

A Molecular Analysis of the Aminopeptidase P-Related Domain of PID-5 from Caenorhabditis elegans


Abstract
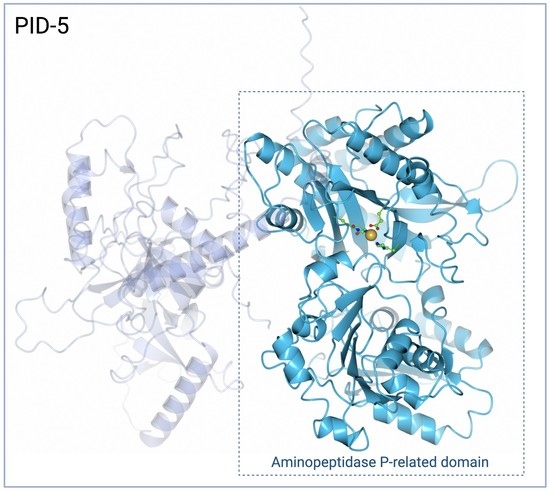
:
1. Introduction
2. Materials and Methods
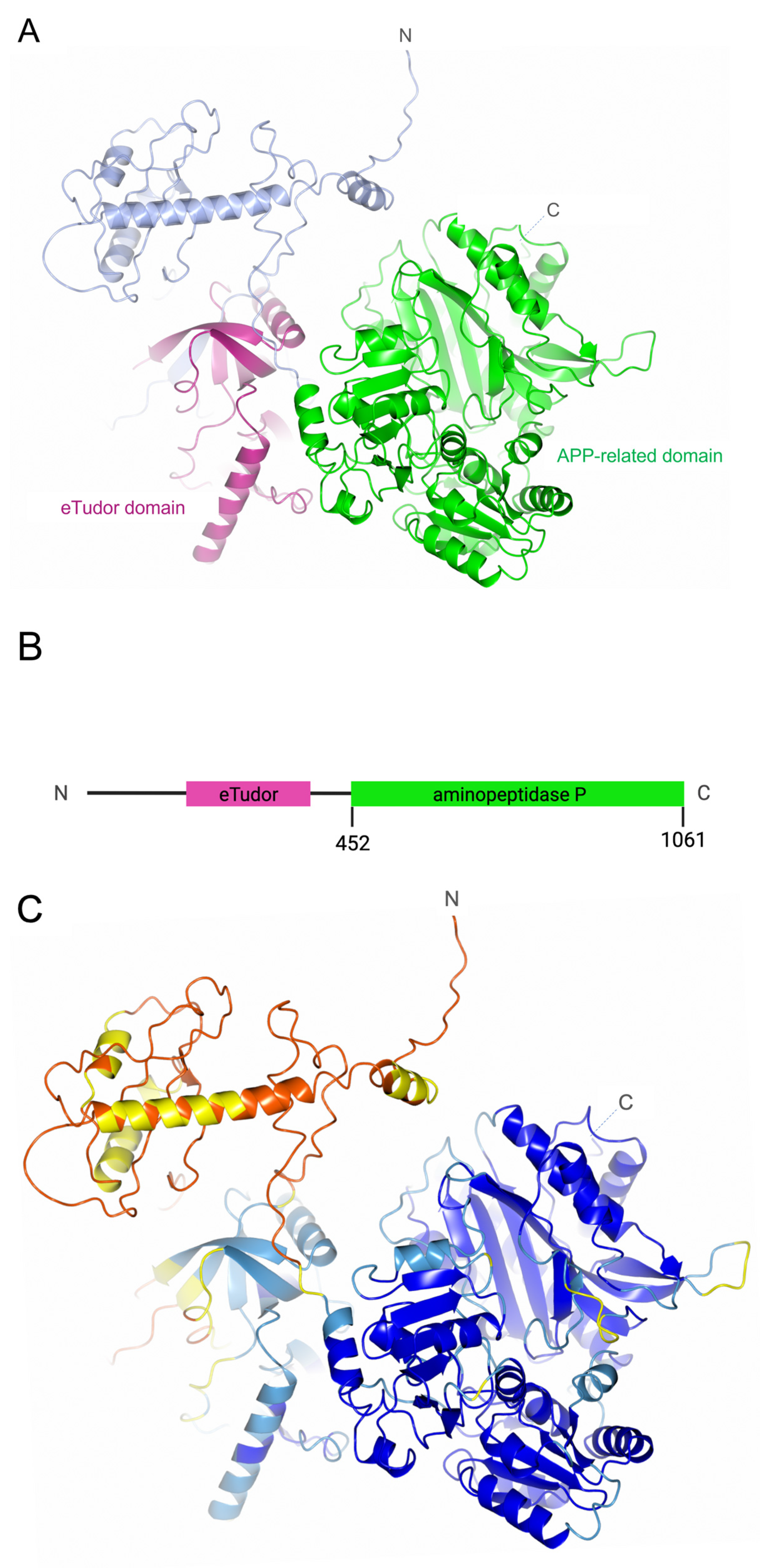
2.1. PID-5 and APP-1 Structures and Homology
2.2. Metal-Coordinating Residues and Composition
2.3. Apstatin Inhibitor Interactions
2.4. Dimerisation Interface Interactions
3. Results
3.1. PID-5 APP-RD and APP-1 Share High Amino Acid Sequence and Structural Homology
3.2. PID-5 APP-RD May Bind a Single Zinc Ion
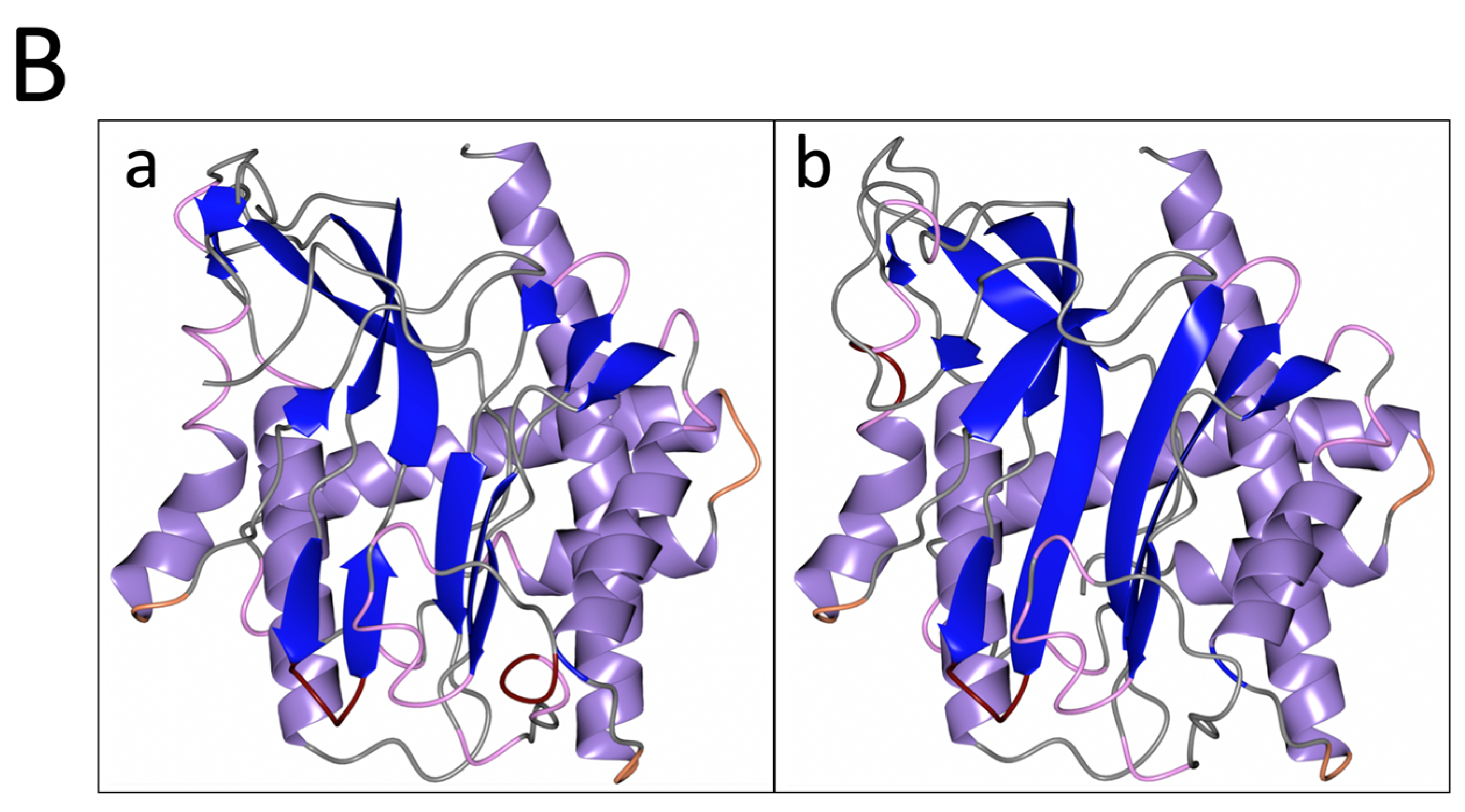
3.3. PID-5 APP-RD Could Bind the APP-1 Inhibitor Apstatin
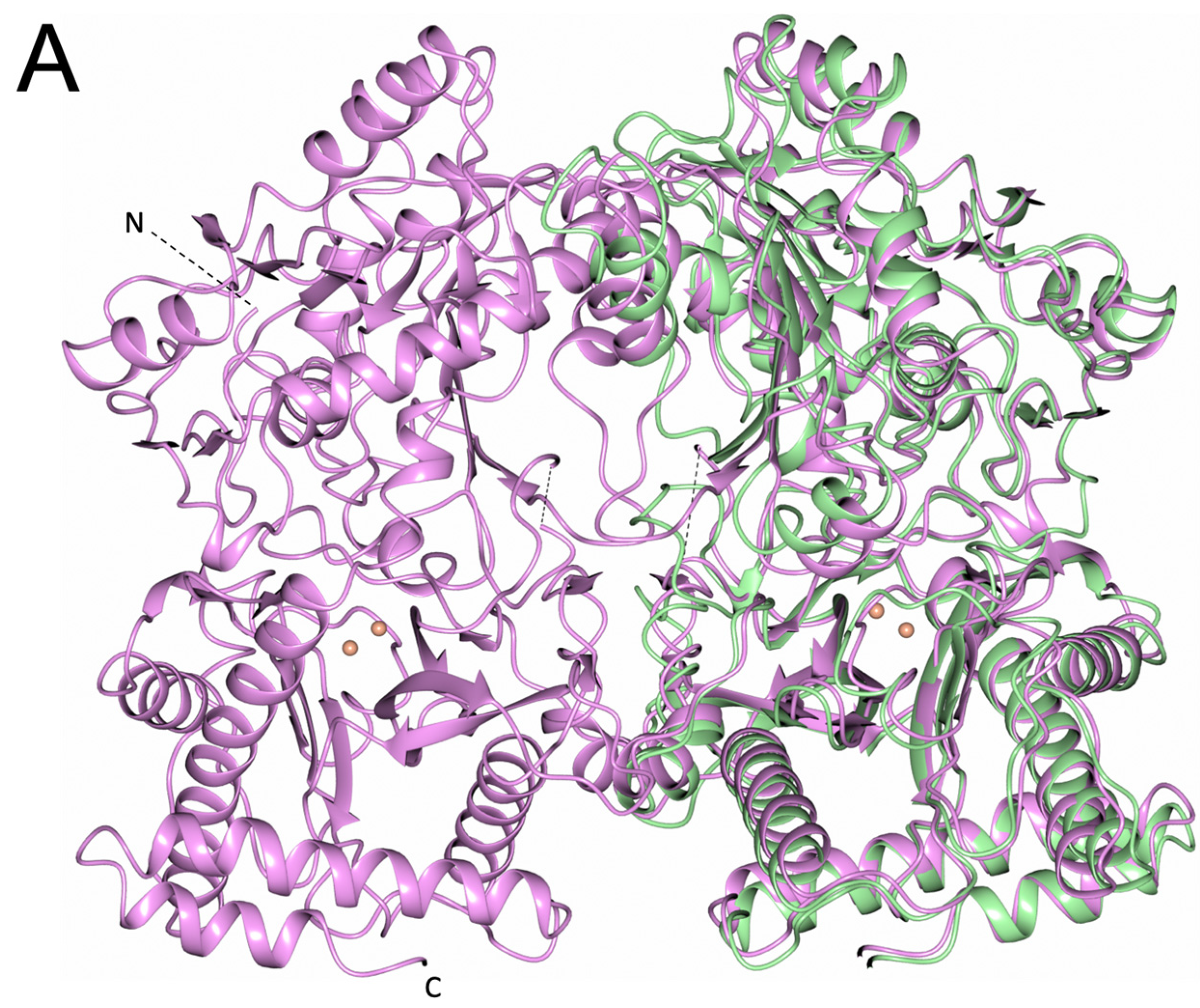
3.4. PID-5 APP-RD Could Heterodimerize with CeAPP-1
4. Discussion
4.1. AlphaFold2 and APP-1 Enabled Homology-Based Annotations
4.2. Mononuclear Zinc Binding PID-5 APP-RD Is Probably Catalytic Inactive
4.3. Apstatin Binding Is Likely to Be Transient in PID-5 APP-RD
4.4. Would Heterodimerisation Result in APP-1 Inactivity?
5. Biological Implications
Therapeutic Translational Research
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meier, B.; Volkova, N.V.; Hong, Y.; Bertolini, S.; Gonzalez-Huici, V.; Petrova, T.; Boulton, S.; Campbell, P.J.; Gerstung, M.; Gartner, A. Protection of the C. elegans germ cell genome depends on diverse DNA repair pathways during normal proliferation. PLoS ONE 2021, 16, e0250291. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.N.; Feschotte, C. A Field Guide to Eukaryotic Transposable Elements. Annu. Rev. Genet. 2020, 54, 539–561. [Google Scholar] [CrossRef] [PubMed]
- Toth, K.F.; Pezic, D.; Stuwe, E.; Webster, A. The piRNA Pathway Guards the Germline Genome Against Transposable Elements. In Non-Coding Rna and the Reproductive System; Wilhelm, D., Bernard, P., Eds.; Advances in Experimental Medicine and Biology; Springer: Dordrecht, Germany, 2016; Volume 886, pp. 51–77. [Google Scholar]
