
Behavioral and Transcriptomic Changes Following Brain-Specific Loss of Noradrenergic Transmission

 , , , , , , ,
, , , , , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Housing and Breeding
2.2. Anxiety- and Depression-Like Parameters
2.2.1. Elevated Plus Maze (EPM)
2.2.2. Novelty-Suppressed Feeding Test (NSF)
2.2.3. Marble Burying Test
2.2.4. Forced Swim Test (FST)
2.2.5. Sucrose Preference Test
2.2.6. Chronic Social Defeat Stress Paradigm (CSDS)
2.2.7. Restrain Stress and Saphenous Blood Collection
2.2.8. Dexamethasone Suppression Test
2.2.9. Corticosterone Level Measurement
2.3. Learning and Memory
2.3.1. Contextual and Cued Fear Conditioning Task
2.3.2. Morris Water Maze (MWM)
2.4. Executive Functions
Attentional Set-Shifting Task (ASST)
2.5. Addiction-Like Behavior
2.5.1. Locomotor Response
2.5.2. Behavioral Sensitization
2.6. Nociception
Tail Immersion Test
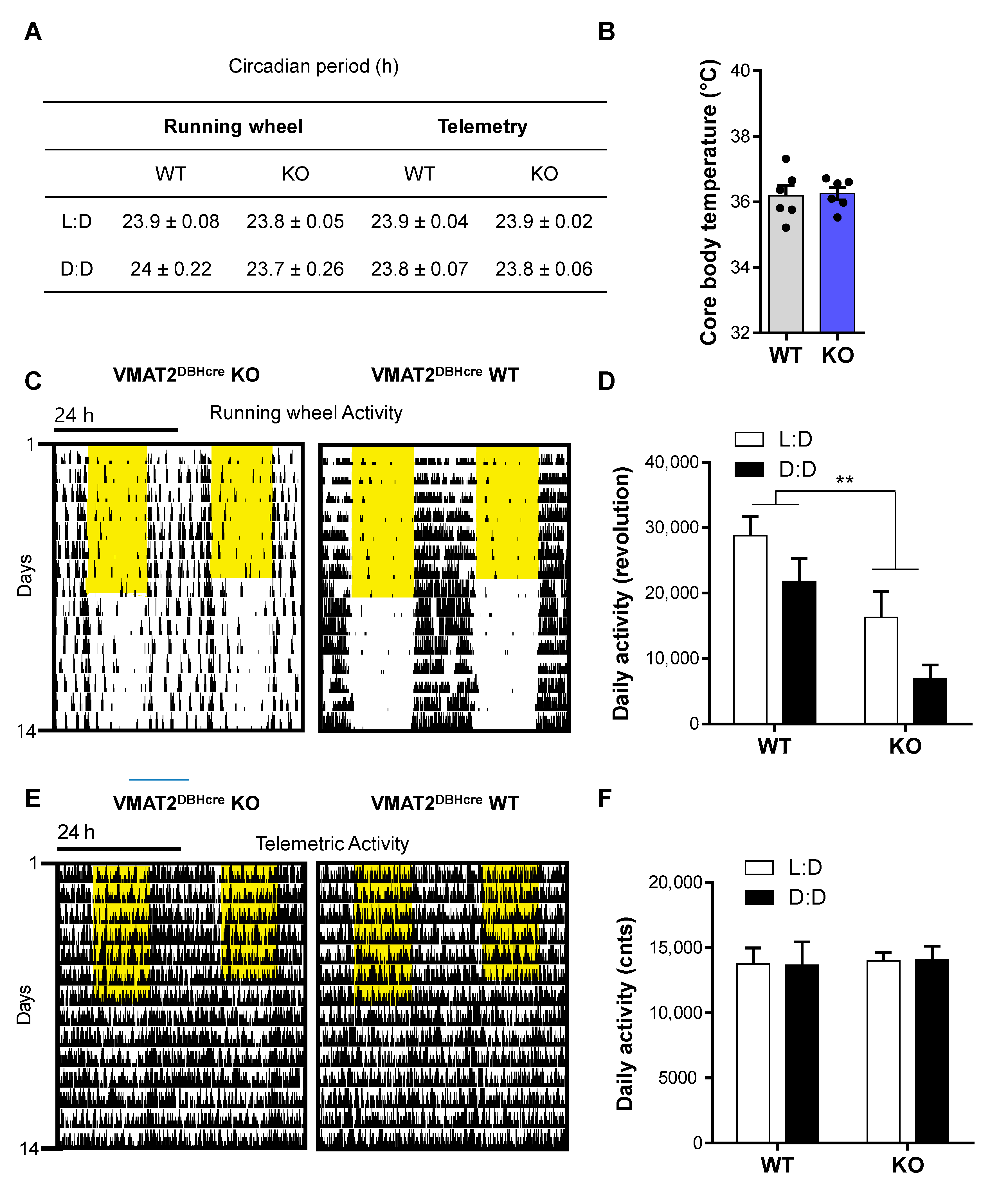
2.7. Circadian Analysis
2.7.1. Running Wheels
2.7.2. Telemetry
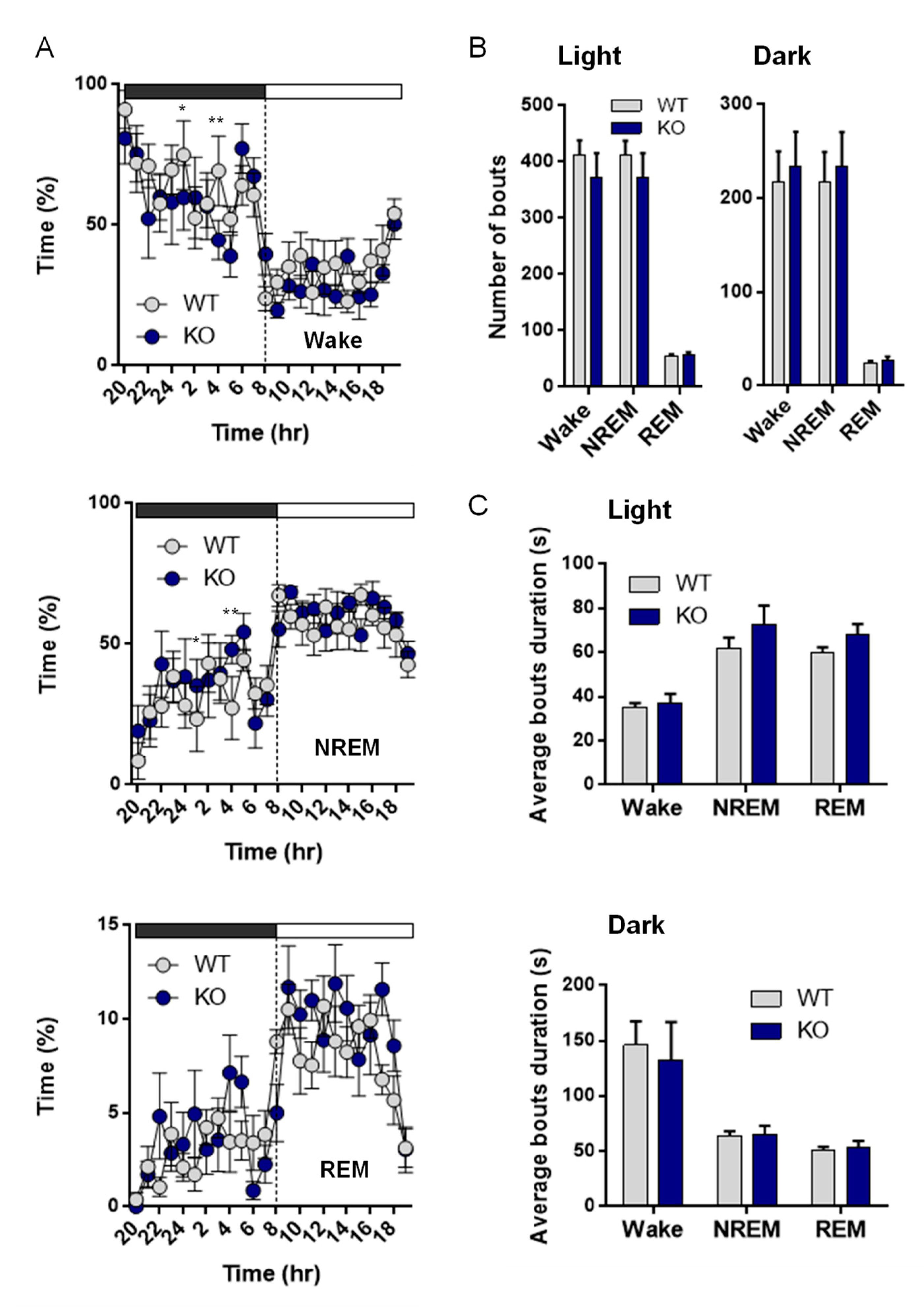
2.8. Sleep Recording
2.8.1. Surgery
2.8.2. Polysomnographic Recording and Data Acquisition
- (1)
- Wake: High-frequency low-amplitude EEG oscillations accompanied by constant EMG activity with phasic bursts.
- (2)
- NREM sleep: Low frequency, high amplitude EEG oscillations with an increase in slow delta wave activity (0.5–4.5 Hz) and a loss of phasic muscle activity.
- (3)
- REM sleep: High frequency, low amplitude EEG oscillations with typical regular theta rhythm (5–9 Hz) and a flat EMG.
