
Chronic Hepatitis B Infection: New Approaches towards Cure


 and
and {kind=link}
{kind=link}
Abstract
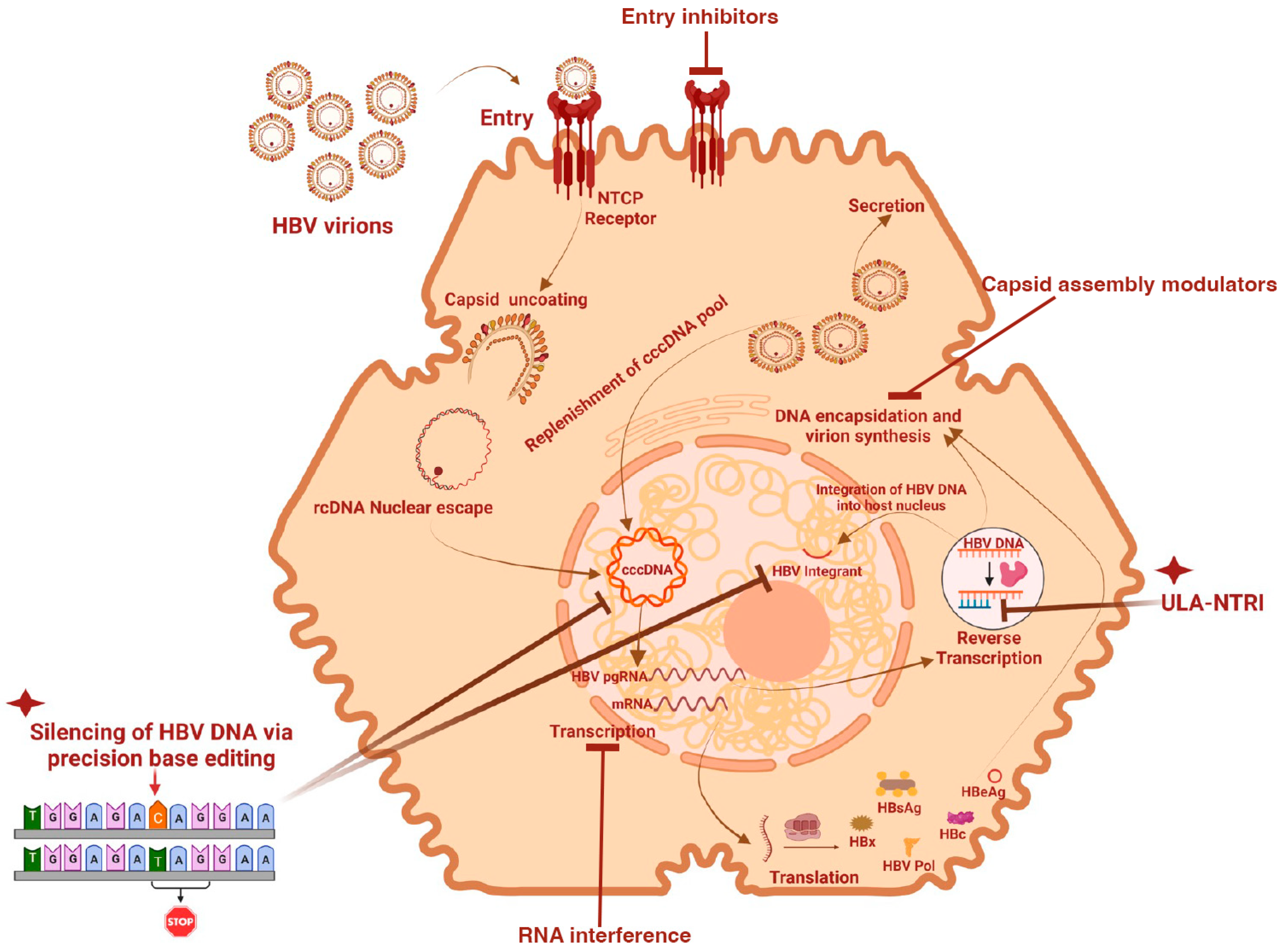
:1. Introduction
2. Chronic Hepatitis B (CHB) Outcomes
3. Available Therapies to Prevent HBV-Induced End-Stage Liver Diseases
3.1. Interferons
3.2. Nucleoside/Nucleotide Analogous
3.3. Classes of Anti-HBV Drugs in Development
3.3.1. Entry Inhibitors
3.3.2. Capsid Assembly Modulators
3.3.3. Immunomodulators
3.3.4. Gene Therapies
3.3.5. Small Molecule Inhibitors
4. Long-Acting Therapeutics
4.1. Long-Acting Injectables for Chronic HBV Treatments
4.2. Implants
4.2.1. TAF Implants
4.2.2. Entecavir Implant
4.2.3. Long-Acting Orals, Patches and Rings
5. Gene Editing Manipulations to Disrupt HBV cccDNA and Integrated Viral Genes
5.1. Application of Gene Editing Technology in Chronic Hepatitis B Infection
5.2. Inactivation of Persistent HBV DNA Forms Using Base Editors
6. Summary
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Littlejohn, M.; Locarnini, S.; Yuen, L. Origins and Evolution of Hepatitis B Virus and Hepatitis D Virus. Cold Spring Harb. Perspect. Med. 2016, 6, a021360. [Google Scholar] [CrossRef] [PubMed]
- Sheena, B.S.; Hiebert, L.; Han, H.; Ippolito, H.; Abbasi-Kangevari, M.; Abbasi-Kangevari, Z.; Abbastabar, H.; Abdoli, A.; Ali, H.A.; Adane, M.M.; et al. Global, regional, and national burden of hepatitis B, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Gastroenterol. Hepatol. 2022, 7, 796–829. [Google Scholar] [CrossRef] [PubMed]
- WHO. World Health Organization. Fact Sheets. Hepatitis B. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (accessed on 3 January 2023).