- Placentino, M.; Domingues, A.M.D.; Schreier, J.; Dietz, S.; Hellmann, S.; de Albuquerque, B.F.M.; Butter, F.; Ketting, R.F. Intrinsically disordered protein PID-2 modulates Z granules and is required for heritable piRNA-induced silencing in the Caenorhabditis elegans embryo. Embo J. 2021, 40, e105280. [Google Scholar] [CrossRef]
- Mani, S.R.; Juliano, C.E. Untangling the Web: The Diverse Functions of the PIWI/piRNA Pathway. Mol. Reprod. Dev. 2013, 80, 632–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, D.F.; O’Connor, B. Proline specific peptidases. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 1997, 1343, 160–186. [Google Scholar] [CrossRef] [PubMed]
- Silva, N.R.; Castellano-Pozo, M.; Matsuzaki, K.J.; Barroso, C.; Roman-Trufero, M.P.; Craig, H.; Brooks, D.P.; Isaac, R.E.; Boulton, S.P.; Martinez-Perez, E. Proline-specific aminopeptidase P prevents replication-associated genome instability. PLoS Genet. 2022, 18, e1010025. [Google Scholar] [CrossRef]
- Mills, B.; Isaac, R.E.; Foster, R. Metalloaminopeptidases of the Protozoan Parasite Plasmodium falciparum as Targets for the Discovery of Novel Antimalarial Drugs. J. Med. Chem. 2021, 64, 1763–1785. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D-Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.Z.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11 Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Holm, L. Dali server: Structural unification of protein families. Nucleic Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef]
- Hekkelman, M.L.; de Vries, I.; Joosten, R.P.; Perrakis, A. AlphaFill: Enriching AlphaFold models with ligands and cofactors. Nat. Methods 2023, 20, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- McNicholas, S.; Potterton, E.; Wilson, K.S.; Noble, M.E.M. Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr. Sect. D-Struct. Biol. 2011, 67, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Zidek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Holm, L. Using Dali for Protein Structure Comparison. In Structural Bioinformatics: Methods and Protocols; Gaspari, Z., Ed.; Methods in Molecular Biology; Humana: New York, NY, USA, 2020; Volume 2112, pp. 29–42. [Google Scholar]
- Peng, C.T.; Liu, L.; Li, C.C.; He, L.H.; Li, T.; Shen, Y.L.; Gao, C.; Wang, N.Y.; Xia, Y.; Zhu, Y.B.; et al. Structure-Function Relationship of Aminopeptidase P from Pseudomonas aeruginosa. Front. Microbiol. 2017, 8, 2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, S.C.; Maher, M.J.; Simmons, W.H.; Freeman, H.C.; Guss, J.M. Structure of Escherichia coli aminopeptidase P in complex with the inhibitor apstatin. Acta Crystallogr. Sect. D-Struct. Biol. 2004, 60, 1770–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mucha, A.; Drag, M.; Dalton, J.P.; Kafarski, P. Metallo-aminopeptidase inhibitors. Biochimie 2010, 92, 1509–1529. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.; La-Borde, P.J.; Payne, K.A.P.; Parsons, M.R.; Turner, A.J.; Isaac, R.E.; Acharya, K.R. Crystal structure of X-prolyl aminopeptidase from Caenorhabditis elegans: A cytosolic enzyme with a di-nuclear active site. FEBS Open Bio 2015, 5, 292–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Lou, Z.Y.; Li, X.M.; Zhou, W.H.; Ma, M.; Cao, Y.J.; Geng, Y.Q.; Bartlam, M.; Zhang, X.C.; Rao, Z.H. Structure of human cytosolic X-prolyl aminopeptidase—A double Mn(II)-dependent dimeric enzyme with a novel three-domain subunit. J. Biol. Chem. 2008, 283, 22858–22866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drinkwater, N.; Sivaraman, K.K.; Bamert, R.S.; Rut, W.; Mohamed, K.; Vinh, N.B.; Scammells, P.J.; Drag, M.; McGowan, S. Structure and substrate fingerprint of aminopeptidase P from Plasmodium falciparum. Biochem. J. 2016, 473, 3189–3204. [Google Scholar] [CrossRef]