2.8.3. Spectral Power Analysis
2.9. Quantitative In Situ Hybridization
2.10. Microarray Transcriptome
2.10.1. Tissue Dissection, RNA Isolation
2.10.2. Normalization, Quality Control, and Filtering of Microarrays Data
2.10.3. Removing Batch Effects
2.10.4. Differential Expression Analysis
2.10.5. Gene Set Enrichment Analysis
2.10.6. Visual Representation
2.10.7. R Session Info
2.11. Statistical Analysis
3. Results
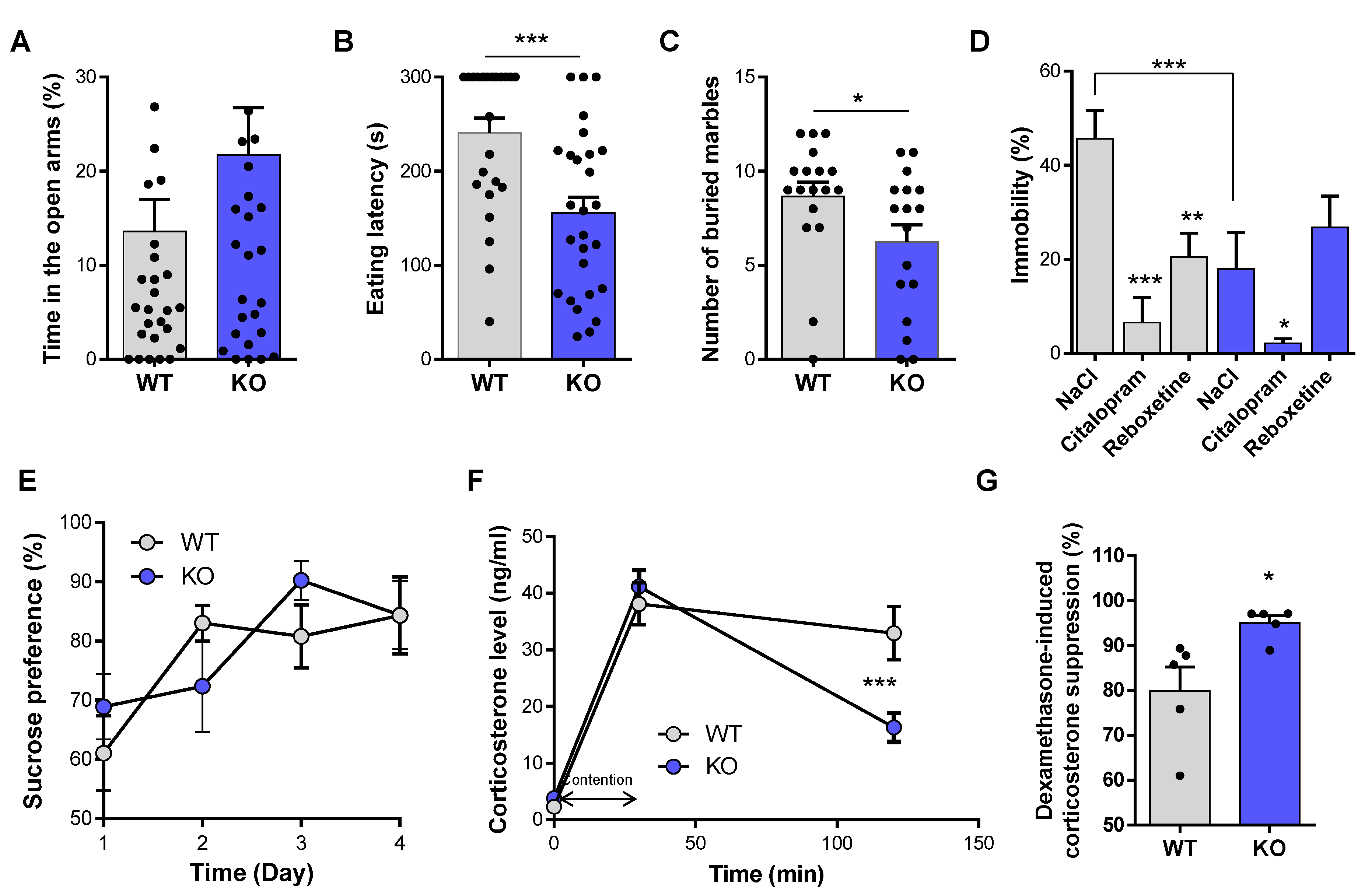
3.1. Role of Central NE System in Anxiety and Depression
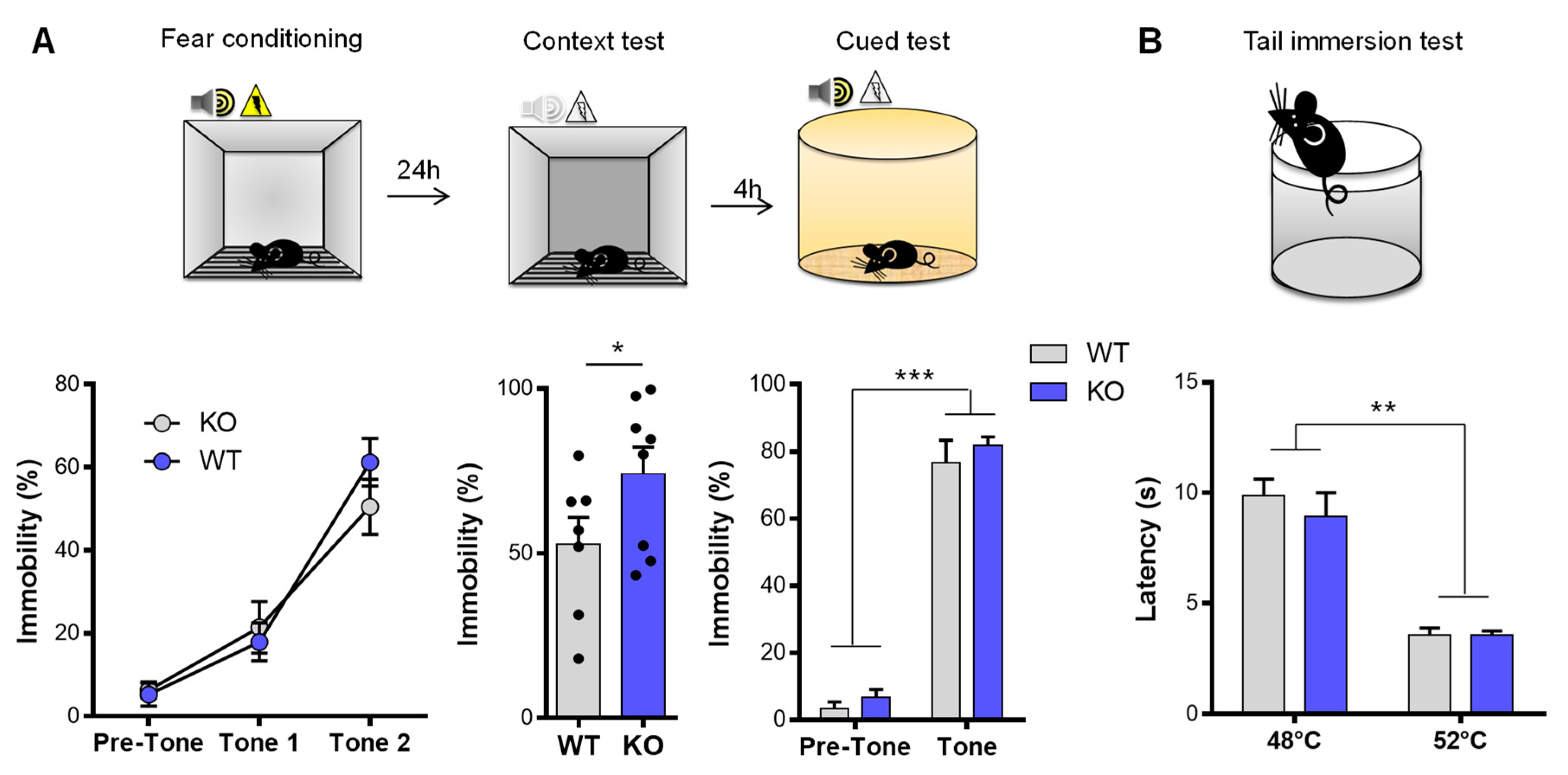
3.2. NE and Fear Conditioning
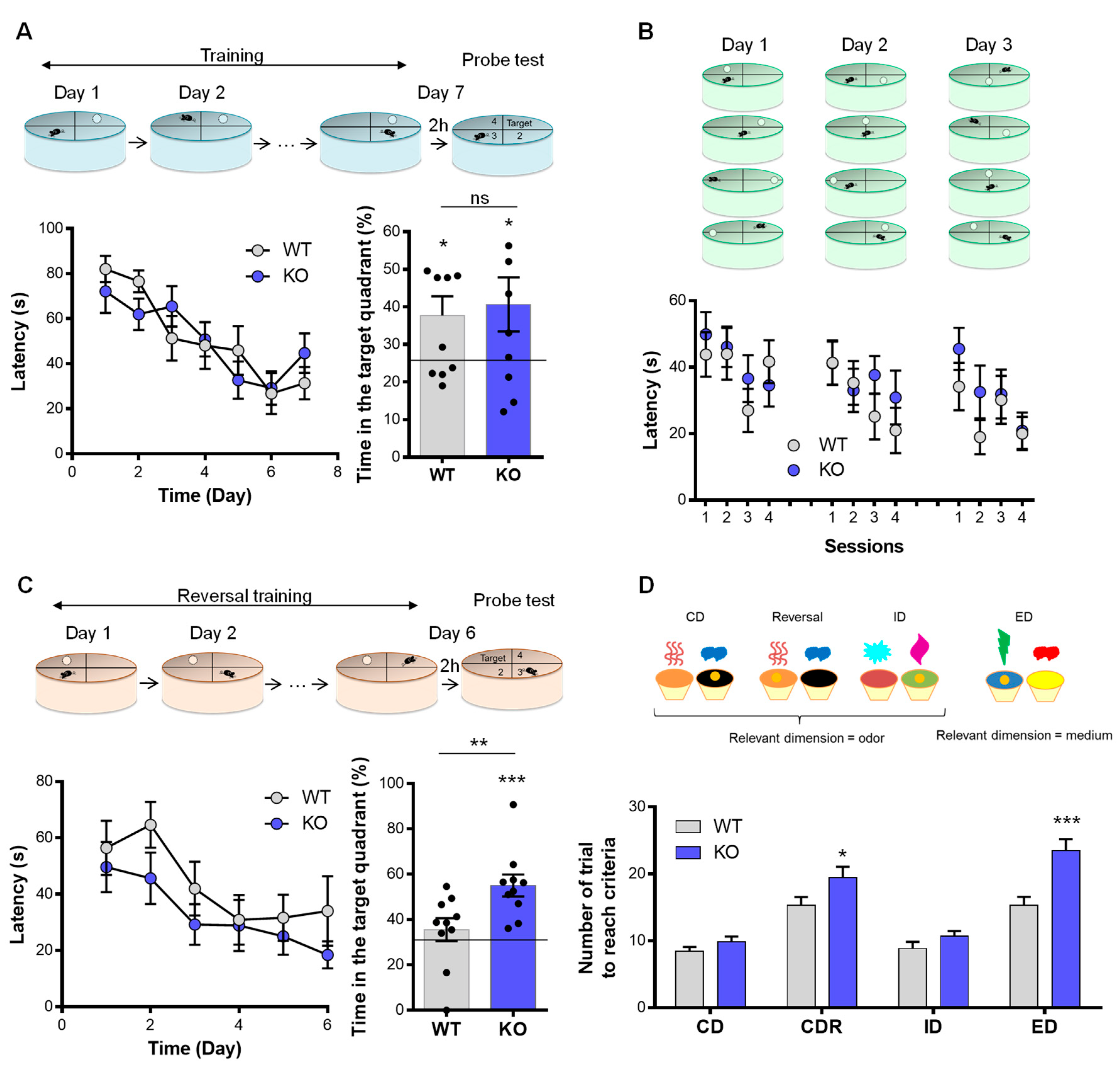
3.3. NE and Memory
3.4. NE Depletion and Behavioral Flexibility
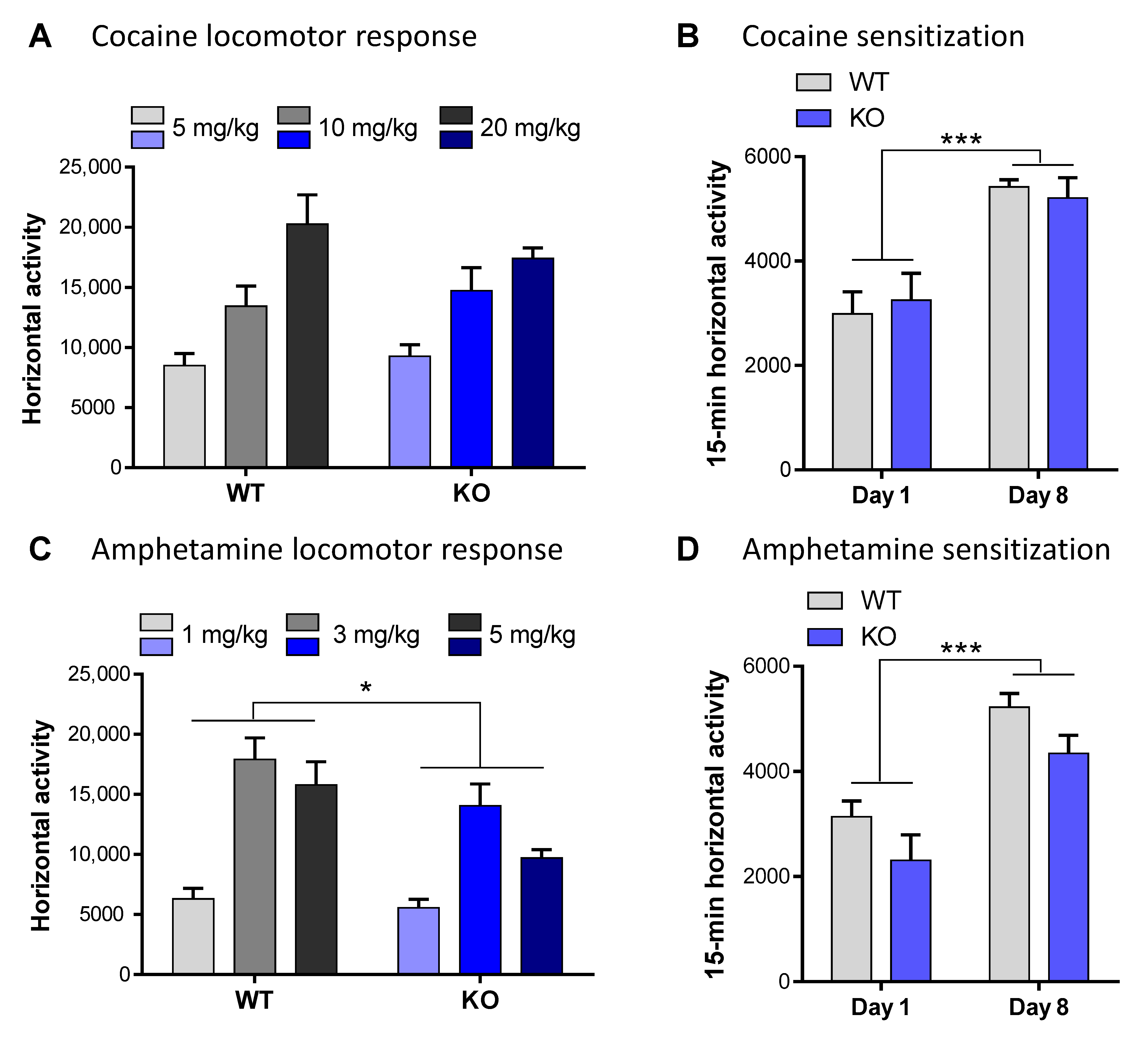
3.5. Role of NE Depletion in Drugs Locomotor Response
3.6. NE and Circadian Rhythms
3.7. NE Control of Sleep and Arousal
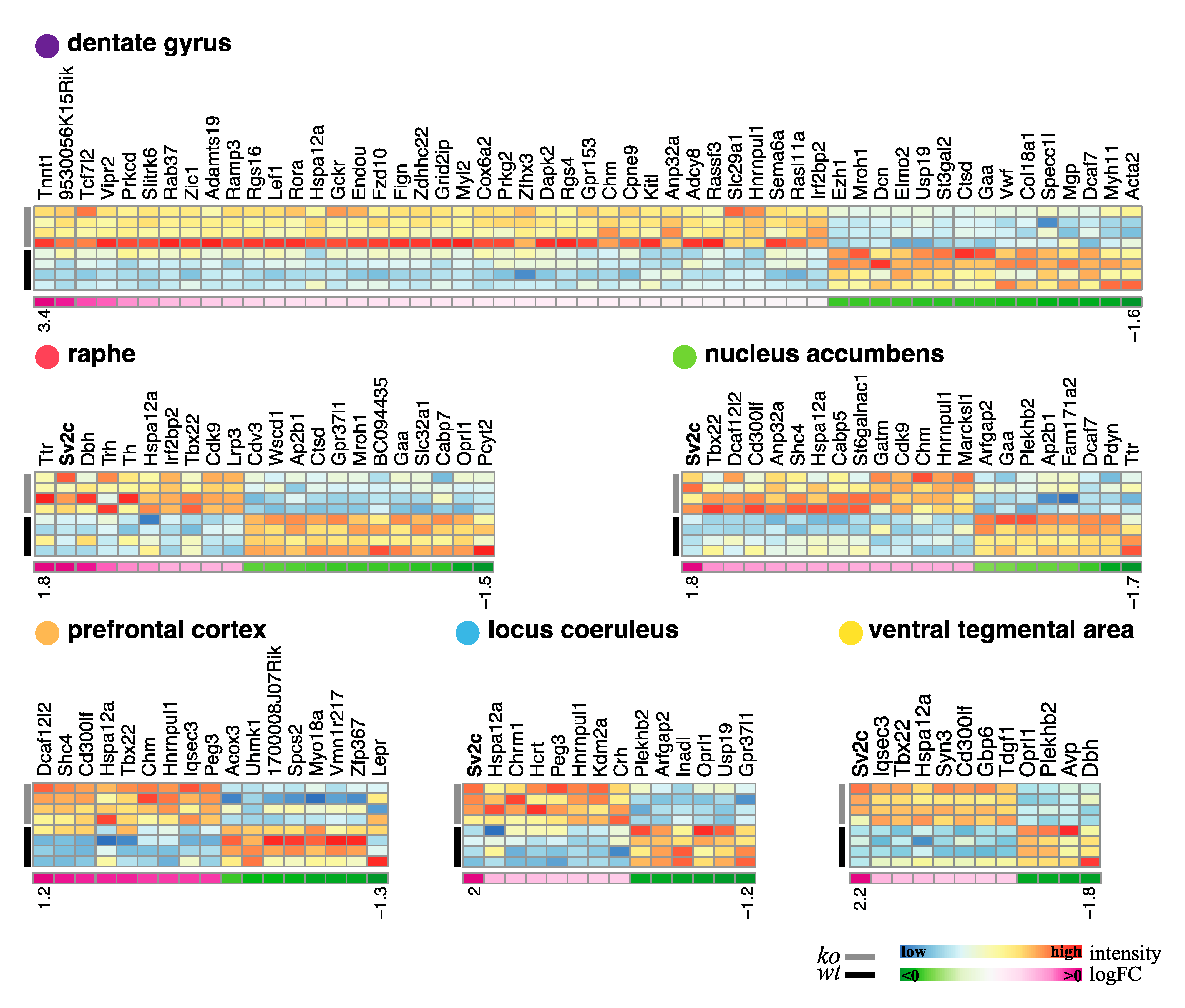
3.8. Transcriptional Effect of NE Differential Gene Expression Signature of Brain-Specific NE Depleted Mice
3.8.1. Profiling Gene Expression Changes across Brain Regions
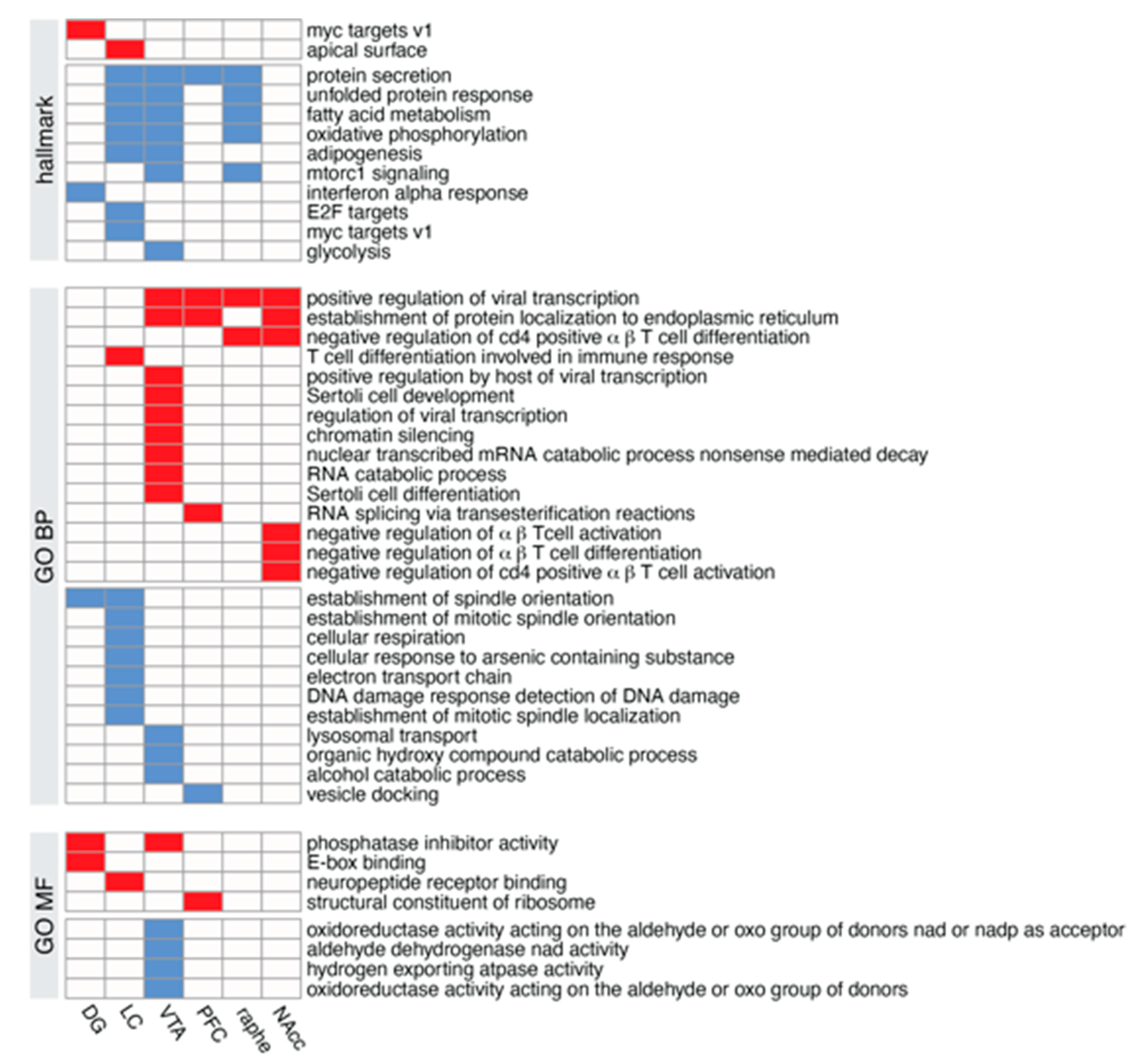
3.8.2. Probing the Biological Significance of NE-Specific DEGs
4. Discussion
4.1. Anxiolytic and Antidepressant Effects of NE Depletion
4.2. NE Regulation of Emotional Memory
4.3. Absence of NE Implication on Spatial and Working Memory
4.4. NE Depletion in Executive Functions and Behavioral Flexibility
4.5. Role of NE Depletion in Locomotor Response and Behavioral Sensitization to Psychostimulants
4.6. Circadian Rhythms
4.7. NE and Sleep and Waves
4.8. Transcriptomic Analyses
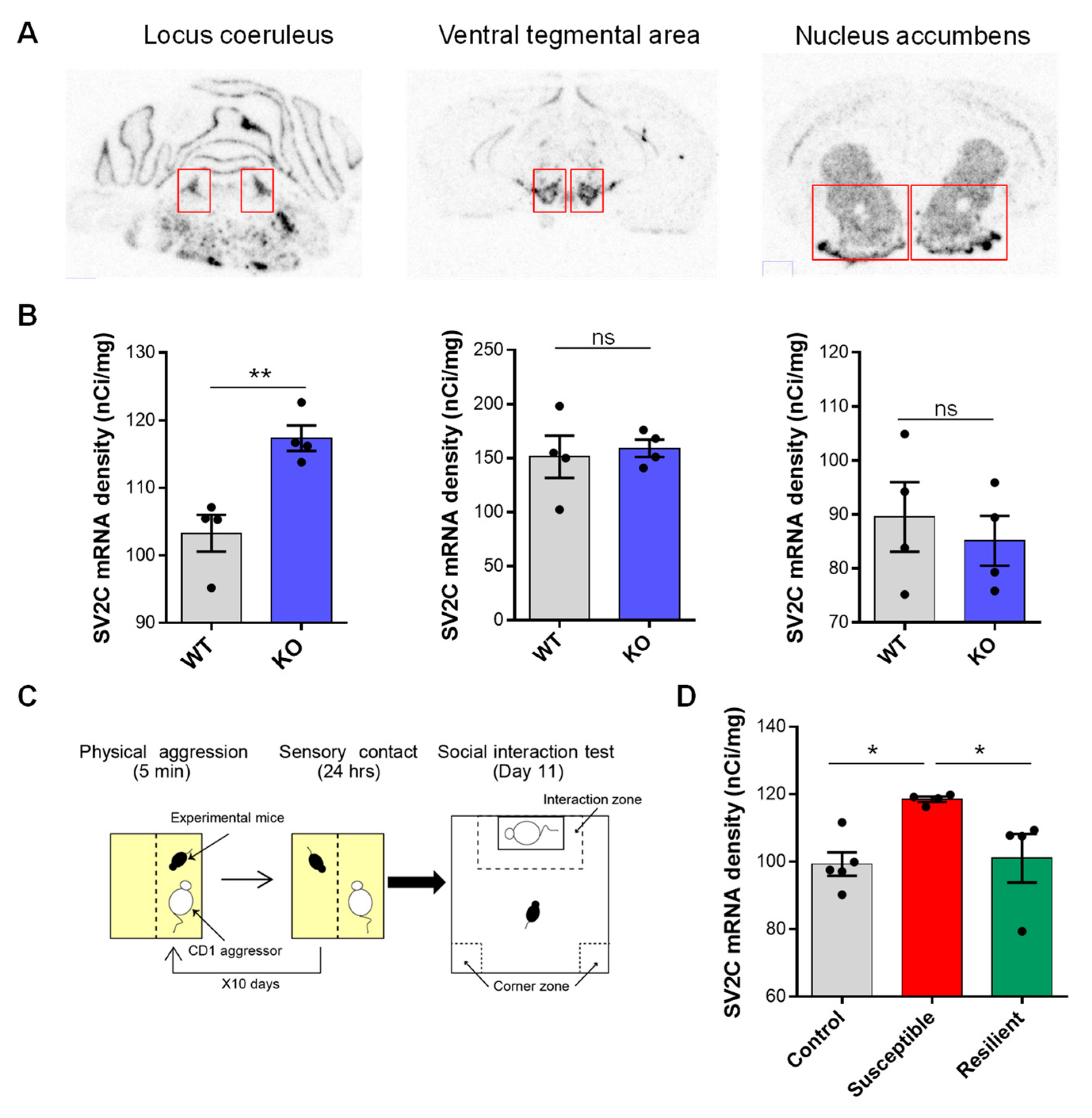
4.9. SV2c Expression Is Regulated by NE Transmission and Chronic Stress
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robbins, T.W. Opinion on monoaminergic contributions to traits and temperament. Philos. Trans. R. Soc. B: Biol. Sci. 2018, 373, 20170153. [Google Scholar] [CrossRef]
- Peter, D.; Liu, Y.; Sternini, C.; de Giorgio, R.; Brecha, N.; Edwards, R.H. Differential expression of two vesicular monoamine transporters. J. Neurosci. 1995, 15, 6179–6188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimer, R.J.; A Fon, E.; Edwards, R.H. Vesicular neurotransmitter transport and the presynaptic regulation of quantal size. Curr. Opin. Neurobiol. 1998, 8, 405–412. [Google Scholar] [CrossRef]
- Weihe, E.; Schäfer, M.K.H.; Erickson, J.D.; Eiden, L.E. Localization of vesicular monoamine transporter isoforms (VMAT1 and VMAT2) to endocrine cells and neurons in rat. J. Mol. Neurosci. 1994, 5, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Blakely, R.D.; Edwards, R.H. Vesicular and Plasma Membrane Transporters for Neurotransmitters. Cold Spring Harb. Perspect. Biol. 2012, 4, a005595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cedarbaum, J.M.; Aghajanian, G.K. Afferent projections to the rat locus coeruleus as determined by a retrograde tracing technique. J. Comp. Neurol. 1978, 178, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.E.; Moore, R.Y. Ascending projections of the locus coeruleus in the rat. II. Autoradiographic study. Brain Res. 1977, 127, 25–53. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.Y.; Bloom, F.E. Central Catecholamine Neuron Systems: Anatomy and Physiology of the Norepinephrine and Epinephrine Systems. Annu. Rev. Neurosci. 1979, 2, 113–168. [Google Scholar] [CrossRef] [PubMed]
- Samuels, E.R.; Szabadi, E. Functional Neuroanatomy of the Noradrenergic Locus Coeruleus: Its Roles in the Regulation of Arousal and Autonomic Function Part II: Physiological and Pharmacological Manipulations and Pathological Alterations of Locus Coeruleus Activity in Humans. Curr. Neuropharmacol. 2008, 6, 254–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, C.W.; Waterhouse, B.D. The locus coeruleus–noradrenergic system: Modulation of behavioral state and state-dependent cognitive processes. Brain Res. Rev. 2003, 42, 33–84. [Google Scholar] [CrossRef] [PubMed]
- Woodward, D.J.; Moises, H.C.; Waterhouse, B.D.; Hoffer, B.J.; Freedman, R. Modulatory actions of norepinephrine in the central nervous system. Fed. Proc. 1979, 38, 2109–2116. [Google Scholar]
- Bouret, S.; Sara, S.J. Locus coeruleus activation modulates firing rate and temporal organization of odour-induced single-cell responses in rat piriform cortex. Eur. J. Neurosci. 2002, 16, 2371–2382. [Google Scholar] [CrossRef]
- Devilbiss, D.M.; Waterhouse, B.D. Phasic and Tonic Patterns of Locus Coeruleus Output Differentially Modulate Sensory Network Function in the Awake Rat. J. Neurophysiol. 2011, 105, 69–87. [Google Scholar] [CrossRef]
- Waterhouse, B.D.; Moises, H.C.; Woodward, D.J. Phasic activation of the locus coeruleus enhances responses of primary sensory cortical neurons to peripheral receptive field stimulation. Brain Res. 1998, 790, 33–44. [Google Scholar] [CrossRef]
- Dahl, D.; Sarvey, J.M. Norepinephrine induces pathway-specific long-lasting potentiation and depression in the hippocampal dentate gyrus. Proc. Natl. Acad. Sci. USA 1989, 86, 4776–4780. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, C.K.; McGaugh, J.L.; Williams, C.L. Interacting brain systems modulate memory consolidation. Neurosci. Biobehav. Rev. 2012, 36, 1750–1762. [Google Scholar] [CrossRef] [Green Version]
- Bouret, S.; Sara, S.J. Network reset: A simplified overarching theory of locus coeruleus noradrenaline function. Trends Neurosci. 2005, 28, 574–582. [Google Scholar] [CrossRef] [PubMed]