- Roberts, H.; Ly, K.N.; Yin, S.; Hughes, E.; Teshale, E.; Jiles, R. Prevalence of HBV Infection, Vaccine-Induced Immunity, and Susceptibility Among At-Risk Populations: US Households, 2013–2018. Hepatology 2021, 74, 2353–2365. [Google Scholar] [CrossRef]
- Weng, M.K.; Doshani, M.; Khan, M.A.; Frey, S.; Ault, K.; Moore, K.L.; Hall, E.W.; Morgan, R.L.; Campos-Outcalt, D.; Wester, C.; et al. Universal Hepatitis B Vaccination in Adults Aged 19–59 Years: Updated Recommendations of the Advisory Committee on Immunization Practices—United States, 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Soriano, V.; Aguilera, A.; Benito, R.; González-Díez, R.; Miró, E.; Liendo, P.; Rodríguez-Diaz, J.C.; Cabezas, T.; Richart, A.; Ramos, J.M.; et al. Susceptibility to hepatitis B virus infection in adults living in Spain. Liver Int. 2023, 43, 1015–1020. [Google Scholar] [CrossRef] [PubMed]
- Ricco, G.; Coco, B.; Colombatto, P.; Oliveri, F.; Cavallone, D.; Bleve, P.; Vianello, B.; Romagnoli, V.; Salvati, A.; Surace, L.; et al. Highly dynamic changes of regional HBV epidemiology over two decades. Dig. Liver Dis. 2022, 55, 519–526. [Google Scholar] [CrossRef]
- Hsu, Y.C.; Huang, D.Q.; Nguyen, M.H. Global burden of hepatitis B virus: Current status, missed opportunities and a call for action. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 524–537. [Google Scholar] [CrossRef]
- Lee, H.M.; Lidofsky, S.D.; Taddei, T.H.; Townshend-Bulson, L.J. Attacking the public health crisis of hepatocellular carcinoma at its roots. Hepatology 2022, 77, 1456–1459. [Google Scholar] [CrossRef]
- Razavi-Shearer, D.; Blach, S.; Gamkrelidze, I.; Estes, C.; Mooneyhan, E.; Razavi-Shearer, K.; Razavi, H. The disease burden of hepatitis B and hepatitis C from 2015 to 2030: The long and winding road. J. Hepatol. 2022, 77, S43. [Google Scholar] [CrossRef]
- Asselah, T.; Rizzetto, M. Hepatitis D Virus Infection. N. Engl. J. Med. 2023, 389, 58–70. [Google Scholar] [CrossRef]
- Iannacone, M.; Guidotti, L.G. Immunobiology and pathogenesis of hepatitis B virus infection. Nat. Rev. Immunol. 2021, 22, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.-Y.; Huang, B.; Huang, D.-P.; Wei, C.-S.; Zhong, W.-C.; Peng, D.-T.; Huang, F.-R.; Tong, G.-D. Long-term follow-up of cumulative incidence of hepatocellular carcinoma in hepatitis B virus patients without antiviral therapy. World J. Gastroenterol. 2021, 27, 1101–1116. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Cheng, H.B.; Wang, H.; Guo, Y. Meta-analysis of the occurrence of hepatocellular carcinoma after the treatment of entecavir and tenofovir for chronic hepatitis B. Medicine 2023, 102, e32894. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-H.; Wu, P.-F.; Chen, T.-I.; Chan, C.; Lin, H.-H.; Huang, Y.-H.; Chen, H.-Y.; Lin, Y.-T.; Chen, C.-J. Tenofovir use is associated with a decreased risk of hepatocellular carcinoma among men with HIV irrespective of coinfection status. JHEP Rep. 2022, 5, 100634. [Google Scholar] [CrossRef]
- Boeijen, L.L.; Hoogeveen, R.C.; Boonstra, A.; Lauer, G.M. Hepatitis B virus infection and the immune response: The big questions. Best Pr. Res. Clin. Gastroenterol. 2017, 31, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Maini, M.K.; Burton, A.R. Restoring, releasing or replacing adaptive immunity in chronic hepatitis B. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 662–675. [Google Scholar] [CrossRef]
- Indolfi, G.; Easterbrook, P.; Dusheiko, G.; Siberry, G.; Chang, M.-H.; Thorne, C.; Bulterys, M.; Chan, P.-L.; El-Sayed, M.H.; Giaquinto, C.; et al. Hepatitis B virus infection in children and adolescents. Lancet Gastroenterol. Hepatol. 2019, 4, 466–476. [Google Scholar] [CrossRef]
- Terrault, N.A.; Lok, A.S.F.; McMahon, B.J.; Chang, K.-M.; Hwang, J.P.; Jonas, M.M.; Brown, R.S., Jr.; Bzowej, N.H.; Wong, J.B. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology 2018, 67, 1560–1599. [Google Scholar] [CrossRef]