- Graham, S.C.; Lee, M.; Freeman, H.C.; Guss, J.M. An orthorhombic form of Escherichia coli aminopeptidase P at 2.4 angstrom resolution. Acta Crystallogr. Sect. D-Struct. Biol. 2003, 59, 897–902. [Google Scholar] [CrossRef] [Green Version]
- Cottrell, G.S.; Hooper, N.M.; Turner, A.J. Cloning, expression, and characterization of human cytosolic aminopeptidase P: A single manganese(II)-dependent enzyme. Biochemistry 2000, 39, 15121–15128. [Google Scholar] [CrossRef]
- McColl, G.; James, S.A.; Mayo, S.; Howard, D.L.; Ryan, C.G.; Kirkham, R.; Moorhead, G.F.; Paterson, D.; de Jonge, M.D.; Bush, A.I. Caenorhabditis elegans Maintains Highly Compartmentalized Cellular Distribution of Metals and Steep Concentration Gradients of Manganese. PLoS ONE 2012, 7, e32685. [Google Scholar] [CrossRef] [Green Version]
- Fukasawa, K.M.; Hata, T.; Ono, Y.; Hirose, J. Metal Preferences of Zinc-Binding Motif on Metalloproteases. J. Amino Acids 2011, 2011, 574816. [Google Scholar] [CrossRef] [Green Version]
- Maggiora, L.L.; Orawski, A.T.; Simmons, W.H. Apstatin analogue inhibitors of aminopeptidase P, a bradykinin-degrading enzyme. J. Med. Chem. 1999, 42, 2394–2402. [Google Scholar] [CrossRef]
- Graham, S.C.; Lilley, P.E.; Lee, M.; Schaeffer, P.M.; Kralicek, A.V.; Dixon, N.E.; Guss, J.M. Kinetic and crystallographic analysis of mutant Escherichia coli aminopeptidase P: Insights into substrate recognition and the mechanism of catalysis. Biochemistry 2006, 45, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.C.; Bond, C.S.; Freeman, H.C.; Guss, J.M. Structural and functional implications of metal ion selection in aminopeptidase P, a metalloprotease with a dinuclear metal center. Biochemistry 2005, 44, 13820–13836. [Google Scholar] [CrossRef] [PubMed]
