.; Bao, Y.; Li, M.; Zhang, Y.; Xi, L.; Guo, J. Computational Insights into the Allosteric Modulation of a Phthalate-Degrading Hydrolase by Distal Mutations. Biomolecules 2023, 13, 443. https://doi.org/10.3390/biom13030443
Xu R, Bao Y, Li M, Zhang Y, Xi L, Guo J. Computational Insights into the Allosteric Modulation of a Phthalate-Degrading Hydrolase by Distal Mutations. Biomolecules. 2023; 13(3):443. https://doi.org/10.3390/biom13030443
Chicago/Turabian StyleXu, Ran, Yiqiong Bao, Mengrong Li, Yan Zhang, Lili Xi, and Jingjing Guo. 2023. "Computational Insights into the Allosteric Modulation of a Phthalate-Degrading Hydrolase by Distal Mutations" Biomolecules 13, no. 3: 443. https://doi.org/10.3390/biom13030443
APA StyleXu, R., Bao, Y., Li, M., Zhang, Y., Xi, L., & Guo, J. (2023). Computational Insights into the Allosteric Modulation of a Phthalate-Degrading Hydrolase by Distal Mutations. Biomolecules, 13(3), 443. https://doi.org/10.3390/biom13030443