- Cornberg, M.; Lok, A.S.-F.; Terrault, N.A.; Zoulim, F. The 2019 EASL-AASLD HBV Treatment Endpoints Conference Faculty Guidance for Design and Endpoints of Clinical Trials in Chronic Hepatitis B—Report From the 2019 EASL-AASLD HBV Treatment Endpoints Conference. Hepatology 2019, 71, 1070–1092. [Google Scholar] [CrossRef]
- Kaur, S.P.; Talat, A.; Karimi-Sari, H.; Grees, A.; Chen, H.W.; Lau, D.T.Y.; Catana, A.M. Hepatocellular Carcinoma in Hepatitis B Virus-Infected Patients and the Role of Hepatitis B Surface Antigen (HBsAg). J. Clin. Med. 2022, 11, 1126. [Google Scholar] [CrossRef]
- Yeo, Y.H.; Ho, H.J.; Yang, H.-I.; Tseng, T.-C.; Hosaka, T.; Trinh, H.N.; Kwak, M.-S.; Park, Y.M.; Fung, J.Y.Y.; Buti, M.; et al. Factors Associated with Rates of HBsAg Seroclearance in Adults with Chronic HBV Infection: A Systematic Review and Meta-analysis. Gastroenterology 2019, 156, 635–646.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, Y.H.; Tseng, T.-C.; Hosaka, T.; Cunningham, C.; Fung, J.Y.Y.; Ho, H.J.; Kwak, M.-S.; Trinh, H.N.; Ungtrakul, T.; Yu, M.-L.; et al. Incidence, Factors, and Patient-Level Data for Spontaneous HBsAg Seroclearance: A Cohort Study of 11,264 Patients. Clin. Transl. Gastroenterol. 2020, 11, e00196. [Google Scholar] [CrossRef] [PubMed]
- Collatuzzo, G.; La Vecchia, C.; Parazzini, F.; Alicandro, G.; Turati, F.; Di Maso, M.; Malvezzi, M.; Pelucchi, C.; Negri, E.; Boffetta, P. Cancers attributable to infectious agents in Italy. Eur. J. Cancer 2023, 183, 69–78. [Google Scholar] [CrossRef]
- Tang, A.; Hallouch, O.; Chernyak, V.; Kamaya, A.; Sirlin, C.B. Epidemiology of hepatocellular carcinoma: Target population for surveillance and diagnosis. Abdom. Imaging 2017, 43, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, E.; Surial, B.; Boillat-Blanco, N.; Günthard, H.F.; Stöckle, M.; Bernasconi, E.; Schmid, P.; Calmy, A.; Suter-Riniker, F.; Rauch, A.; et al. Hepatitis B Virus (HBV) Replication During Tenofovir Therapy Is Frequent in Human Immunodeficiency Virus/HBV Coinfection. Clin. Infect. Dis. 2022, 76, 730–733. [Google Scholar] [CrossRef]
- Dezanet, L.N.C.; Kassime, R.; Miailhes, P.; Lascoux-Combe, C.; Chas, J.; Maylin, S.; Gabassi, A.; Rougier, H.; Delaugerre, C.; Lacombe, K.; et al. Effect of Viral Replication and Liver Fibrosis on All-Cause Mortality in Human Immunodeficiency Virus–Hepatitis B Virus–Coinfected Individuals: A Retrospective Analysis of a 15-Year Longitudinal Cohort. Clin. Infect. Dis. 2021, 74, 1012–1021. [Google Scholar] [CrossRef]
- Boyd, A.; Moh, R.; Badje, A.; Gabillard, D.; Ouattara, E.; Ntakpe, J.-B.; Maylin, S.; Chekaraou, M.A.; Zoulim, F.; Lacombe, K.; et al. Higher Mortality Despite Early Antiretroviral Therapy in Human Immunodeficiency Virus and Hepatitis B Virus (HBV)–Coinfected Patients with High HBV Replication. Clin. Infect. Dis. 2017, 66, 112–120. [Google Scholar] [CrossRef] [Green Version]
- Belloni, L.; Allweiss, L.; Guerrieri, F.; Pediconi, N.; Volz, T.; Pollicino, T.; Petersen, J.; Raimondo, G.; Dandri, M.; Levrero, M. IFN-α inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J. Clin. Investig. 2012, 122, 529–537. [Google Scholar] [CrossRef] [Green Version]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Perrillo, R. Benefits and risks of interferon therapy for hepatitis B. Hepatology 2009, 49, S103–S111. [Google Scholar] [CrossRef]
- Ren, H.; Huang, Y. Effects of pegylated interferon-α based therapies on functional cure and the risk of hepatocellular carcinoma development in patients with chronic hepatitis B. J. Viral Hepat. 2019, 26, 5–31. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wu, X.; Wei, W.; You, H.; Jia, J.; Kong, Y. A Meta-Analysis of the Efficacy of Interferon Monotherapy or Combined with Different Nucleos(t)ide Analogues for Chronic Hepatitis B. Int. J. Environ. Res. Public Health 2016, 13, 730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, P.; Cao, Z.; Mo, R.; Liu, Y.; Chen, L.; Honglian, G.; Zhou, T.; Lu, J.; Liu, Y.; Guo, Q.; et al. Interferon-based treatment is superior to nucleos(t)ide analog in reducing HBV-related hepatocellular carcinoma for chronic hepatitis B patients at high risk. Expert Opin. Biol. Ther. 2018, 18, 1085–1094. [Google Scholar] [CrossRef]
