

Quality Control—A Stepchild in Quantitative Proteomics: A Case Study for the Human CSF Proteome

 , , ,
, , ,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Standard In-Solution Digestion
2.3. Rapid In-Solution Digestion
2.4. Standard Filter-Aided Sample Preparation (FASP)
2.5. Rapid FASP
2.6. Label-Free NanoLC-MS with DDA Aquisition
2.7. Data Analysis
3. Results
3.1. Impact of Sample Preparation on Peptide Recovery
3.2. Raw Data Quality Assessment (Stage 1)
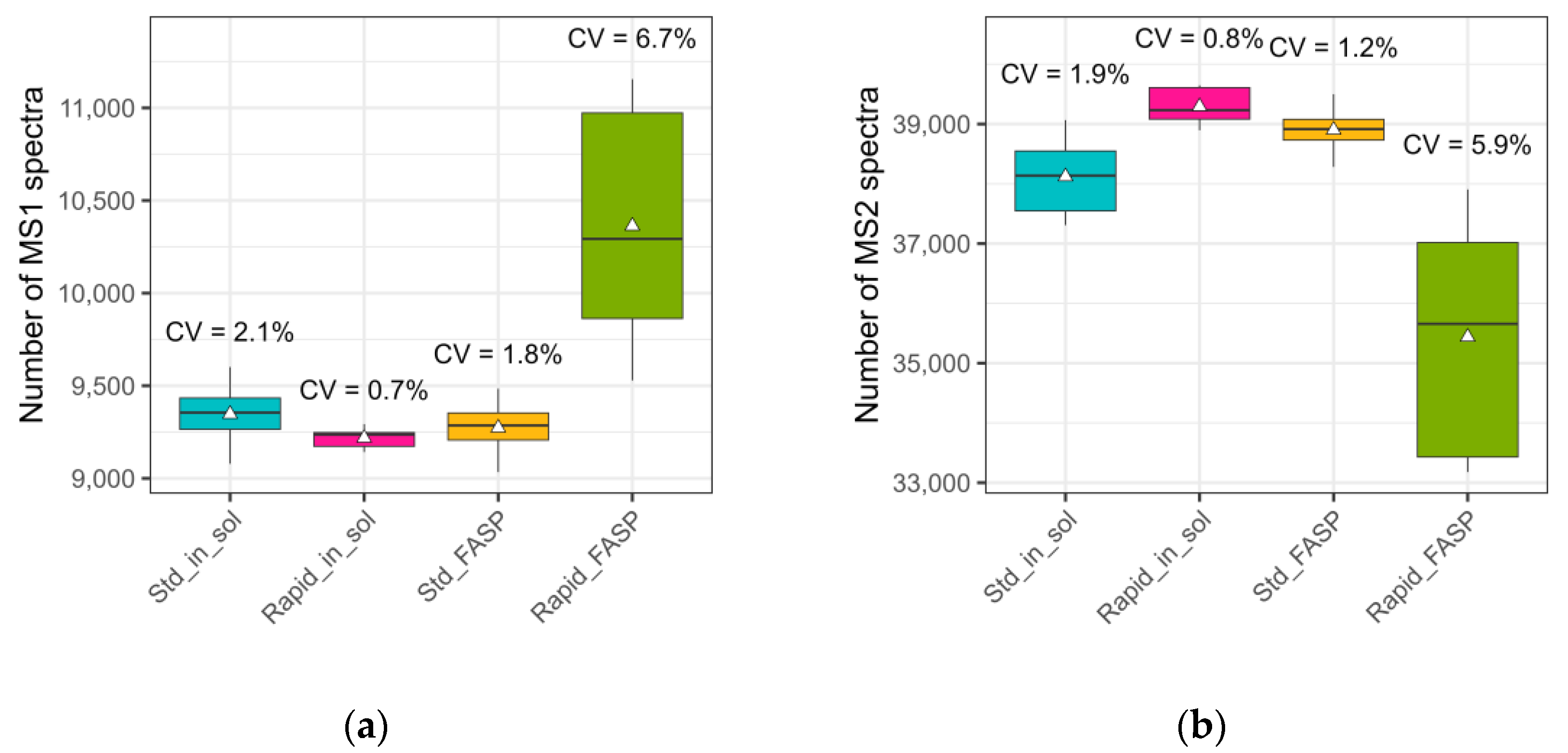
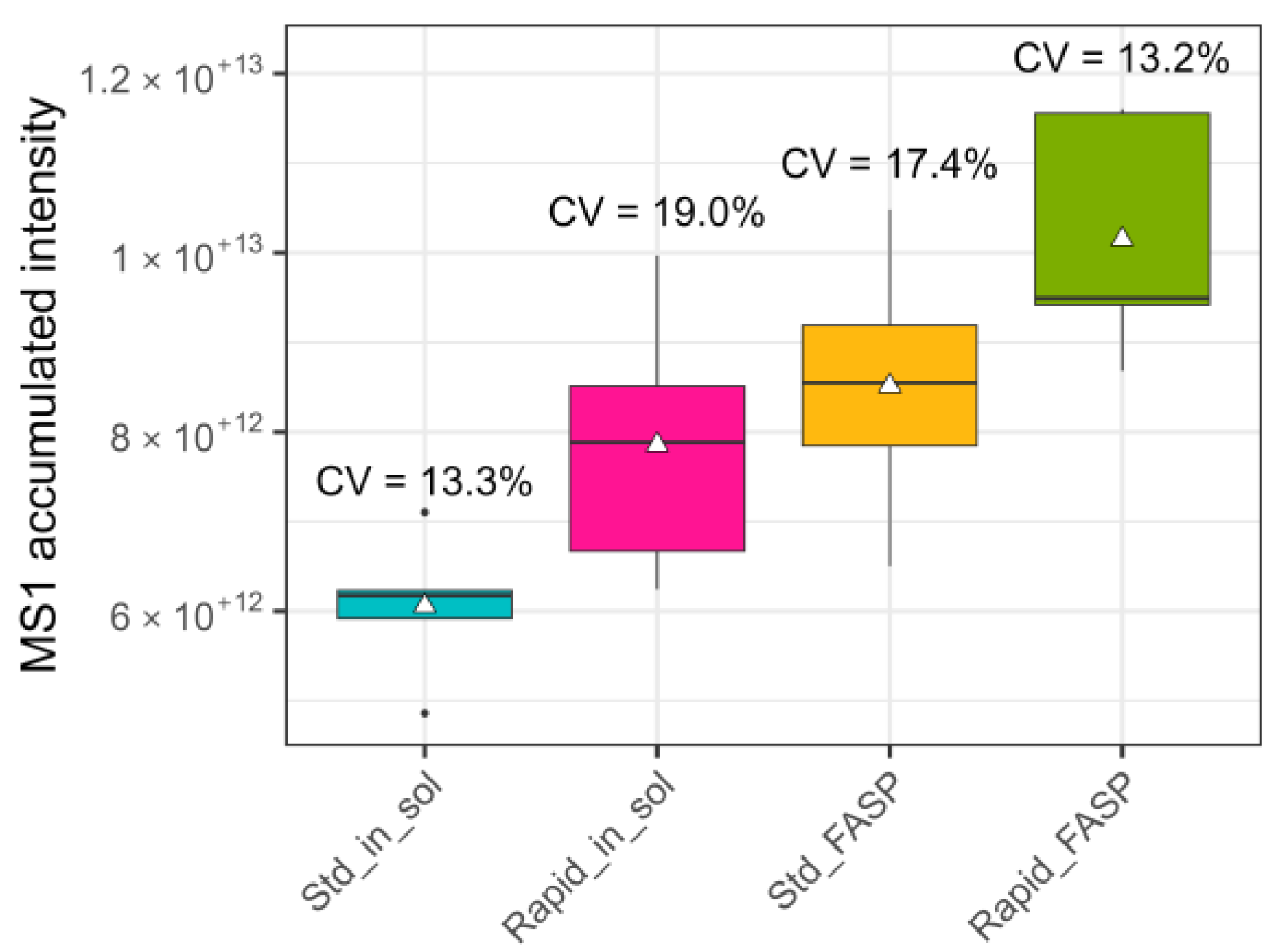
- Number of acquired MS1 and MS2 spectra;
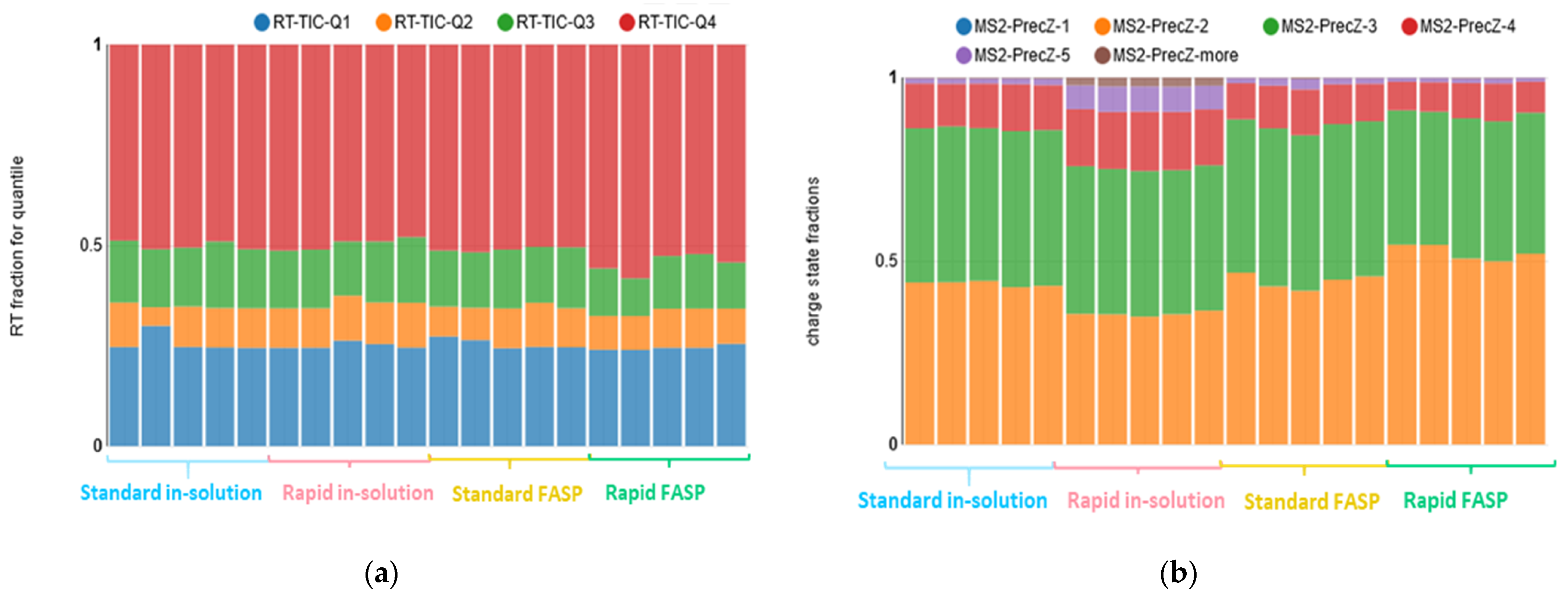
- Ion chromatogram profile;
- Distribution of precursor charge states.
3.2.1. NanoLC-MS: Total Number of Scan Events
3.2.2. NanoLC-MS: Ion Chromatogram and Charge State Distribution Evaluation
3.3. Identification Quality Assessment
- Number of FDR-controlled peptide spectrum matches (PSMs), peptides, and protein groups (PGs);