- McGaughy, J.; Ross, R.S.; Eichenbaum, H. Noradrenergic, but not cholinergic, deafferentation of prefrontal cortex impairs attentional set-shifting. Neuroscience 2008, 153, 63–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, A.J.; Dayan, P. Uncertainty, Neuromodulation, and Attention. Neuron 2005, 46, 681–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCall, J.G.; Al-Hasani, R.; Siuda, E.R.; Hong, D.Y.; Norris, A.J.; Ford, C.P.; Bruchas, M.R. CRH Engagement of the Locus Coeruleus Noradrenergic System Mediates Stress-Induced Anxiety. Neuron 2015, 87, 605–620. [Google Scholar] [CrossRef] [Green Version]
- Morilak, D.A.; Barrera, G.; Echevarria, D.J.; Garcia, A.S.; Hernandez, A.; Ma, S.; Petre, C.O. Role of brain norepinephrine in the behavioral response to stress. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 1214–1224. [Google Scholar] [CrossRef]
- Berridge, C.W.; Isaac, S.O.; España, R.A. Additive wake-promoting actions of medial basal forebrain noradrenergic α1- and β-receptor stimulation. Behav. Neurosci. 2003, 117, 350–359. [Google Scholar] [CrossRef] [Green Version]
- Carter, M.E.; Yizhar, O.; Chikahisa, S.; Nguyen, H.; Adamantidis, A.; Nishino, S.; Deisseroth, K.; de Lecea, L. Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nat. Neurosci. 2010, 13, 1526–1533. [Google Scholar] [CrossRef] [Green Version]
- Manns, I.D.; Lee, M.G.; Modirrousta, M.; Hou, Y.P.; Jones, B.E. Alpha 2 adrenergic receptors on GABAergic, putative sleep-promoting basal forebrain neurons. Eur. J. Neurosci. 2003, 18, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Abercrombie, E.D.; Zigmond, M.J. Partial injury to central noradrenergic neurons: Reduction of tissue norepinephrine content is greater than reduction of extracellular norepinephrine measured by microdialysis. J. Neurosci. 1989, 9, 4062–4067. [Google Scholar] [CrossRef] [Green Version]
- Cryan, J.F.; Page, M.E.; Lucki, I. Noradrenergic lesions differentially alter the antidepressant-like effects of reboxetine in a modified forced swim test. Eur. J. Pharmacol. 2002, 436, 197–205. [Google Scholar] [CrossRef]
- Bezard, E.; Przedborski, S. A tale on animal models of Parkinson’s disease. Mov. Disord. 2011, 26, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Fritschy, J.-M.; Grzanna, R. Selective effects of DSP-4 on locus coeruleus axons: Are there pharmacologically different types of noradrenergic axons in the central nervous system? Prog. Brain Res. 1991, 88, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, A.K.; Galambos, E. The effect of reserpine, alpha-methyl-m-tyrosine, prenylamine, and guanethidine on metrazol-convulsions and the brain monoamine level in mice. Arch. Int. Pharmacodyn. Ther. 1967, 165, 201–211. [Google Scholar] [PubMed]
- Kobayashi, K.; Morita, S.; Sawada, H.; Mizuguchi, T.; Yamada, K.; Nagatsu, I.; Hata, T.; Watanabe, Y.; Fujita, K.; Nagatsu, T. Targeted Disruption of the Tyrosine Hydroxylase Locus Results in Severe Catecholamine Depletion and Perinatal Lethality in Mice. J. Biol. Chem. 1995, 270, 27235–27243. [Google Scholar] [CrossRef] [Green Version]
- Schank, J.R.; Ventura, R.; Puglisi-Allegra, S.; Alcaro, A.; Cole, C.D.; Liles, L.C.; Seeman, P.; Weinshenker, D. Dopamine β-Hydroxylase Knockout Mice have Alterations in Dopamine Signaling and are Hypersensitive to Cocaine. Neuropsychopharmacology 2006, 31, 2221–2230. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.A.; Matsumoto, A.M.; Palmiter, R.D. Noradrenaline is essential for mouse fetal development. Nature 1995, 374, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Isingrini, E.; Perret, L.; Rainer, Q.; Sagueby, S.; Moquin, L.; Gratton, A.; Giros, B. Selective genetic disruption of dopaminergic, serotonergic and noradrenergic neurotransmission: Insights into motor, emotional and addictive behaviour. J. Psychiatry Neurosci. 2016, 41, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Blum, I.D.; Zhu, L.; Moquin, L.; Kokoeva, M.V.; Gratton, A.; Giros, B.; Storch, K.-F. A highly tunable dopaminergic oscillator generates ultradian rhythms of behavioral arousal. Elife 2014, 3, e02809. [Google Scholar] [CrossRef]
- Jego, S.; Glasgow, S.; Herrera, C.G.; Ekstrand, M.; Reed, S.J.; Boyce, R.; Friedman, J.; Burdakov, D.; Adamantidis, A.R. Optogenetic identification of a rapid eye movement sleep modulatory circuit in the hypothalamus. Nat. Neurosci. 2013, 16, 1637–1643. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Franklin, K.B.J. The Mouse Brain in Stereotaxic Coordinates; Academic Press: San Diego, CA, USA, 2001. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. In R Foundation for Statistical Computing; The R Foundation: Vienna, Austria, 2017. [Google Scholar]
- Khatri, P.; Sirota, M.; Butte, A.J. Ten Years of Pathway Analysis: Current Approaches and Outstanding Challenges. PLOS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef]
- Väremo, L.; Nielsen, J.; Nookaew, I. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 2013, 41, 4378–4391. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Isingrini, E.; Perret, L.; Rainer, Q.; Amilhon, B.; Guma, E.; Tanti, A.; Martin, G.; Robinson, J.; Moquin, L.; Marti, F.; et al. Resilience to chronic stress is mediated by noradrenergic regulation of dopamine neurons. Nat. Neurosci. 2016, 19, 560–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breton-Provencher, V.; Drummond, G.T.; Feng, J.; Li, Y.; Sur, M. Spatiotemporal dynamics of noradrenaline during learned behaviour. Nature 2022, 606, 732–738. [Google Scholar] [CrossRef]