- Mao, Q.-G.; Liang, H.-Q.; Yin, Y.-L.; Tang, J.-M.; Yang, J.-E.; Wu, C.-C.; Chen, Y.; Zhang, M.-Y.; Liu, Y.-Y.; Zheng, X.-T.; et al. Comparison of Interferon-α-based therapy and nucleos(t)ide analogs in preventing adverse outcomes in patients with chronic hepatitis B. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101758. [Google Scholar] [CrossRef] [PubMed]
- Degasperi, E.; Vigano, M.; Aghemo, A.; Lampertico, P.; Colombo, M. PegIFN-alpha2a for the treatment of chronic hepatitis B and C: A 10-year history. Expert. Rev. Anti Infect. Ther. 2013, 11, 459–474. [Google Scholar] [CrossRef]
- Xia, Y.; Schlapschy, M.; Morath, V.; Roeder, N.; Vogt, E.I.; Stadler, D.; Cheng, X.; Dittmer, U.; Sutter, K.; Heikenwalder, M.; et al. PASylated interferon α efficiently suppresses hepatitis B virus and induces anti-HBs seroconversion in HBV-transgenic mice. Antivir. Res. 2018, 161, 134–143. [Google Scholar] [CrossRef]
- Negro, F. Adverse effects of drugs in the treatment of viral hepatitis. Best Pr. Res. Clin. Gastroenterol. 2010, 24, 183–192. [Google Scholar] [CrossRef]
- Menéndez-Arias, L.; Álvarez, M.; Pacheco, B. Nucleoside/nucleotide analog inhibitors of hepatitis B virus polymerase: Mechanism of action and resistance. Curr. Opin. Virol. 2014, 8, 1–9. [Google Scholar] [CrossRef]
- Dienstag, J.L. Benefits and risks of nucleoside analog therapy for hepatitis B. Hepatology 2009, 49, S112–S121. [Google Scholar] [CrossRef]
- Kayaaslan, B.; Guner, R. Adverse effects of oral antiviral therapy in chronic hepatitis B. World J. Hepatol. 2017, 9, 227–241. [Google Scholar] [CrossRef]
- Xie, G.-J.; Zhang, H.-Y.; Chen, Q.; Liu, H.-M.; You, J.-P.; Yang, S.; Mao, Q.; Zhang, X.-Q. Changing etiologies and outcome of liver failure in Southwest China. Virol. J. 2016, 13, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Xiao, G.; Liao, M.; Zheng, L.; Jie, Y.; Lin, G.; Chong, Y. Inappropriate cessation of nucleos(t)ide analog associated with reduced liver transplant-free survival in patients with HBV-related acute on chronic liver failure. Biomed. Pharmacother. 2020, 134, 111118. [Google Scholar] [CrossRef]
- Kim, W.R.; Loomba, R.; Berg, T.; Schall, R.E.A.; Yee, L.J.; Dinh, P.V.; Flaherty, J.F.; Martins, E.B.; Therneau, T.M.; Jacobson, I.; et al. Impact of long-term tenofovir disoproxil fumarate on incidence of hepatocellular carcinoma in patients with chronic hepatitis B. Cancer 2015, 121, 3631–3638. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, T.; Suzuki, F.; Kobayashi, M.; Seko, Y.; Kawamura, Y.; Sezaki, H.; Akuta, N.; Suzuki, Y.; Saitoh, S.; Arase, Y.; et al. Long-term entecavir treatment reduces hepatocellular carcinoma incidence in patients with hepatitis B virus infection. Hepatology 2012, 58, 98–107. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, Y.; Yang, J.; Hu, K.; Huang, Y. The effectiveness of TDF versus ETV on incidence of HCC in CHB patients: A meta analysis. BMC Cancer 2019, 19, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Choi, W.-M.; Choi, J.; Lim, Y.-S. Effects of Tenofovir vs Entecavir on Risk of Hepatocellular Carcinoma in Patients with Chronic HBV Infection: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2020, 19, 246–258.e9. [Google Scholar] [CrossRef]
- Tseng, C.-H.; Hsu, Y.-C.; Chen, T.-H.; Ji, F.; Chen, I.-S.; Tsai, Y.-N.; Hai, H.; Thuy, L.T.T.; Hosaka, T.; Sezaki, H.; et al. Hepatocellular carcinoma incidence with tenofovir versus entecavir in chronic hepatitis B: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2020, 5, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Li, R.; Huo, R.; Li, M.; Papatheodoridis, G.; Zhong, J. Lower risk of hepatocellular carcinoma with tenofovir than entecavir treatment in subsets of chronic hepatitis B patients: An updated meta-analysis. J. Gastroenterol. Hepatol. 2022, 37, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Yue-Meng, W.; Li, Y.-H.; Wu, H.-M.; Yang, J.; Xu, Y.; Yang, L.-H.; Yang, J.-H. Telbivudine versus lamivudine and entecavir for treatment-naïve decompensated hepatitis B virus-related cirrhosis. Clin. Exp. Med. 2016, 17, 233–241. [Google Scholar] [CrossRef]