- Percentage of missed cleavage states.
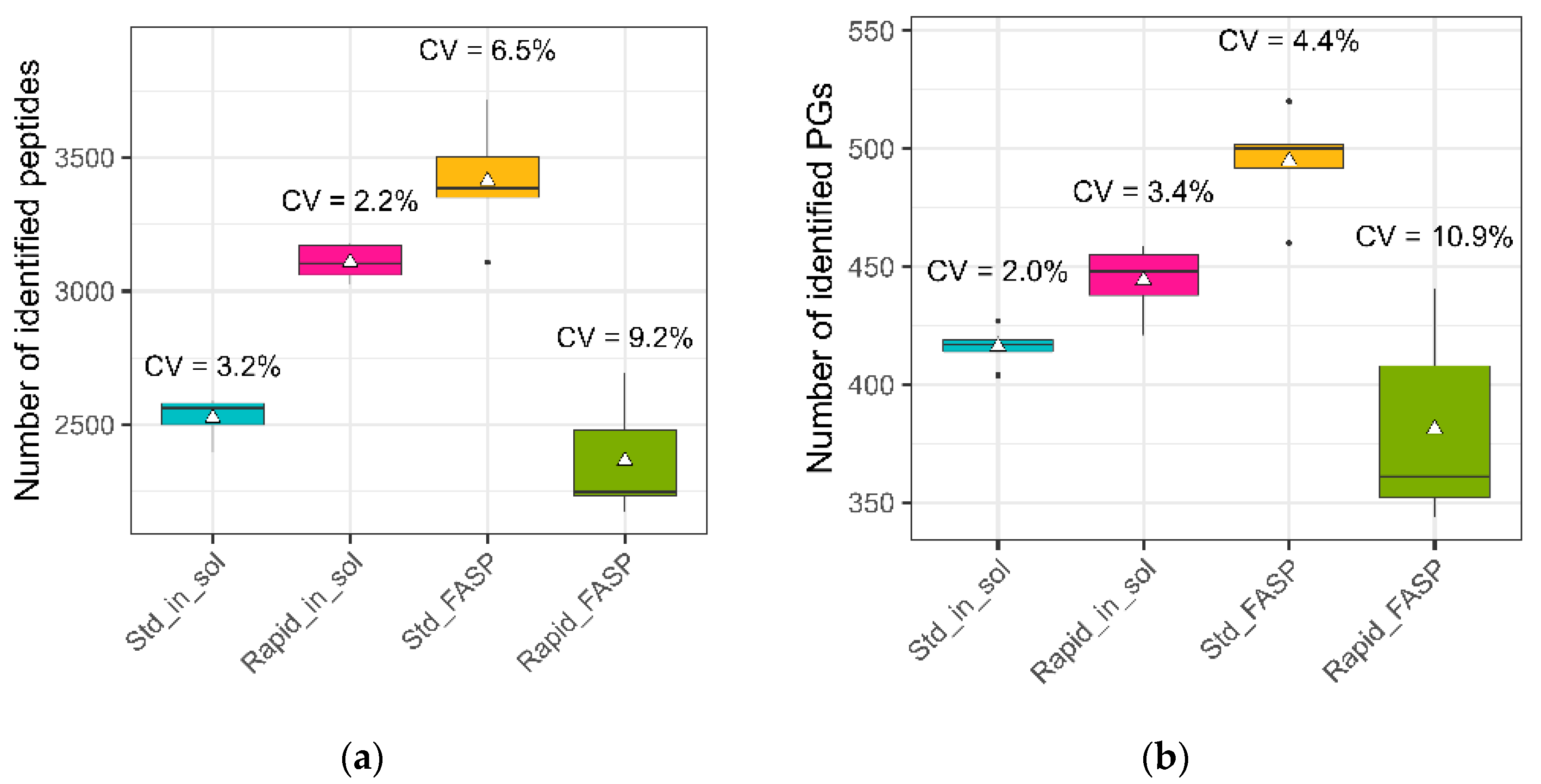
3.3.1. Number of Identified Peptides/Protein Groups (PGs)
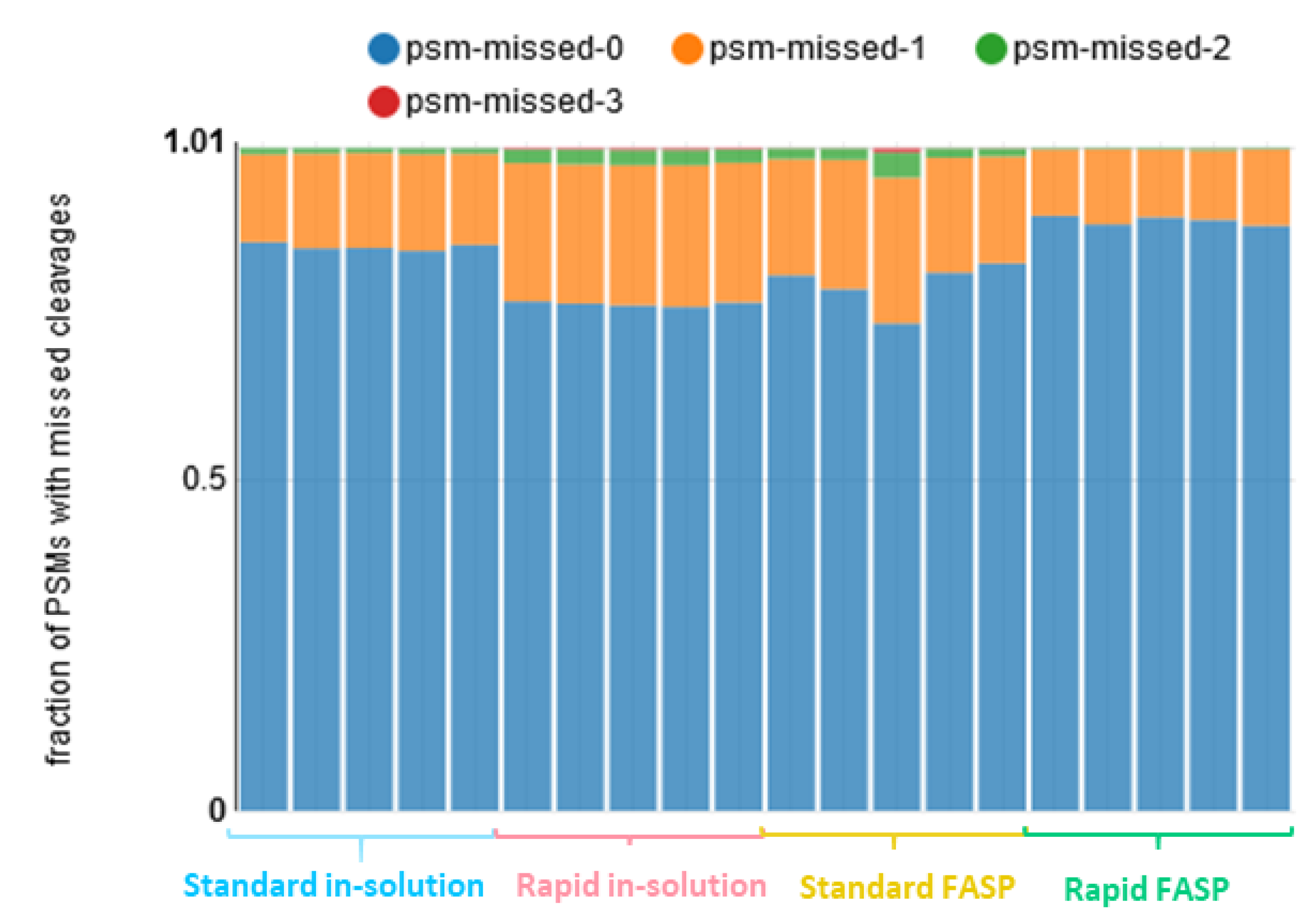
3.3.2. Peptide Identification Quality Assessment: Missed Cleavages
3.4. QC for Label-Free Quantification
- Number of quantified peptides/protein groups and data completeness;
- Similarity of the replicates based on clustering;
- Efficiency of data normalization.