- Pek, J.W.; Anand, A.; Kai, T. Tudor domain proteins in development. Development 2012, 139, 2255–2266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Feng, X.Z.; Chen, X.Y.; Weng, C.C.; Yan, Q.; Xu, T.; Hong, M.J.; Guang, S.H. A Cytoplasmic Argonaute Protein Promotes the Inheritance of RNAi. Cell Rep. 2018, 23, 2482–2494. [Google Scholar] [CrossRef] [Green Version]
- Ishidate, T.; Ozturk, A.R.; Durning, D.J.; Sharma, R.; Shen, E.Z.; Chen, H.; Seth, M.; Shirayama, M.; Mello, C.C. ZNFX-1 Functions within Perinuclear Nuage to Balance Epigenetic Signals. Mol. Cell 2018, 70, 639–649.e6. [Google Scholar] [CrossRef] [Green Version]
- Phillips, C.M.; Updike, D.L. Germ granules and gene regulation in the Caenorhabditis elegans germline. Genetics 2022, 220, iyab195. [Google Scholar] [CrossRef]
- Laurent, V.; Brooks, D.R.; Coates, D.; Isaac, R.E. Functional expression and characterization of the cytoplasmic aminopeptidase P of Caenorhabditis elegans. Eur. J. Biochem. 2001, 268, 5430–5438. [Google Scholar] [CrossRef]
- Wan, G.; Fields, B.D.; Spracklin, G.; Shukla, A.; Phillips, C.M.; Kennedy, S. Spatiotemporal regulation of liquid-like condensates in epigenetic inheritance. Nature 2018, 557, 679–683. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Code | Identity (%) | Similarity (%) | |
|---|---|---|---|
| CeAPP-1 | 4S2R | 41.4 | 61.4 |
| HuAPP-1 | 3CTZ | 33.7 | 51.7 |
| PfAPP-1 | 5JQK | 25.0 | 45.7 |
| EcAPP-1 | 1WL9 | 16.6 | 27.8 |
| Rank | Protein | Z-Score | RMSD (Å) | ID (%) | Chain |
|---|---|---|---|---|---|
| 1 | CeAPP-1 | 47.9 | 1.8 | 44 | 4S2R-Q |
| 2 | CeAPP-1:Apstatin | 47.9 | 1.7 | 44 | 4S2T-Q |
| 3 | HuAPP-1 | 43.3 | 2.1 | 34 | 3CTZ-A |
| 4 | PfAPP-1:Apstatin | 41.9 | 2.0 | 26 | 5JR6-A |
| 5 | PfAPP-1 | 41.8 | 1.8 | 26 | 5JQK-A |
| 6 | EcAPP-1:Apstatin | 21.0 | 3.2 | 18 | 1N51-A |
| 7 | EcAPP-1 | 20.9 | 3.4 | 16 | 1WL9-A |
| PID-5 APP-RD | CeAPP-1 | HuAPP-1 | PfAPP-1 | EcAPP-1 | |