- Hou, J.-L.; Xu, D.; Shi, G.; Wan, M.; Goodman, Z.; Tan, D.; Xie, Q.; Chen, C.; Wei, L.; Niu, J.; et al. Long-Term Telbivudine Treatment Results in Resolution of Liver Inflammation and Fibrosis in Patients with Chronic Hepatitis B. Adv. Ther. 2015, 32, 727–741. [Google Scholar] [CrossRef] [Green Version]
- Yuen, M.-F.; Seto, W.-K.; Chow, D.H.-F.; Tsui, K.; Wong, D.K.-H.; Ngai, V.W.-S.; Wong, B.C.-Y.; Fung, J.; Yuen, J.C.-H.; Lai, C.-L. Long-term lamivudine therapy reduces the risk of long-term complications of chronic hepatitis B infection even in patients without advanced disease. Antivir. Ther. 2007, 12, 1295–1304. [Google Scholar] [CrossRef]
- Singal, A.K.; Salameh, H.; Kuo, Y.-F.; Fontana, R.J. Meta-analysis: The impact of oral anti-viral agents on the incidence of hepatocellular carcinoma in chronic hepatitis B. Aliment. Pharmacol. Ther. 2013, 38, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoulim, F.; Locarnini, S. Hepatitis B Virus Resistance to Nucleos(t)ide Analogues. Gastroenterology 2009, 137, 1593–1608.e2. [Google Scholar] [CrossRef]
- World Health Organization. Consolidated Guidelines on HIV Prevention, Testing, Treatment, Service Delivery and Monitoring: Recommendations for a Public Health Approach; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Mizushima, D.; Takano, M.; Aoki, T.; Ando, N.; Uemura, H.; Yanagawa, Y.; Watanabe, K.; Gatanaga, H.; Kikuchi, Y.; Oka, S. Effect of tenofovir-based HIV pre-exposure prophylaxis against HBV infection in men who have sex with men. Hepatology 2023, 77, 2084–2092. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Xia, H.; He, Y.; Zeng, S.; Shen, Z.; Huang, W. The progress of molecules and strategies for the treatment of HBV infection. Front. Cell. Infect. Microbiol. 2023, 13, 1128807. [Google Scholar] [CrossRef]
- Donkers, J.M.; Zehnder, B.; van Westen, G.J.P.; Kwakkenbos, M.J.; Ijzerman, A.P.; Oude Elferink, R.P.J.; Beuers, U.; Urban, S.; Van De Graaf, S.F.J. Reduced hepatitis B and D viral entry using clinically applied drugs as novel inhibitors of the bile acid transporter NTCP. Sci. Rep. 2017, 7, 15307. [Google Scholar] [CrossRef] [Green Version]
- Blank, A.; Markert, C.; Hohmann, N.; Carls, A.; Mikus, G.; Lehr, T.; Alexandrov, A.; Haag, M.; Schwab, M.; Urban, S.; et al. First-in-human application of the novel hepatitis B and hepatitis D virus entry inhibitor myrcludex B. J. Hepatol. 2016, 65, 483–489. [Google Scholar] [CrossRef]
- Schieck, A.; Schulze, A.; Gähler, C.; Müller, T.; Haberkorn, U.; Alexandrov, A.; Urban, S.; Mier, W. Hepatitis B virus hepatotropism is mediated by specific receptor recognition in the liver and not restricted to susceptible hosts. Hepatology 2013, 58, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Berke, J.M.; Dehertogh, P.; Vergauwen, K.; Van Damme, E.; Mostmans, W.; Vandyck, K.; Pauwels, F. Capsid Assembly Modulators Have a Dual Mechanism of Action in Primary Human Hepatocytes Infected with Hepatitis B Virus. Antimicrob. Agents Chemother. 2017, 61, e00560-17. [Google Scholar] [CrossRef] [Green Version]
- Zhao, N.; Jia, B.; Zhao, H.; Xu, J.; Sheng, X.; Luo, L.; Huang, Z.; Wang, X.; Ren, Q.; Zhang, Y.; et al. A First-in-Human Trial of GLS4, a Novel Inhibitor of Hepatitis B Virus Capsid Assembly, following Single- and Multiple-Ascending-Oral-Dose Studies with or without Ritonavir in Healthy Adult Volunteers. Antimicrob. Agents Chemother. 2019, 64, e01686-19. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, F.; Zhu, X.; Chen, Y.; Chen, H.; Li, X.; Wu, M.; Li, C.; Liu, J.; Zhang, Y.; et al. Antiviral Activity and Pharmacokinetics of the Hepatitis B Virus (HBV) Capsid Assembly Modulator GLS4 in Patients with Chronic HBV Infection. Clin. Infect. Dis. 2020, 73, 175–182. [Google Scholar] [CrossRef]
- Yuen, M.F.; Gane, E.J.; Kim, D.J.; Weilert, F.; Chan, H.L.Y.; Lalezari, J.; Hwang, S.G.; Nguyen, T.; Flores, O.; Hartman, G.; et al. Antiviral Activity, Safety, and Pharmacokinetics of Capsid Assembly Modulator NVR 3-778 in Patients with Chronic HBV Infection. Gastroenterology 2019, 156, 1392–1403.e7. [Google Scholar] [CrossRef] [Green Version]