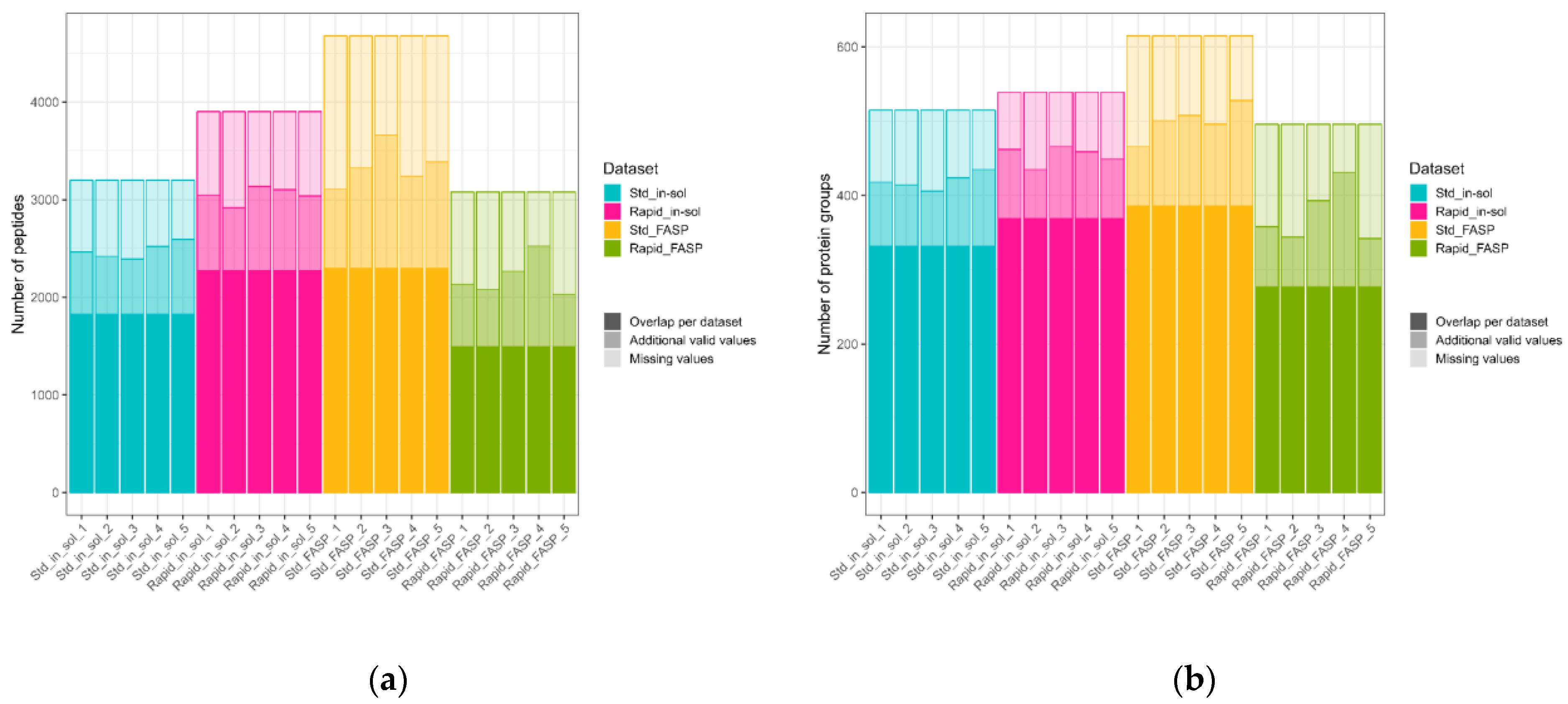
3.4.1. Number of Quantified Peptides/Protein Groups
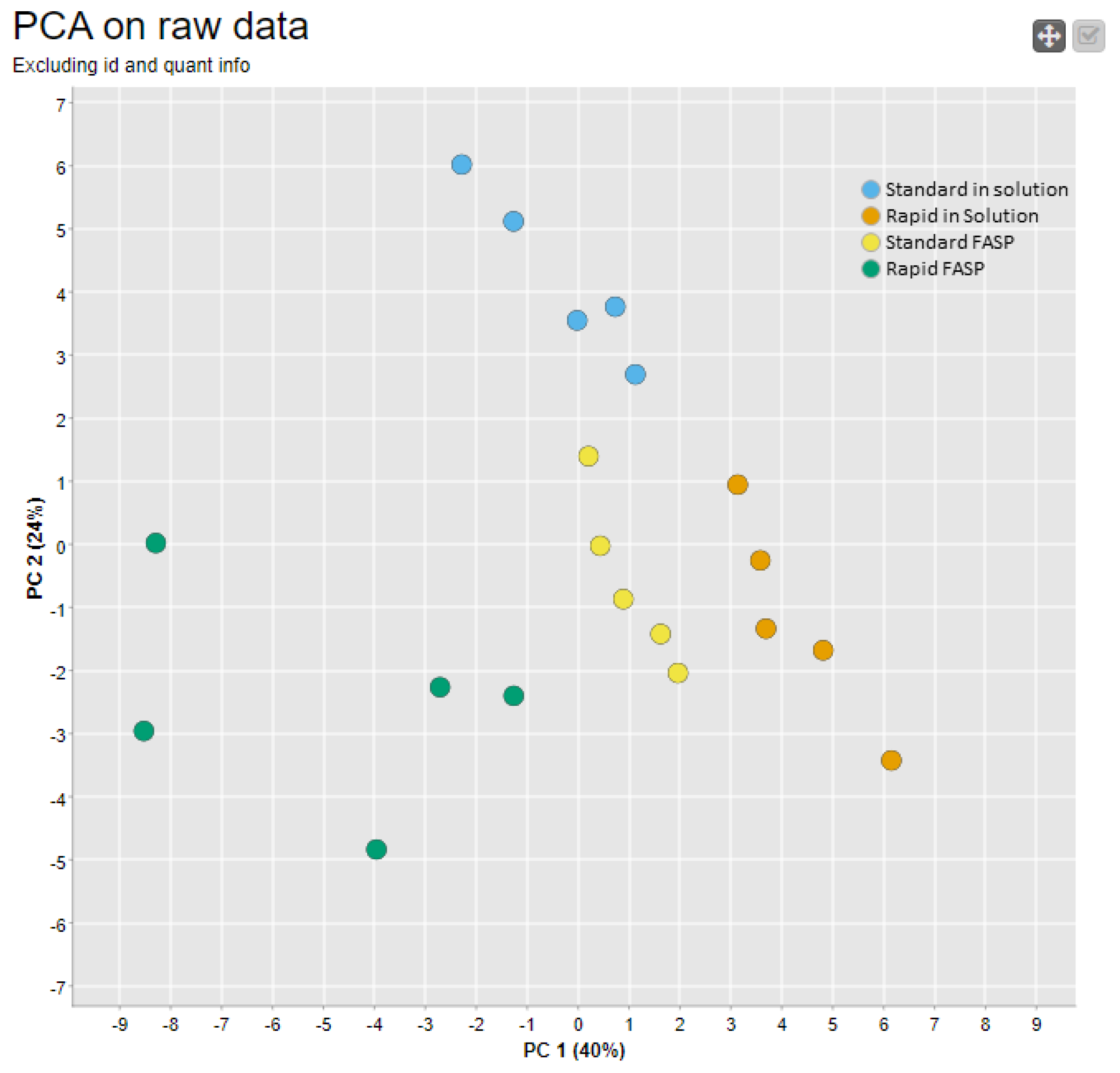
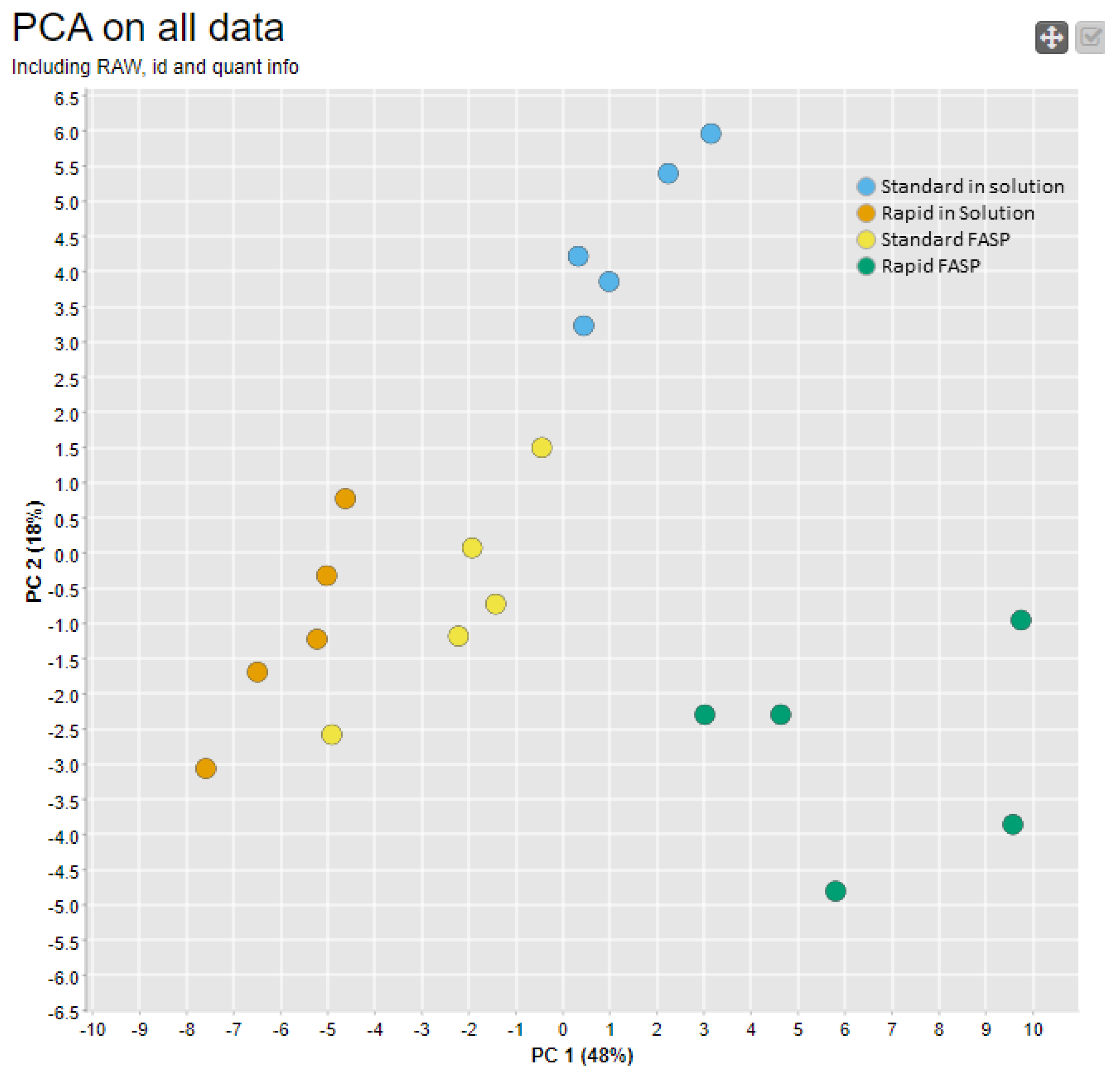
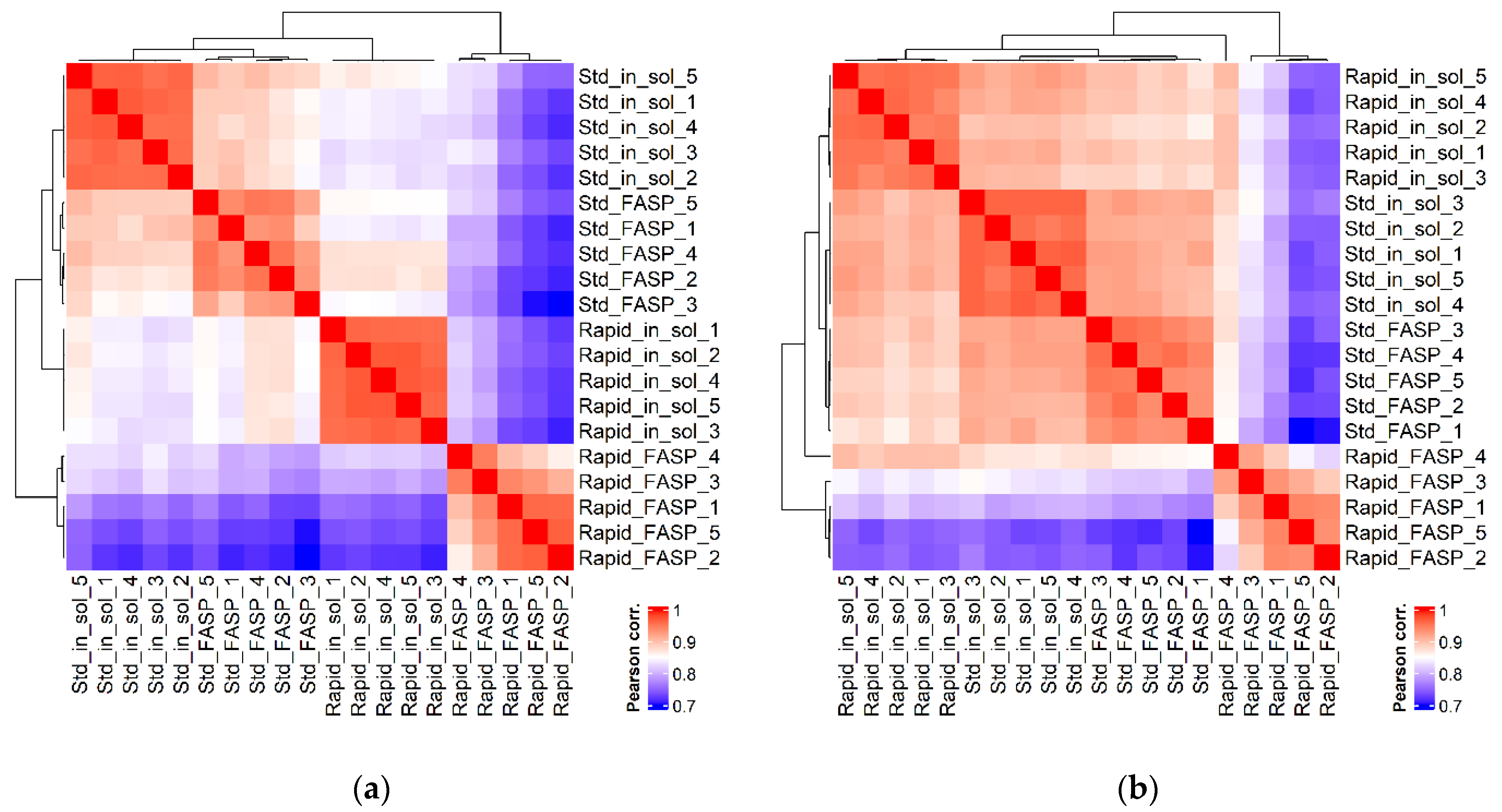
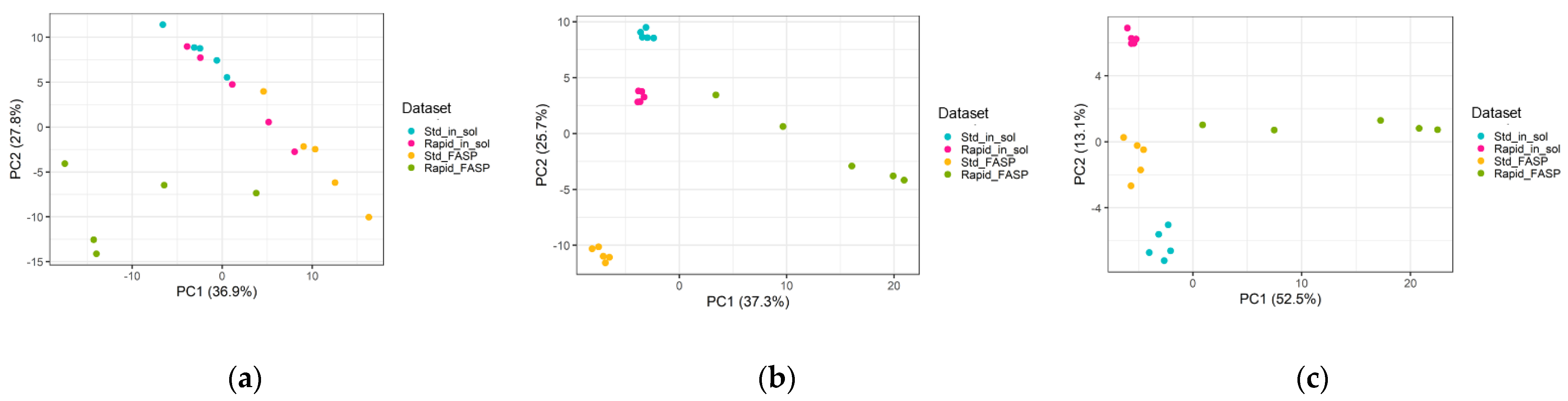
3.4.2. Replicate Similarity Based on Correlation
3.4.3. Assessment of Data Normalization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McDonald, W.H.; Yates, J.R. Shotgun proteomics and biomarker discovery. Dis. Mrk. 2002, 18, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajic, T.; Liu, Y.; Aebersold, R. Using data-independent, high-resolution mass spectrometry in protein biomarker research: Perspectives and clinical applications. Proteom. Clin. Appl. 2015, 9, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef]
- Rozanova, S.; Barkovits, K.; Nikolov, M.; Schmidt, C.; Urlaub, H.; Marcus, K. Quantitative Mass Spectrometry-Based Proteomics: An Overview. Methods Mol. Biol. 2021, 2228, 85–116. [Google Scholar] [CrossRef]
- Dupree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A Critical Review of Bottom-Up Proteomics: The Good, the Bad, and the Future of this Field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Salvagno, G.L.; Danese, E.; Lippi, G. Preanalytical variables for liquid chromatography-mass spectrometry (LC-MS) analysis of human blood specimens. Clin. Biochem. 2017, 50, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, K.R.; Schroll, M.M.; Hummon, A.B. Comparison of In-Solution, FASP, and S-Trap Based Digestion Methods for Bottom-Up Proteomic Studies. J. Proteome Res. 2018, 17, 2480–2490. [Google Scholar] [CrossRef]
- Piehowski, P.D.; Petyuk, V.A.; Orton, D.J.; Xie, F.; Moore, R.J.; Ramirez-Restrepo, M.; Engel, A.; Lieberman, A.P.; Albin, R.L.; Camp, D.G.; et al. Sources of technical variability in quantitative LC-MS proteomics: Human brain tissue sample analysis. J. Proteome Res. 2013, 12, 2128–2137. [Google Scholar] [CrossRef] [Green Version]
- Bittremieux, W.; Tabb, D.L.; Impens, F.; Staes, A.; Timmerman, E.; Martens, L.; Laukens, K. Quality control in mass spectrometry-based proteomics. Mass Spectrom. Rev. 2018, 37, 697–711. [Google Scholar] [CrossRef]
- Hühmer, A.F.; Biringer, R.G.; Amato, H.; Fonteh, A.N.; Harrington, M.G. Protein analysis in human cerebrospinal fluid: Physiological aspects, current progress and future challenges. Dis. Mrk. 2006, 22, 3–26. [Google Scholar] [CrossRef] [Green Version]
- Schulenborg, T.; Schmidt, O.; van Hall, A.; Meyer, H.E.; Hamacher, M.; Marcus, K. Proteomics in neurodegeneration--disease driven approaches. J. Neural Transm. 2006, 113, 1055–1073. [Google Scholar] [CrossRef] [PubMed]
- van Steenoven, I.; Koel-Simmelink, M.J.A.; Vergouw, L.J.M.; Tijms, B.M.; Piersma, S.R.; Pham, T.V.; Bridel, C.; Ferri, G.L.; Cocco, C.; Noli, B.; et al. Identification of novel cerebrospinal fluid biomarker candidates for dementia with Lewy bodies: A proteomic approach. Mol. Neurodegener. 2020, 15, 36. [Google Scholar] [CrossRef] [PubMed]
- Li, K.W.; Ganz, A.B.; Smit, A.B. Proteomics of neurodegenerative diseases: Analysis of human post-mortem brain. J. Neurochem. 2019, 151, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Schilde, L.M.; Kösters, S.; Steinbach, S.; Schork, K.; Eisenacher, M.; Galozzi, S.; Turewicz, M.; Barkovits, K.; Mollenhauer, B.; Marcus, K.; et al. Protein variability in cerebrospinal fluid and its possible implications for neurological protein biomarker research. PLoS ONE 2018, 13, e0206478. [Google Scholar] [CrossRef] [Green Version]