|---|---|---|---|---|---|
| MA | N868 | D424 | D426 | D581 | D271 |
| E967 | E522 | E523 | E676 | E383 | |
| G981 | E536 | E537 | E690 | E406 | |
| H932 | H487 | H489 | H644 | H354 | |
| MB | Q857 | D413 | D415 | D570 | D260 |
| N868 | D424 | D426 | D581 | D271 | |
| G981 | E536 | E537 | E690 | E406 |
| Compound | Identity (%) | PDB.Chain | Global RMSD (Å) | Local RMSD (Å) | TCS |
|---|---|---|---|---|---|
| Zn | 40 | 4S2R.A | 2.12 | 1.13 | 0.16 |
| Mn | 30 | 3CTZ.A | 3.66 | 0.33 | 0.19 |
| 25 | 5CDL.A | 2.97 | 0.39 | 0.00 | |
| Ca | 30 | 3CTZ.A | 3.66 | 1.36 | 0.05 |
| 25 | 3Q6D.A | 2.10 | 0.34 | 0.47 | |
| Na | 30 | 3CTZ.A | 3.66 | 0.37 | 0.11 |
| 25 | 5CDV.A | 2.98 | 0.41 | 0.00 | |
| 25 | 2ZSG.A | 9.33 | 0.02 | 0.28 | |
| 25 | 5GIU.A | 2.98 | 0.37 | 0.06 | |
| 25 | 2ZSG.B | 9.81 | 2.34 | 0.89 | |
| Co | 25 | 1WN1.A | 2.55 | 0.36 | 0.28 |
| 25 | 1WN1.A | 2.55 | 0.42 | 0.00 |
| PID-5 APP-RD | CeAPP-1 | PfAPP-1 | EcAPP-1 | HuAPP-1 |
|---|---|---|---|---|
| L492 | Y43 | I163 | - | Y42 |
| D525 | D76 | N196 | - | D75 |
| G527 | R78 | L198 | - | R77 |
| Q825 | F378 | F537 | Y229 | F381 |
| P826 | D379 | S538 | N230 | P382 |
| I828 | I381 | I540 | I232 | I384 |
| A839 | H392 | H551 | H243 | H395 |
| Q857 | D413 | D570 | D260 | D415 |
| N868 | D424 | D581 | D271 | D426 |
| H928 | H483 | H640 | H350 | H485 |
| E929 | G484 | G641 | G351 | G486 |
| G931 | G486 | G643 | S353 | G488 |
| H932 | H487 | H644 | H354 | H489 |
| I940 | V495 | V652 | V360 | V497 |
| R941 | H496 | H653 | H361 | H498 |
| E950 | R505 | V662 | R370 | K507 |
| E967 | E522 | E676 | E383 | E523 |
| R979 | R534 | R688 | R404 | R535 |
| G981 | E536 | E690 | E406 | E537 |
| CeAPP-1 Chain Q | CeAPP-1 Chain P | PID-5 APP-RD (Chain Q Equivalent) | PID-5 APP-RD (Chain P Equivalent) | ||
|---|---|---|---|---|---|
| Hydrogen bonds | 1 | K195 | V110 * | K642 | A557 |
| 2 | K195 | R111 * | K642 | K558 | |