- Gane, E.J.; Kim, H.J.; Visvanathan, K.; Kim, Y.J.; Nguyen, A.; Wallin, J.J.; Chen, D.Y.; McDonald, C.; Arora, P.; Tan, S.K.; et al. Safety, Pharmacokinetics, and Pharmacodynamics of the Oral TLR8 Agonist Selgantolimod in Chronic Hepatitis B. Hepatology 2021, 74, 1737–1749. [Google Scholar] [CrossRef]
- Janssen, H.L.; Brunetto, M.R.; Kim, Y.J.; Ferrari, C.; Massetto, B.; Nguyen, A.-H.; Joshi, A.; Woo, J.; Lau, A.H.; Gaggar, A.; et al. Safety, efficacy and pharmacodynamics of vesatolimod (GS-9620) in virally suppressed patients with chronic hepatitis B. J. Hepatol. 2018, 68, 431–440. [Google Scholar] [CrossRef]
- Lok, A.S.; McMahon, B.J.; Brown, R.S.; Wong, J.B.; Ahmed, A.T.; Farah, W.; Almasri, J.; Alahdab, F.; Benkhadra, K.; Mouchli, M.A.; et al. Antiviral therapy for chronic hepatitis B viral infection in adults: A systematic review and meta-analysis. Hepatology 2015, 63, 284–306. [Google Scholar] [CrossRef]
- Boni, C.; Vecchi, A.; Rossi, M.; Laccabue, D.; Giuberti, T.; Alfieri, A.; Lampertico, P.; Grossi, G.; Facchetti, F.; Brunetto, M.R.; et al. TLR7 Agonist Increases Responses of Hepatitis B Virus–Specific T Cells and Natural Killer Cells in Patients with Chronic Hepatitis B Treated with Nucleos(T)Ide Analogues. Gastroenterology 2018, 154, 1764–1777.e7. [Google Scholar] [CrossRef]
- Boni, C.; Janssen, H.L.; Rossi, M.; Yoon, S.K.; Vecchi, A.; Barili, V.; Yoshida, E.M.; Trinh, H.; Rodell, T.C.; Laccabue, D.; et al. Combined GS-4774 and Tenofovir Therapy Can Improve HBV-Specific T-Cell Responses in Patients with Chronic Hepatitis. Gastroenterology 2019, 157, 227–241.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Lim, T.H.; Leerapun, A.; Weltman, M.; Jia, J.; Lim, Y.-S.; Tangkijvanich, P.; Sukeepaisarnjaroen, W.; Ji, Y.; Le Bert, N.; et al. Therapeutic vaccine BRII-179 restores HBV-specific immune responses in patients with chronic HBV in a phase Ib/IIa study. JHEP Rep. 2021, 3, 100361. [Google Scholar] [CrossRef] [PubMed]
- Zoulim, F.; Fournier, C.; Habersetzer, F.; Sprinzl, M.; Pol, S.; Coffin, C.S.; Leroy, V.; Ma, M.; Wedemeyer, H.; Lohse, A.W.; et al. Safety and immunogenicity of the therapeutic vaccine TG1050 in chronic hepatitis B patients: A phase 1b placebo-controlled trial. Hum. Vaccines Immunother. 2019, 16, 388–399. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.; Khurana, A.; Allawadhi, P.; Banothu, A.K.; Bharani, K.K.; Weiskirchen, R. Emerging Role of PD-1/PD-L1 Inhibitors in Chronic Liver Diseases. Front. Pharmacol. 2021, 12, 790963. [Google Scholar] [CrossRef] [PubMed]
- Pu, D.; Yin, L.; Zhou, Y.; Li, W.; Huang, L.; Cai, L.; Zhou, Q. Safety and efficacy of immune checkpoint inhibitors in patients with HBV/HCV infection and advanced-stage cancer: A systematic review. Medicine 2020, 99, e19013. [Google Scholar] [CrossRef]
- Michel, M.-L.; Deng, Q.; Mancini-Bourgine, M. Therapeutic vaccines and immune-based therapies for the treatment of chronic hepatitis B: Perspectives and challenges. J. Hepatol. 2011, 54, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- Hui, R.W.-H.; Mak, L.-Y.; Seto, W.-K.; Yuen, M.-F. RNA interference as a novel treatment strategy for chronic hepatitis B infection. Clin. Mol. Hepatol. 2022, 28, 408–424. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.-F.; Lim, S.-G.; Plesniak, R.; Tsuji, K.; Janssen, H.L.; Pojoga, C.; Gadano, A.; Popescu, C.P.; Stepanova, T.; Asselah, T.; et al. Efficacy and Safety of Bepirovirsen in Chronic Hepatitis B Infection. N. Engl. J. Med. 2022, 387, 1957–1968. [Google Scholar] [CrossRef] [PubMed]