- Mollenhauer, B.; Locascio, J.J.; Schulz-Schaeffer, W.; Sixel-Döring, F.; Trenkwalder, C.; Schlossmacher, M.G. α-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: A cohort study. Lancet Neurol. 2011, 10, 230–240. [Google Scholar] [CrossRef]
- Barkovits, K.; Kruse, N.; Linden, A.; Tönges, L.; Pfeiffer, K.; Mollenhauer, B.; Marcus, K. Blood Contamination in CSF and Its Impact on Quantitative Analysis of Alpha-Synuclein. Cells 2020, 9, 370. [Google Scholar] [CrossRef] [Green Version]
- Boja, E.S.; Fales, H.M. Overalkylation of a protein digest with iodoacetamide. Anal. Chem. 2001, 73, 3576–3582. [Google Scholar] [CrossRef]
- Krokhin, O.V.; Antonovici, M.; Ens, W.; Wilkins, J.A.; Standing, K.G. Deamidation of -Asn-Gly- sequences during sample preparation for proteomics: Consequences for MALDI and HPLC-MALDI analysis. Anal. Chem. 2006, 78, 6645–6650. [Google Scholar] [CrossRef]
- Proc, J.L.; Kuzyk, M.A.; Hardie, D.B.; Yang, J.; Smith, D.S.; Jackson, A.M.; Parker, C.E.; Borchers, C.H. A quantitative study of the effects of chaotropic agents, surfactants, and solvents on the digestion efficiency of human plasma proteins by trypsin. J. Proteome Res. 2010, 9, 5422–5437. [Google Scholar] [CrossRef] [Green Version]
- Nitride, C.; Nørgaard, J.; Omar, J.; Emons, H.; Esteso, M.M.; O’Connor, G. An assessment of the impact of extraction and digestion protocols on multiplexed targeted protein quantification by mass spectrometry for egg and milk allergens. Anal. Bioanal. Chem. 2019, 411, 3463–3475. [Google Scholar] [CrossRef] [Green Version]
- Chick, J.M.; Kolippakkam, D.; Nusinow, D.P.; Zhai, B.; Rad, R.; Huttlin, E.L.; Gygi, S.P. A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides. Nat. Biotechnol. 2015, 33, 743–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.C.; Yates, J.R. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, A.W.; Deutsch, E.W.; Au, C.E.; Kearney, R.E.; Beavis, R.; Sechi, S.; Nilsson, T.; Bergeron, J.J.; Group, H.T.S.W. A HUPO test sample study reveals common problems in mass spectrometry-based proteomics. Nat. Methods 2009, 6, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Sandin, M.; Teleman, J.; Malmström, J.; Levander, F. Data processing methods and quality control strategies for label-free LC-MS protein quantification. Biochim. Biophys. Acta 2014, 1844, 29–41. [Google Scholar] [CrossRef]
- Välikangas, T.; Suomi, T.; Elo, L.L. A systematic evaluation of normalization methods in quantitative label-free proteomics. Brief. Bioinform. 2018, 19, 1–11. [Google Scholar] [CrossRef]
- Čuklina, J.; Lee, C.H.; Williams, E.G.; Sajic, T.; Collins, B.C.; Rodríguez Martínez, M.; Sharma, V.S.; Wendt, F.; Goetze, S.; Keele, G.R.; et al. Diagnostics and correction of batch effects in large-scale proteomic studies: A tutorial. Mol. Syst. Biol. 2021, 17, e10240. [Google Scholar] [CrossRef]
- Abbatiello, S.; Ackermann, B.L.; Borchers, C.; Bradshaw, R.A.; Carr, S.A.; Chalkley, R.; Choi, M.; Deutsch, E.; Domon, B.; Hoofnagle, A.N.; et al. New Guidelines for Publication of Manuscripts Describing Development and Application of Targeted Mass Spectrometry Measurements of Peptides and Proteins. Mol. Cell. Proteom. 2017, 16, 327–328. [Google Scholar] [CrossRef] [Green Version]