| 3 | K195 | L113 * | K642 | L560 | |
| 4 | K136 | E301 | Q583 | E748 | |
| 5 | T468 | R465 * | I913 | R910 | |
| 6 | M547 * | L477 * | S992 | A922 | |
| 7 | S548 * | L477 * | Q993 | A922 | |
| 8 | S548 | G478 * | Q993 | G923 | |
| 9 | V110 * | K195 | A557 | K642 | |
| 10 | R111 * | K195 | K558 | K642 | |
| 11 | E301 | K136 | E748 | Q583 | |
| 12 | R465 * | T468 | R910 | I913 | |
| 13 | D467 | R465 | D912 | R910 | |
| 14 | L477 * | M547 * | A922 | S992 | |
| 15 | L477 * | S548 * | A922 | Q993 | |
| 16 | G478 * | S548 | G923 | Q993 | |
| Salt bridges | 1 | K136 | E301 | Q583 | E748 |
| 2 | R465 | D467 | R910 | D912 | |
| 3 | E301 | K136 | E748 | Q583 | |
| 4 | D467 | R465 | D912 | R910 |
| Protein Dimer | Solvent-Accessible Interface Area | |
|---|---|---|
| (Å2) | (%) | |
| CeAPP-1: CeAPP-1 | 2005.3 | 7.7 |
| CeAPP-1: PID-5 APP-RD | 1782.6 | 6.7 |
| PID-5 APP-RD: PID-5 APP-RD | 1574.4 | 5.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lloyd, A.C.; Gregory, K.S.; Isaac, R.E.; Acharya, K.R. A Molecular Analysis of the Aminopeptidase P-Related Domain of PID-5 from Caenorhabditis elegans. Biomolecules 2023, 13, 1132. https://doi.org/10.3390/biom13071132
Lloyd AC, Gregory KS, Isaac RE, Acharya KR. A Molecular Analysis of the Aminopeptidase P-Related Domain of PID-5 from Caenorhabditis elegans. Biomolecules. 2023; 13(7):1132. https://doi.org/10.3390/biom13071132
Chicago/Turabian StyleLloyd, Anna C., Kyle S. Gregory, R. Elwyn Isaac, and K. Ravi Acharya. 2023. "A Molecular Analysis of the Aminopeptidase P-Related Domain of PID-5 from Caenorhabditis elegans" Biomolecules 13, no. 7: 1132. https://doi.org/10.3390/biom13071132
APA StyleLloyd, A. C., Gregory, K. S., Isaac, R. E., & Acharya, K. R. (2023). A Molecular Analysis of the Aminopeptidase P-Related Domain of PID-5 from Caenorhabditis elegans. Biomolecules, 13(7), 1132. https://doi.org/10.3390/biom13071132