- Breton-Provencher, V.; Sur, M. Active control of arousal by a locus coeruleus GABAergic circuit. Nat. Neurosci. 2019, 22, 218–228. [Google Scholar] [CrossRef]
- Uematsu, A.; Tan, B.Z.; A Ycu, E.; Cuevas, J.S.; Koivumaa, J.; Junyent, F.; Kremer, E.; Witten, I.B.; Deisseroth, K.; Johansen, J.P. Modular organization of the brainstem noradrenaline system coordinates opposing learning states. Nat. Neurosci. 2017, 20, 1602–1611. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.; Luchetti, A.; Fernandes, G.; Filho, D.A.; Kastellakis, G.; Tzilivaki, A.; Ramirez, E.M.; Tran, M.Y.; Poirazi, P.; Silva, A.J. A locus coeruleus-dorsal CA1 dopaminergic circuit modulates memory linking. Neuron 2022, 110, 3374–3388.e8. [Google Scholar] [CrossRef]
- Kempadoo, K.A.; Mosharov, E.V.; Choi, S.J.; Sulzer, D.; Kandel, E.R. Dopamine release from the locus coeruleus to the dorsal hippocampus promotes spatial learning and memory. Proc. Natl. Acad. Sci. USA 2016, 113, 14835–14840. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, T.; Duszkiewicz, A.J.; Sonneborn, A.; Spooner, P.A.; Yamasaki, M.; Watanabe, M.; Smith, C.C.; Fernández, G.; Deisseroth, K.; Greene, R.W.; et al. Locus coeruleus and dopaminergic consolidation of everyday memory. Nature 2016, 537, 357–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beas, B.S.; Wright, B.J.; Skirzewski, M.; Leng, Y.; Hyun, J.H.; Koita, O.; Ringelberg, N.; Kwon, H.-B.; Buonanno, A.; Penzo, M.A. The locus coeruleus drives disinhibition in the midline thalamus via a dopaminergic mechanism. Nat. Neurosci. 2018, 21, 963–973. [Google Scholar] [CrossRef]
- Glavin, G.B. Stress and brain noradrenaline: A review. Neurosci. Biobehav. Rev. 1985, 9, 233–243. [Google Scholar] [CrossRef]
- McEwen, B.S. The neurobiology of stress: From serendipity to clinical relevance. Brain Res. 2000, 886, 172–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lake, C.R.; Pickar, D.; Ziegler, M.G.; Lipper, S.; Slater, S.; Murphy, D.L. High plasma norepinephrine levels in patients with major affective disorder. Am. J. Psychiatry 1982, 139, 1315–1318. [Google Scholar] [CrossRef]
- Sevy, S.; Papadimitriou, G.N.; Surmont, D.W.; Goldman, S.; Mendiewicz, J. Noradrenergic function in generalized anxiety disorder, major depressive disorder, and healthy subjects. Biol. Psychiatry 1989, 25, 141–152. [Google Scholar] [CrossRef]
- Roy, A.; Pickar, D.; De Jong, J.; Karoum, F.; Linnoila, M. Suicidal behavior in depression: Relationship to noradrenergic function. Biol. Psychiatry 1989, 25, 341–350. [Google Scholar] [CrossRef]
- Secunda, S.K.; Cross, C.K.; Koslow, S.; Katz, M.M.; Kocsis, J.; Maas, J.W.; Landis, H. Biochemistry and suicidal behavior in depressed patients. Biol. Psychiatry 1986, 21, 756–767. [Google Scholar] [CrossRef]
- Cryan, J.F.; Dalvi, A.; Jin, S.H.; Hirsch, B.R.; Lucki, I.; Thomas, S.A. Use of dopamine-beta-hydroxylase-deficient mice to determine the role of norepinephrine in the mechanism of action of antidepressant drugs. J. Pharmacol. And. Exp. Ther. 2001, 298, 651–657. [Google Scholar]
- Cryan, J.F.; Page, M.E.; Lucki, I. Differential behavioral effects of the antidepressants reboxetine, fluoxetine, and moclobemide in a modified forced swim test following chronic treatment. Psychopharmacology 2005, 182, 335–344. [Google Scholar] [CrossRef]
- Marino, M.D.; Bourdélat-Parks, B.N.; Liles, L.C.; Weinshenker, D. Genetic reduction of noradrenergic function alters social memory and reduces aggression in mice. Behav. Brain Res. 2005, 161, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Redrobe, J.P.; Bourin, M.; Colombel, M.C.; Baker, G.B. Dose-dependent noradrenergic and serotonergic properties of venlafaxine in animal models indicative of antidepressant activity. Psychopharmacology 1998, 138, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Semba, J.-I.; Takahashi, R. Effect of monoamine precursors on the forced-swimming test in mice. Psychopharmacology 1988, 95, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Tillage, R.P.; Foster, S.L.; Lustberg, D.; Liles, L.C.; McCann, K.E.; Weinshenker, D. Co-released norepinephrine and galanin act on different timescales to promote stress-induced anxiety-like behavior. Neuropsychopharmacology 2021, 46, 1535–1543. [Google Scholar] [CrossRef]
- Venault, P.; Jacquot, F.; Save, E.; Sara, S.; Chapouthier, G. Anxiogenic-like effects of yohimbine and idazoxan in two behavioral situations in mice. Life Sci. 1993, 52, 639–645. [Google Scholar] [CrossRef]
- Montoya, A.; Bruins, R.; Katzman, M.A.; Blier, P. The noradrenergic paradox: Implications in the management of depression and anxiety. Neuropsychiatr. Dis. Treat. 2016, 12, 541–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCall, J.G.; Siuda, E.R.; Bhatti, D.L.; A Lawson, L.; A McElligott, Z.; Stuber, G.D.; Bruchas, M.R. Locus coeruleus to basolateral amygdala noradrenergic projections promote anxiety-like behavior. Elife 2017, 6, e18247. [Google Scholar] [CrossRef] [PubMed]
- Willner, P.; Towell, A.; Sampson, D.; Sophokleous, S.; Muscat, R. Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology 1987, 93, 358–364. [Google Scholar] [CrossRef]