- Chiva, C.; Ortega, M.; Sabidó, E. Influence of the digestion technique, protease, and missed cleavage peptides in protein quantitation. J. Proteome Res. 2014, 13, 3979–3986. [Google Scholar] [CrossRef]
- Rudnick, P.A.; Clauser, K.R.; Kilpatrick, L.E.; Tchekhovskoi, D.V.; Neta, P.; Blonder, N.; Billheimer, D.D.; Blackman, R.K.; Bunk, D.M.; Cardasis, H.L.; et al. Performance metrics for liquid chromatography-tandem mass spectrometry systems in proteomics analyses. Mol. Cell. Proteom. 2010, 9, 225–241. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.Q.; Polzin, K.O.; Dasari, S.; Chambers, M.C.; Schilling, B.; Gibson, B.W.; Tran, B.Q.; Vega-Montoto, L.; Liebler, D.C.; Tabb, D.L. QuaMeter: Multivendor performance metrics for LC-MS/MS proteomics instrumentation. Anal. Chem. 2012, 84, 5845–5850. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.M.; Dance, J.; Taylor, R.J.; Prince, J.T. Metriculator: Quality assessment for mass spectrometry-based proteomics. Bioinformatics 2013, 29, 2948–2949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichler, P.; Mazanek, M.; Dusberger, F.; Weilnböck, L.; Huber, C.G.; Stingl, C.; Luider, T.M.; Straube, W.L.; Köcher, T.; Mechtler, K. SIMPATIQCO: A server-based software suite which facilitates monitoring the time course of LC-MS performance metrics on Orbitrap instruments. J. Proteome Res. 2012, 11, 5540–5547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Bartolomé, S.; Medina-Aunon, J.A.; López-García, M.; González-Tejedo, C.; Prieto, G.; Navajas, R.; Salazar-Donate, E.; Fernández-Costa, C.; Yates, J.R.; Albar, J.P. PACOM: A Versatile Tool for Integrating, Filtering, Visualizing, and Comparing Multiple Large Mass Spectrometry Proteomics Data Sets. J. Proteome Res. 2018, 17, 1547–1558. [Google Scholar] [CrossRef]
- Olivella, R.; Chiva, C.; Serret, M.; Mancera, D.; Cozzuto, L.; Hermoso, A.; Borràs, E.; Espadas, G.; Morales, J.; Pastor, O.; et al. QCloud2: An Improved Cloud-based Quality-Control System for Mass-Spectrometry-based Proteomics Laboratories. J. Proteome Res. 2021, 20, 2010–2013. [Google Scholar] [CrossRef]
- Chiva, C.; Olivella, R.; Borràs, E.; Espadas, G.; Pastor, O.; Solé, A.; Sabidó, E. QCloud: A cloud-based quality control system for mass spectrometry-based proteomics laboratories. PLoS ONE 2018, 13, e0189209. [Google Scholar] [CrossRef] [Green Version]
- Bereman, M.S.; Johnson, R.; Bollinger, J.; Boss, Y.; Shulman, N.; MacLean, B.; Hoofnagle, A.N.; MacCoss, M.J. Implementation of statistical process control for proteomic experiments via LC MS/MS. J. Am. Soc. Mass Spectrom. 2014, 25, 581–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanfill, B.A.; Nakayasu, E.S.; Bramer, L.M.; Thompson, A.M.; Ansong, C.K.; Clauss, T.R.; Gritsenko, M.A.; Monroe, M.E.; Moore, R.J.; Orton, D.J.; et al. Quality Control Analysis in Real-time (QC-ART): A Tool for Real-time Quality Control Assessment of Mass Spectrometry-based Proteomics Data. Mol. Cell. Proteom. 