- Turton, K.L.; Meier-Stephenson, V.; Badmalia, M.D.; Coffin, C.S.; Patel, T.R. Host Transcription Factors in Hepatitis B Virus RNA Synthesis. Viruses 2020, 12, 160. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.-Q.; Ding, Q.-Y.; Tao, N.-N.; Tan, M.; Zhang, Y.; Li, F.; Zhou, Y.-J.; Dong, M.-L.; Cheng, S.-T.; Ren, F.; et al. SIRT2 Promotes HBV Transcription and Replication by Targeting Transcription Factor p53 to Increase the Activities of HBV Enhancers and Promoters. Front. Microbiol. 2022, 13, 836446. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, Q.; Zhang, J.D.; Zhang, Y.; Ni, X.; Xiang, K.; Jiang, J.; Li, B.; Yu, Y.; Hu, H.; et al. Discovery of a first-in-class orally available HBV cccDNA inhibitor. J. Hepatol. 2022, 78, 742–753. [Google Scholar] [CrossRef]
- Gorsuch, C.L.; Nemec, P.; Yu, M.; Xu, S.; Han, D.; Smith, J.; Lape, J.; van Buuren, N.; Ramirez, R.; Muench, R.C.; et al. Targeting the hepatitis B cccDNA with a sequence-specific ARCUS nuclease to eliminate hepatitis B virus in vivo. Mol. Ther. 2022, 30, 2909–2922. [Google Scholar] [CrossRef]
- Mueller, H.; Wildum, S.; Luangsay, S.; Walther, J.; Lopez, A.; Tropberger, P.; Ottaviani, G.; Lu, W.; Parrott, N.J.; Zhang, J.D.; et al. A novel orally available small molecule that inhibits hepatitis B virus expression. J. Hepatol. 2018, 68, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Ge, X.; Jin, H.; Lu, D.; Chen, S.; Zhang, Y.; Wang, X.; Xu, H.; Ao, W.; Zhang, Y. Discovery, optimization and biological evaluation of novel HBsAg production inhibitors. Eur. J. Med. Chem. 2023, 256, 115387. [Google Scholar] [CrossRef]
- Zoulim, F.; Testoni, B. Eliminating cccDNA to cure hepatitis B virus infection. J. Hepatol. 2023, 78, 677–680. [Google Scholar] [CrossRef]
- United Nations. Contraceptive Use by Method 2019: Data Booklet; United Nations: New York, NY, USA, 2019. [Google Scholar]
- Taylor, M.; Perera, U. NICE CG178 Psychosis and Schizophrenia in Adults: Treatment and Management—An evidence-based guideline? Br. J. Psychiatry 2015, 206, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llorca, P.M.; Abbar, M.; Courtet, P.; Guillaume, S.; Lancrenon, S.; Samalin, L. Guidelines for the use and management of long-acting injectable antipsychotics in serious mental illness. BMC Psychiatry 2013, 13, 340. [Google Scholar] [CrossRef] [Green Version]
- Cobb, D.A.; Smith, N.A.; Edagwa, B.J.; McMillan, J.M. Long-acting approaches for delivery of antiretroviral drugs for prevention and treatment of HIV: A review of recent research. Expert Opin. Drug Deliv. 2020, 17, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Paik, J. Lenacapavir: First Approval. Drugs 2022, 82, 1499–1504. [Google Scholar] [CrossRef]
- Markham, A. Cabotegravir Plus Rilpivirine: First Approval. Drugs 2020, 80, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Healthcare, V. Cabenuva®® Prescribing Information; Viiv Healthcare: Research Triangle Park, NC, USA, 2021. [Google Scholar]
- Delany-Moretlwe, S.; Hughes, J.P.; Bock, P.; Ouma, S.G.; Hunidzarira, P.; Kalonji, D.; Kayange, N.; Makhema, J.; Mandima, P.; Mathew, C.; et al. Cabotegravir for the prevention of HIV-1 in women: Results from HPTN 084, a phase 3, randomised clinical trial. Lancet 2022, 399, 1779–1789. [Google Scholar] [CrossRef]
- Venkatesan, P. Long-acting injectable ART for HIV: A (cautious) step forward. Lancet Microbe 2022, 3, e94. [Google Scholar] [CrossRef]
- Cobb, D.A.; Smith, N.; Deodhar, S.; Bade, A.N.; Gautam, N.; Shetty, B.L.D.; McMillan, J.; Alnouti, Y.; Cohen, S.M.; Gendelman, H.E.; et al. Transformation of tenofovir into stable ProTide nanocrystals with long-acting pharmacokinetic profiles. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Ho, M.J.; Lee, D.R.; Im, S.H.; A Yoon, J.; Shin, C.Y.; Kim, H.J.; Jang, S.W.; Choi, Y.W.; Han, Y.T.; Kang, M.J. Design and in vivo evaluation of entecavir-3-palmitate microcrystals for subcutaneous sustained delivery. Eur. J. Pharm. Biopharm. 2018, 130, 143–151. [Google Scholar] [CrossRef]
- Soni, D.; Bade, A.N.; Gautam, N.; Herskovitz, J.; Ibrahim, I.M.; Smith, N.; Wojtkiewicz, M.S.; Shetty, B.L.D.; Alnouti, Y.; McMillan, J.; et al. Synthesis of a long acting nanoformulated emtricitabine ProTide. Biomaterials 2019, 222, 119441. [Google Scholar] [CrossRef]