- Purdy, R.H.; Morrow, A.L.; Moore, P.H., Jr.; Paul, S.M. Stress-induced elevations of gamma-aminobutyric acid type A receptor-active steroids in the rat brain. Proc. Natl. Acad. Sci. USA 1991, 88, 4553–4557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, H.M.; Davis, M.C.; Otte, C.; Mohr, D.C. Depression and cortisol responses to psychological stress: A meta-analysis. Psychoneuroendocrinology 2005, 30, 846–856. [Google Scholar] [CrossRef]
- Davies, M.; Tsui, J.Y.; Flannery, J.A.; Li, X.; DeLorey, T.M.; Hoffman, B.B. Augmentation of the noradrenergic system in alpha-2 adrenergic receptor deficient mice: Anatomical changes associated with enhanced fear memory. Brain Res. 2003, 986, 157–165. [Google Scholar] [CrossRef]
- Kim, J.J.; Shih, J.C.; Chen, K.; Chen, L.; Bao, S.; Maren, S.; Anagnostaras, S.G.; Fanselow, M.S.; De Maeyer, E.; Seif, I.; et al. Selective enhancement of emotional, but not motor, learning in monoamine oxidase A-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 5929–5933. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Kobayashi, T. Genetic evidence for noradrenergic control of long-term memory consolidation. Brain Dev. 2001, 23 (Suppl. S1), S16–S23. [Google Scholar] [CrossRef]
- Murchison, C.F.; Schutsky, K.; Jin, S.-H.; Thomas, S.A. Norepinephrine and ß1-adrenergic signaling facilitate activation of hippocampal CA1 pyramidal neurons during contextual memory retrieval. Neuroscience 2011, 181, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murchison, C.F.; Zhang, X.-Y.; Zhang, W.-P.; Ouyang, M.; Lee, A.; Thomas, S.A. A Distinct Role for Norepinephrine in Memory Retrieval. Cell 2004, 117, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, M.; Thomas, S.A. A requirement for memory retrieval during and after long-term extinction learning. Proc. Natl. Acad. Sci. USA 2005, 102, 9347–9352. [Google Scholar] [CrossRef] [Green Version]
- Schutsky, K.; Ouyang, M.; Thomas, S.A. Xamoterol impairs hippocampus-dependent emotional memory retrieval via Gi/o-coupled β2-adrenergic signaling. Learn. Mem. 2011, 18, 598–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.; Redmond, D.E., Jr.; Baraban, J.M. Noradrenergic agonists and antagonists: Effects on conditioned fear as measured by the potentiated startle paradigm. Psychopharmacology 1979, 65, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Laitman, B.M.; Gajewski, N.D.; Mann, G.L.; Kubin, L.; Morrison, A.R.; Ross, R.J. The α1 adrenoceptor antagonist prazosin enhances sleep continuity in fear-conditioned Wistar–Kyoto rats. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2014, 49, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Mataix, L.; Piper, W.T.; Schiff, H.C.; Roberts, C.H.; Campese, V.D.; Sears, R.M.; LeDoux, J.E. Characterization of the amplificatory effect of norepinephrine in the acquisition of Pavlovian threat associations. Learn. Mem. 2017, 24, 432–439. [Google Scholar] [CrossRef] [Green Version]
- Branco, R.C.; Burkett, J.P.; Black, C.A.; Winokur, E.; Ellsworth, W.; Dhamsania, R.K.; Lohr, K.M.; Schroeder, J.P.; Weinshenker, D.; Jovanovic, T.; et al. Vesicular monoamine transporter 2 mediates fear behavior in mice. Genes Brain Behav. 2020, 19, e12634. [Google Scholar] [CrossRef]
- Selden, N.R.; Everitt, B.J.; Robbins, T.W. Telencephalic but not diencephalic noradrenaline depletion enhances behavioural but not endocrine measures of fear conditioning to contextual stimuli. Behav. Brain Res. 1991, 43, 139–154. [Google Scholar] [CrossRef]
- Selden, N.R.; Robbins, T.W.; Everitt, B.J. Enhanced behavioral conditioning to context and impaired behavioral and neuroendocrine responses to conditioned stimuli following ceruleocortical noradrenergic lesions: Support for an attentional hypothesis of central noradrenergic function. J. Neurosci. 1990, 10, 531–539. [Google Scholar] [CrossRef]
- Sara, S.J.; Roullet, P.; Przybyslawski, J. Consolidation of memory for odor-reward association: Beta-adrenergic receptor involvement in the late phase. Learn. Mem. 1999, 6, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Tronel, S.; Feenstra, M.G.; Sara, S.J. Noradrenergic Action in Prefrontal Cortex in the Late Stage of Memory Consolidation. Learn. Mem. 2004, 11, 453–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, B.J.; Robbins, T.W. Dissociable effects of lesions to the dorsal or ventral noradrenergic bundle on the acquisition, performance, and extinction of aversive conditioning. Behav. Neurosci. 1987, 101, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Fibiger, H.C.; Mason, S.T. The Effects of Dorsal Bundle Injections of 6-Hydroxydopamine on Avoidance Responding in Rats. Br. J. Pharmacol. 1978, 64, 601–605. [Google Scholar] [CrossRef] [Green Version]
- Harris, G.C.; Fitzgerald, R.D. Locus coeruleus involvement in the learning of classically conditioned bradycardia. J. Neurosci. 1991, 11, 2314–2320. [Google Scholar] [CrossRef] [Green Version]
- Langlais, P.J.; Connor, D.J.; Thal, L. Comparison of the effects of single and combined neurotoxic lesions of the nucleus basalis magnocellularis and dorsal noradrenergic bundle on learning and memory in the rat. Behav. Brain Res. 1993, 54, 81–90. [Google Scholar] [CrossRef]
- Mason, S.T.; Iversen, S.D. Learning in the absence of forebrain noradrenaline. Nature 1975, 258, 422–424. [Google Scholar] [CrossRef]
- Neophytou, S.I.; Aspley, S.; Butler, S.; Beckett, S.; Marsden, C.A. Effects of lesioning noradrenergic neurones in the locus coeruleus on conditioned and unconditioned aversive behaviour in the rat. Prog. Neuro. Psychopharmacol. Biol. Psychiatry 2001, 25, 1307–1321. [Google Scholar] [CrossRef]