2018, 17, 1824–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratton, K.G.; Webb-Robertson, B.M.; McCue, L.A.; Stanfill, B.; Claborne, D.; Godinez, I.; Johansen, T.; Thompson, A.M.; Burnum-Johnson, K.E.; Waters, K.M.; et al. pmartR: Quality Control and Statistics for Mass Spectrometry-Based Biological Data. J. Proteome Res. 2019, 18, 1418–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielow, C.; Mastrobuoni, G.; Kempa, S. Proteomics Quality Control: Quality Control Software for MaxQuant Results. J. Proteome Res. 2016, 15, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Barkovits, K.; Linden, A.; Galozzi, S.; Schilde, L.; Pacharra, S.; Mollenhauer, B.; Stoepel, N.; Steinbach, S.; May, C.; Uszkoreit, J.; et al. Characterization of Cerebrospinal Fluid via Data-Independent Acquisition Mass Spectrometry. J. Proteome Res. 2018, 17, 3418–3430. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Plum, S.; Helling, S.; Theiss, C.; Leite, R.E.P.; May, C.; Jacob-Filho, W.; Eisenacher, M.; Kuhlmann, K.; Meyer, H.E.; Riederer, P.; et al. Combined enrichment of neuromelanin granules and synaptosomes from human substantia nigra pars compacta tissue for proteomic analysis. J. Proteom. 2013, 94, 202–206. [Google Scholar] [CrossRef] [Green Version]
- May, C.; Serschnitzki, B.; Marcus, K. Good Old-Fashioned Protein Concentration Determination by Amino Acid Analysis. Methods Mol. Biol. 2021, 2228, 21–28. [Google Scholar] [CrossRef]
- Trezzi, J.P.; Galozzi, S.; Jaeger, C.; Barkovits, K.; Brockmann, K.; Maetzler, W.; Berg, D.; Marcus, K.; Betsou, F.; Hiller, K.; et al. Distinct metabolomic signature in cerebrospinal fluid in early parkinson’s disease. Mov. Disord. 2017, 32, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.L.; Xue, L.L.; Chen, Y.J.; Du, R.L.; Wang, Q.; Wen, S.; Zhou, L.; Liu, T.; Wang, T.H.; Yu, C.Y. Proteomics Study on the Cerebrospinal Fluid of Patients with Encephalitis. ACS Omega 2021, 6, 16288–16296. [Google Scholar] [CrossRef] [PubMed]
- Bader, J.M.; Geyer, P.E.; Müller, J.B.; Strauss, M.T.; Koch, M.; Leypoldt, F.; Koertvelyessy, P.; Bittner, D.; Schipke, C.G.; Incesoy, E.I.; et al. Proteome profiling in cerebrospinal fluid reveals novel biomarkers of Alzheimer’s disease. Mol. Syst. Biol. 2020, 16, e9356. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Sieb, C.; Thiel, K.; Wiswedel, B. KNIME: The Konstanz Information Miner. In Proceedings of the Data Analysis, Machine Learning and Applications; Springer: Berlin, Germany, 2008; pp. 319–326. [Google Scholar]
- Perkins, D.N.; Pappin, D.J.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar] [CrossRef]
- Uszkoreit, J.; Maerkens, A.; Perez-Riverol, Y.; Meyer, H.E.; Marcus, K.; Stephan, C.; Kohlbacher, O.; Eisenacher, M. PIA: An Intuitive Protein Inference Engine with a Web-Based User Interface. J. Proteome Res. 2015, 14, 2988–2997. [Google Scholar] [CrossRef]
- Uszkoreit, J.; Perez-Riverol, Y.; Eggers, B.; Marcus, K.; Eisenacher, M. Protein Inference Using PIA Workflows and PSI Standard File Formats. J. Proteome Res. 2019, 18, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Pfeuffer, J.; Sachsenberg, T.; Alka, O.; Walzer, M.; Fillbrunn, A.; Nilse, L.; Schilling, O.; Reinert, K.; Kohlbacher, O. OpenMS—A platform for reproducible analysis of mass spectrometry data. J. Biotechnol. 2017, 261, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Consortium, U. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [Green Version]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 7 February 2023).