- Płaźnik, A.; Kostowski, W. Locus coeruleus lesions and avoidance behavior in rats. Acta Neurobiol. Exp. 1980, 40, 217–225. [Google Scholar]
- Tsaltas, E.; Schugens, M.M.; Gray, J.A. Effects of lesions of the dorsal noradrenergic bundle on conditioned suppression to a CS and to a contextual background stimulus. Behav. Brain Res. 1989, 31, 243–256. [Google Scholar] [CrossRef]
- Amaral, D.G.; Foss, J.A. Locus Coeruleus Lesions and Learning. Science 1975, 188, 377–378. [Google Scholar] [CrossRef] [Green Version]
- Koob, G.F.; Kelley, A.E.; Mason, S.T. Locus coeruleus lesions: Learning and extinction. Physiol. Behav. 1978, 20, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Tsaltas, E.; Gray, J.A.; Fillenz, M. Alleviation of response suppression to conditioned aversive stimuli by lesions of the dorsal noradrenergic bundle. Behav. Brain Res. 1984, 13, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Holets, V.; Hökfelt, T.; Rökaeus, Å.; Terenius, L.; Goldstein, M. Locus coeruleus neurons in the rat containing neuropeptide Y, tyrosine hydroxylase or galanin and their efferent projections to the spinal cord, cerebral cortex and hypothalamus. Neuroscience 1988, 24, 893–906. [Google Scholar] [CrossRef]
- Birrell, J.M.; Brown, V.J. Medial frontal cortex mediates perceptual attentional set shifting in the rat. J. Neurosci. 2000, 20, 4320–4324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, L.; Cools, R.; Robbins, T. The neuropsychology of ventral prefrontal cortex: Decision-making and reversal learning. Brain Cogn. 2004, 55, 41–53. [Google Scholar] [CrossRef]
- Floresco, S.B.; Magyar, O.; Ghods-Sharifi, S.; Vexelman, C.; Tse, M.T.L. Multiple Dopamine Receptor Subtypes in the Medial Prefrontal Cortex of the Rat Regulate Set-Shifting. Neuropsychopharmacology 2006, 31, 297–309. [Google Scholar] [CrossRef]
- Aston-Jones, G.; Cohen, J.D. Adaptive gain and the role of the locus coeruleus-norepinephrine system in optimal performance. J. Comp. Neurol. 2005, 493, 99–110. [Google Scholar] [CrossRef]
- Aston-Jones, G.; Cohen, J.D. An Integrative Theory of Locus Coeruleus-Norepinephrine Function: Adaptive Gain and Optimal Performance. Annu. Rev. Neurosci. 2005, 28, 403–450. [Google Scholar] [CrossRef] [Green Version]
- Montague, P.R.; Dayan, P.; Sejnowski, T.J. A framework for mesencephalic dopamine systems based on predictive Hebbian learning. J. Neurosci. 1996, 16, 1936–1947. [Google Scholar] [CrossRef] [Green Version]
- Janitzky, K.; Lippert, M.T.; Engelhorn, A.; Tegtmeier, J.; Goldschmidt, J.; Heinze, H.-J.; Ohl, F.W. Optogenetic silencing of locus coeruleus activity in mice impairs cognitive flexibility in an attentional set-shifting task. Front. Behav. Neurosci. 2015, 9, 286. [Google Scholar] [CrossRef] [Green Version]
- Amara, S.G.; Kuhar, M.J. Neurotransmitter Transporters: Recent Progress. Annu. Rev. Neurosci. 1993, 16, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Giros, B.; Caron, M.G. Molecular characterization of the dopamine transporter. Trends Pharmacol. Sci. 1993, 14, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Giros, B.; Jaber, M.; Jones, S.R.; Wightman, R.M.; Caron, M.G. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 1996, 379, 606–612. [Google Scholar] [CrossRef]
- Pacholczyk, T.; Blakely, R.D.; Amara, S.G. Expression cloning of a cocaine-and antidepressant-sensitive human noradrenaline transporter. Nature 1991, 350, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Gainetdinov, R.R.; Wetsel, W.C.; Jones, S.R.; Bohn, L.M.; Miller, G.W.; Wang, Y.-M.; Caron, M.G. Mice lacking the norepinephrine transporter are supersensitive to psychostimulants. Nat. Neurosci. 2000, 3, 465–471. [Google Scholar] [CrossRef]
- Sulzer, D.; Sonders, M.S.; Poulsen, N.W.; Galli, A. Mechanisms of neurotransmitter release by amphetamines: A review. Prog. Neurobiol. 2005, 75, 406–433. [Google Scholar] [CrossRef]
- Darracq, L.; Blanc, G.; Glowinski, J.; Tassin, J.-P. Importance of the Noradrenaline–Dopamine Coupling in the Locomotor Activating Effects of d-Amphetamine. J. Neurosci. 1998, 18, 2729–2739. [Google Scholar] [CrossRef] [Green Version]
- Ecke, L.E.; Elmer, G.I.; Suto, N. Cocaine self-administration is not dependent upon mesocortical α1 noradrenergic signaling. Neuroreport 2012, 23, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Mitrano, D.; Pare, J.-F.; Smith, Y.; Weinshenker, D. D1-dopamine and α1-adrenergic receptors co-localize in dendrites of the rat prefrontal cortex. Neuroscience 2014, 258, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Begemann, K.; Neumann, A.M.; Oster, H. Regulation and function of extra-SCN circadian oscillators in the brain. Acta Physiol. 2020, 229, e13446. [Google Scholar] [CrossRef] [Green Version]
- Guilding, C.; Piggins, H.D. Challenging the omnipotence of the suprachiasmatic timekeeper: Are circadian oscillators present throughout the mammalian brain? Eur. J. Neurosci. 2007, 25, 3195–3216. [Google Scholar] [CrossRef] [PubMed]
- Kalsbeek, A.; Buijs, R.M. Organization of the neuroendocrine and autonomic hypothalamic paraventricular nucleus. Handb. Clin. Neurol. 2021, 180, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Kalsbeek, A.; van der Spek, R.; Lei, J.; Endert, E.; Buijs, R.M.; Fliers, E. Circadian rhythms in the hypothalamo–pituitary–adrenal (HPA) axis. Mol. Cell Endocrinol. 2012, 349, 20–29. [Google Scholar] [CrossRef]
- Nader, N.; Chrousos, G.P.; Kino, T. Interactions of the circadian CLOCK system and the HPA axis. Trends Endocrinol. Metab. 2010, 21, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Nicolaides, N.C.; Charmandari, E.; Chrousos, G.P.; Kino, T. Circadian endocrine rhythms: The hypothalamic-pituitary-adrenal axis and its actions. Ann. N. Y. Acad. Sci. 2014, 1318, 71–80. [Google Scholar] [CrossRef]
- Matsumura, T.; Nakagawa, H.; Suzuki, K.; Ninomiya, C.; Ishiwata, T. Influence of circadian disruption on neurotransmitter levels, physiological indexes, and behaviour in rats. Chronobiol. Int. 2015, 32, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Menon, J.M.L.; Nolten, C.; Achterberg, E.J.M.; Joosten, R.; Dematteis, M.; Feenstra, M.G.P.; Drinkenburg, W.H.P.; Leenaars, C.H.C. Brain Microdialysate Monoamines in Relation to Circadian Rhythms, Sleep, and Sleep Deprivation–a Systematic Review, Network Meta-analysis, and New Primary Data. J. Circadian Rhythm. 2019, 17, 1. [Google Scholar] [CrossRef] [Green Version]
- Aston-Jones, G.; Bloom, F. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J. Neurosci. 1981, 1, 876–886. [Google Scholar] [CrossRef] [Green Version]
- Hunsley, M.S.; Palmiter, R.D. Altered sleep latency and arousal regulation in mice lacking norepinephrine. Pharmacol. Biochem. Behav. 2004, 78, 765–773. [Google Scholar] [CrossRef]
- Ouyang, M.; Hellman, K.; Abel, T.; Thomas, S.A. Adrenergic Signaling Plays a Critical Role in the Maintenance of Waking and in the Regulation of REM Sleep. J. Neurophysiol. 2004, 92, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.A.; Palmiter, R.D. Examining Adrenergic Roles in Development, Physiology, and Behavior through Targeted Disruption of the Mouse Dopamine β-Hydroxylase Gene. Adv. Pharmacol. 1998, 42, 57–60. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Hopf, F.W.; Li, S.-B.; de Lecea, L. In vivo cell type-specific CRISPR knockdown of dopamine beta hydroxylase reduces locus coeruleus evoked wakefulness. Nat. Commun. 2018, 9, 5211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayat, H.; Regev, N.; Matosevich, N.; Sales, A.; Paredes-Rodriguez, E.; Krom, A.J.; Bergman, L.; Li, Y.; Lavigne, M.; Kremer, E.J.; et al. Locus coeruleus norepinephrine activity mediates sensory-evoked awakenings from sleep. Sci. Adv. 2020, 6, eaaz4232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, B.E.; Halaris, A.E.; McIlhany, M.; Moore, R.Y. Ascending projections of the locus coeruleus in the rat. I. axonal transport in central noradrenaline neurons. Brain Res. 1977, 127, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, L.A.; Miyamichi, K.; Gao, X.J.; Beier, K.T.; Weissbourd, B.; Deloach, K.E.; Ren, J.; Ibanes, S.; Malenka, R.C.; Kremer, E.J.; et al. Viral-genetic tracing of the input–output organization of a central noradrenaline circuit. Nature 2015, 524, 88–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, L.W.; Hartman, B.K. The central adrenergic system. An immunofluorescence study of the location of cell bodies and their efferent connections in the rat utilizing dopamine-B-hydroxylase as a marker. J. Comp. Neurol. 1975, 163, 467–505. [Google Scholar] [CrossRef]
- Isingrini, E.; Guinaudie, C.; Perret, L.C.; Rainer, Q.; Moquin, L.; Gratton, A.; Giros, B. Genetic elimination of dopamine vesicular stocks in the nigrostriatal pathway replicates Parkinson’s disease motor symptoms without neuronal degeneration in adult mice. Sci. Rep. 2017, 7, 12432. [Google Scholar] [CrossRef] [Green Version]
- Narboux-Nême, N.; Angenard, G.; Mosienko, V.; Klempin, F.; Pitychoutis, P.M.; Deneris, E.; Bader, M.; Giros, B.; Alenina, N.; Gaspar, P. Postnatal Growth Defects in Mice with Constitutive Depletion of Central Serotonin. ACS Chem. Neurosci. 2013, 4, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Narboux-Nême, N.; Sagné, C.; Doly, S.; Diaz, S.L.; Martin, C.B.P.; Angenard, G.; Martres, M.-P.; Giros, B.; Hamon, M.; Lanfumey, L.; et al. Severe Serotonin Depletion after Conditional Deletion of the Vesicular Monoamine Transporter 2 Gene in Serotonin Neurons: Neural and Behavioral Consequences. Neuropsychopharmacology 2011, 36, 2538–2550. [Google Scholar] [CrossRef] [Green Version]
- Robertson, D.; Haile, V.; Perry, S.E.; Robertson, R.M.; A Phillips, J.A., 3rd; Biaggioni, I. Dopamine beta-hydroxylase deficiency. A genetic disorder of cardiovascular regulation. Hypertension 1991, 18, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janz, R.; Südhof, T. SV2C is a synaptic vesicle protein with an unusually restricted localization: Anatomy of a synaptic vesicle protein family. Neuroscience 1999, 94, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Yeh, F.; Tepp, W.H.; Dean, C.; Johnson, E.A.; Janz, R.; Chapman, E.R. SV2 Is the Protein Receptor for Botulinum Neurotoxin A. Science 2006, 312, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Dardou, D.; Dassesse, D.; Cuvelier, L.; Deprez, T.; De Ryck, M.; Schiffmann, S.N. Distribution of SV2C mRNA and protein expression in the mouse brain with a particular emphasis on the basal ganglia system. Brain Res. 2011, 1367, 130–145. [Google Scholar] [CrossRef]
- Dunn, A.R.; Hoffman, C.A.; Stout, K.A.; Ozawa, M.; Dhamsania, R.K.; Miller, G.W. Immunochemical analysis of the expression of SV2C in mouse, macaque and human brain. Brain Res. 2019, 1702, 85–95. [Google Scholar] [CrossRef]