- Hadley, W. ggplot2 Elegant Graphics for Data Analysis, 2nd ed.; Springer: Cham, Switzerland; New York, NY, USA, 2016; p. 260. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Callister, S.J.; Barry, R.C.; Adkins, J.N.; Johnson, E.T.; Qian, W.J.; Webb-Robertson, B.J.; Smith, R.D.; Lipton, M.S. Normalization approaches for removing systematic biases associated with mass spectrometry and label-free proteomics. J. Proteome Res. 2006, 5, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Taouatas, N.; Cappadona, S.; van Breukelen, B.; Mohammed, S.; Scholten, A.; Heck, A.J. Protease bias in absolute protein quantitation. Nat. Methods 2012, 9, 524–525. [Google Scholar] [CrossRef]
- Varnavides, G.; Madern, M.; Anrather, D.; Hartl, N.; Reiter, W.; Hartl, M. In Search of a Universal Method: A Comparative Survey of Bottom-Up Proteomics Sample Preparation Methods. J. Proteome Res. 2022, 21, 2397–2411. [Google Scholar] [CrossRef]
- Sun, S.; Zhou, J.Y.; Yang, W.; Zhang, H. Inhibition of protein carbamylation in urea solution using ammonium-containing buffers. Anal. Biochem. 2014, 446, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Cole, E.G.; Mecham, D.K. Cyanate formation and electrophoretic behavior of proteins in gels containing urea. Anal. Biochem. 1966, 14, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Tenga, M.J.; Lazar, I.M. Impact of peptide modifications on the isobaric tags for relative and absolute quantitation method accuracy. Anal. Chem. 2011, 83, 701–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loroch, S.; Kopczynski, D.; Schneider, A.C.; Schumbrutzki, C.; Feldmann, I.; Panagiotidis, E.; Reinders, Y.; Sakson, R.; Solari, F.A.; Vening, A.; et al. Toward Zero Variance in Proteomics Sample Preparation: Positive-Pressure FASP in 96-Well Format (PF96) Enables Highly Reproducible, Time- and Cost-Efficient Analysis of Sample Cohorts. J. Proteome Res. 2022, 21, 1181–1188. [Google Scholar] [CrossRef]
- Chawade, A.; Alexandersson, E.; Levander, F. Normalyzer: A tool for rapid evaluation of normalization methods for omics data sets. J. Proteome Res. 2014, 13, 3114–3120. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rozanova, S.; Uszkoreit, J.; Schork, K.; Serschnitzki, B.; Eisenacher, M.; Tönges, L.; Barkovits-Boeddinghaus, K.; Marcus, K. Quality Control—A Stepchild in Quantitative Proteomics: A Case Study for the Human CSF Proteome. Biomolecules 2023, 13, 491. https://doi.org/10.3390/biom13030491
Rozanova S, Uszkoreit J, Schork K, Serschnitzki B, Eisenacher M, Tönges L, Barkovits-Boeddinghaus K, Marcus K. Quality Control—A Stepchild in Quantitative Proteomics: A Case Study for the Human CSF Proteome. Biomolecules. 2023; 13(3):491. https://doi.org/10.3390/biom13030491
Chicago/Turabian StyleRozanova, Svitlana, Julian Uszkoreit, Karin Schork, Bettina Serschnitzki, Martin Eisenacher, Lars Tönges, Katalin Barkovits-Boeddinghaus, and Katrin Marcus. 2023. "Quality Control—A Stepchild in Quantitative Proteomics: A Case Study for the Human CSF Proteome" Biomolecules 13, no. 3: 491. https://doi.org/10.3390/biom13030491
APA StyleRozanova, S., Uszkoreit, J., Schork, K., Serschnitzki, B., Eisenacher, M., Tönges, L., Barkovits-Boeddinghaus, K., & Marcus, K. (2023). Quality Control—A Stepchild in Quantitative Proteomics: A Case Study for the Human CSF Proteome. Biomolecules, 13(3), 491. https://doi.org/10.3390/biom13030491