- Dardou, D.; Monlezun, S.; Foerch, P.; Courade, J.-P.; Cuvelier, L.; De Ryck, M.; Schiffmann, S. A role for Sv2c in basal ganglia functions. Brain Res. 2013, 1507, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Dunn, A.R.; Stout, K.A.; Ozawa, M.; Lohr, K.M.; Hoffman, C.A.; Bernstein, A.I.; Li, Y.; Wang, M.; Sgobio, C.; Sastry, N.; et al. Synaptic vesicle glycoprotein 2C (SV2C) modulates dopamine release and is disrupted in Parkinson disease. Proc. Natl. Acad. Sci. USA 2017, 114, E2253–E2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, V.; Han, M.-H.; Graham, D.L.; Berton, O.; Renthal, W.; Russo, S.J.; LaPlant, Q.; Graham, A.; Lutter, M.; Lagace, D.C.; et al. Molecular Adaptations Underlying Susceptibility and Resistance to Social Defeat in Brain Reward Regions. Cell 2007, 131, 391–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | DEG | Adj.p.value < 0.05 | Adj.p.value < 0.05 & l logFC l > 0.5 | Adj.p.value < 0.05 & l logFC l > 1 |
|---|---|---|---|---|
| Locus coeruleus | UP | 982 | 387 | 8 |
| DOWN | 1019 | 222 | 6 | |
| TOTAL | 2001 | 616 | 14 | |
| Nucleus accumbens | UP | 1462 | 345 | 14 |
| DOWN | 1627 | 343 | 8 | |
| TOTAL | 3089 | 688 | 22 | |
| Ventral tegmental area | UP | 1034 | 242 | 8 |
| DOWN | 1190 | 210 | 4 | |
| TOTAL | 2224 | 452 | 12 | |
| Prefrontal cortex | UP | 1006 | 186 | 9 |
| DOWN | 946 | 164 | 8 | |
| TOTAL | 1952 | 350 | 17 | |
| Raphe | UP | 1985 | 186 | 10 |
| DOWN | 2048 | 164 | 12 | |
| TOTAL | 4033 | 350 | 22 | |
| Dentate girus | UP | 784 | 382 | 38 |
| DOWN | 545 | 230 | 15 | |
| TOTAL | 1329 | 612 | 53 |
| Tested Modalities | Behavioral Paradigms | Measured Outcome | NE-Depletion Effects | ||
|---|---|---|---|---|---|
| Behavioral Results | Interpretations | ||||
| Anxiety- and depression-like parameters | Elevated plus maze | Time spend in the open arm | Ø | Anxiolitic and antidepressant effect | |
| Novelty suppressed feeding test | Eating latency | ↘ eating latency | |||
| Marble burying | Number of buried marbles | ↘ number of buried marbles | |||
| Force swim test | Immobility time | ↘ immobility time | |||
| Sucrose preference | Percentage of preference | Ø | |||
| CORT level | CORT level (ng/mL) | ↗ CORT level return | |||
| Dexamethasone suppression | Percentage of CORT level suppression | ↗ suppression of CORT level | |||
| Analgesia | Tail immersion | Latency of tail removal | Ø | No effect on analgesia | |
| Learning and memory | Contextual fear conditioning | Freezing time | ↗ freezing time | Enhance contextual fear memory | |
| Cued fear conditioning | Freezing time | Ø | |||
| Morris water maze | Spatial | - Latency to find the platform - Time spend in the active quadrant | Ø Ø | ||
| Reversal | - Latency to find the platform - Time spend in the target quadrant | Ø ↗ time in target quadrant | |||
| Rapid place learning | - Latency to find the platform - Time spend in the active quadrant | Ø Ø | |||
| Executive function | Attentional set shifting | Reversal | Number of trials to reach criteria | Ø | Alteration of cognitive flexibility |
| ID | Ø | ||||
| ED | ↗ trials number | ||||
| Addiction | Locomotor drug’s response | Cocaine | Horizontal activity | Ø | Decrease amphetamine-induced hyperlocomotion |
| Amphetamine | ↘ locomotor response | ||||
| Drug’s sensitization | Cocaine | 15-min Horizontal activity | Ø | ||
| Amphetamine | Ø | ||||
| Circadian cycle | Running wheel | - Circadian period - Body temperature - Daily activity | Ø Ø ↘ revolution | Decrease running wheel activity | |
| Telemetry | - Circadian period - Body temperature - Daily activity | Ø Ø Ø | |||
| Sleep analysis | % Time | Awake | ↘ time awake (Dark phase) | Changes in sleep quality | |
| NREM | ↗ time in NREM (Dark phase) | ||||
| REM | Ø | ||||
| Bouts number and duration | Awake | Ø | |||
| NREM | Ø | ||||
| REM | Ø | ||||
| Power spectrum | Awake | ↘ Delta (Dark phase) | |||
| NREM | ↗ Delta (Dark and light phases) | ||||
| REM | ↗ Theta (Light phase) | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isingrini, E.; Guinaudie, C.; Perret, L.; Guma, E.; Gorgievski, V.; Blum, I.D.; Colby-Milley, J.; Bairachnaya, M.; Mella, S.; Adamantidis, A.; et al. Behavioral and Transcriptomic Changes Following Brain-Specific Loss of Noradrenergic Transmission. Biomolecules 2023, 13, 511. https://doi.org/10.3390/biom13030511
Isingrini E, Guinaudie C, Perret L, Guma E, Gorgievski V, Blum ID, Colby-Milley J, Bairachnaya M, Mella S, Adamantidis A, et al. Behavioral and Transcriptomic Changes Following Brain-Specific Loss of Noradrenergic Transmission. Biomolecules. 2023; 13(3):511. https://doi.org/10.3390/biom13030511
Chicago/Turabian StyleIsingrini, Elsa, Chloé Guinaudie, Léa Perret, Elisa Guma, Victor Gorgievski, Ian D. Blum, Jessica Colby-Milley, Maryia Bairachnaya, Sébastien Mella, Antoine Adamantidis, and et al. 2023. "Behavioral and Transcriptomic Changes Following Brain-Specific Loss of Noradrenergic Transmission" Biomolecules 13, no. 3: 511. https://doi.org/10.3390/biom13030511
APA StyleIsingrini, E., Guinaudie, C., Perret, L., Guma, E., Gorgievski, V., Blum, I. D., Colby-Milley, J., Bairachnaya, M., Mella, S., Adamantidis, A., Storch, K. -F., & Giros, B. (2023). Behavioral and Transcriptomic Changes Following Brain-Specific Loss of Noradrenergic Transmission. Biomolecules, 13(3), 511. https://doi.org/10.3390/biom13030511