- Mandal, S.; Kang, G.; Prathipati, P.K.; Zhou, Y.; Fan, W.; Li, Q.; Destache, C.J. Nanoencapsulation introduces long-acting phenomenon to tenofovir alafenamide and emtricitabine drug combination: A comparative pre-exposure prophylaxis efficacy study against HIV-1 vaginal transmission. J. Control. Release 2019, 294, 216–225. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Bade, A.N.; Lin, Z.; Soni, D.; Wojtkiewicz, M.; Shetty, B.L.D.; Gautam, N.; McMillan, J.M.; Alnouti, Y.; Edagwa, B.J.; et al. Synthesis and characterization of a long-acting emtricitabine prodrug nanoformulation. Int. J. Nanomed. 2019, ume 14, 6231–6247. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Smith, N.; Makarov, E.; Sun, Y.; Gebhart, C.L.; Ganesan, M.; Osna, N.A.; Gendelman, H.E.; Edagwa, B.J.; Poluektova, L.Y. A long-acting 3TC ProTide nanoformulation suppresses HBV replication in humanized mice. Nanomed. Nanotechnol. Biol. Med. 2020, 28, 102185. [Google Scholar] [CrossRef]
- Smith, N.; Bade, A.N.; Soni, D.; Gautam, N.; Alnouti, Y.; Herskovitz, J.; Ibrahim, I.M.; Wojtkiewicz, M.S.; Shetty, B.L.D.; McMillan, J.; et al. A long acting nanoformulated lamivudine ProTide. Biomaterials 2019, 223, 119476. [Google Scholar] [CrossRef] [PubMed]
- McConnachie, L.A.; Kinman, L.M.; Koehn, J.; Kraft, J.C.; Lane, S.; Lee, W.; Collier, A.C.; Ho, R.J. Long-Acting Profile of 4 Drugs in 1 Anti-HIV Nanosuspension in Nonhuman Primates for 5 Weeks After a Single Subcutaneous Injection. J. Pharm. Sci. 2018, 107, 1787–1790. [Google Scholar] [CrossRef]
- Guo, D.; Zhou, T.; Arainga, M.; Palandri, D.; Gautam, N.; Bronich, T.; Alnouti, Y.; McMillan, J.; Edagwa, B.; Gendelman, H.E. Creation of a Long-Acting Nanoformulated 2′,3′-Dideoxy-3′-Thiacytidine. J. Acquir. Immune Defic. Syndr. 2017, 74, e75–e83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castor, D.; Meyers, K.; Allen, S. The only way is up: Priorities for implementing long-acting antiretrovirals for HIV prevention and treatment. Curr. Opin. HIV AIDS 2020, 15, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weld, E.D.; Rana, S.; Dallas, R.H.; Camacho-Gonzalez, A.F.; Ryscavage, P.; Gaur, A.; Chakraborty, R.; Swindells, S.; Flexner, C.; Agwu, A.L. Interest of Youth Living with HIV in Long-Acting Antiretrovirals. Am. J. Ther. 2019, 80, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Mustafayev, K.; Torres, H. Hepatitis B virus and hepatitis C virus reactivation in cancer patients receiving novel anticancer therapies. Clin. Microbiol. Infect. 2022, 28, 1321–1327. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Hsu, C.-M.; Hsiao, S.Y.; Hsiao, H.-H. Hepatitis B Virus Infection in Patients Receiving Allogeneic Hematopoietic Stem Cell Transplantation. J. Pers. Med. 2021, 11, 1108. [Google Scholar] [CrossRef]
- Doyle, J.; Raggatt, M.; Slavin, M.; McLachlan, S.; Strasser, S.I.; Sasadeusz, J.J.; Howell, J.; Hajkowicz, K.; Nandurkar, H.; Johnston, A.; et al. Hepatitis B management during immunosuppression for haematological and solid organ malignancies: An Australian consensus statement. Med. J. Aust. 2019, 210, 462–468. [Google Scholar] [CrossRef]
- Soriano, V.; Edagwa, B.; de Mendoza, C.; Barreiro, P.; Corral, O.; Treviño, A.; Gendelman, H.E. Ultra-long-acting antivirals as chemical vaccines to prevent viral diseases. Futur. Microbiol. 2022, 17, 887–897. [Google Scholar] [CrossRef]
- Thomas, D.L.; Kiser, J.J.; Baum, M.M. Long-Acting Treatments for Hepatitis B. Clin. Infect. Dis. 2022, 75, S517–S524. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Guidelines for the Prevention, Care and Treatment of Persons with Chronic Hepatitis B Infection; WHO: Geneva, Switzerland, 2015. [Google Scholar]
- Das, S.; Wang, W.; Ganesan, M.; Fonseca-Lanza, F.; Cobb, D.A.; Bybee, G.; Sun, Y.; Guo, L.; Hanson, B.; Cohen, S.M.; et al. An ultralong-acting tenofovir ProTide nanoformulation achieves monthslong HBV suppression. Sci. Adv. 2022, 8, eade9582. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, A.; Wang, H.; Yan, M.; Liang, R.; He, X.; Fu, F.; Mu, H.; Sun, K. Entecavir-loaded poly (lactic-co-glycolic acid) microspheres for long-term therapy of chronic hepatitis-B: Preparation and in vitro and in vivo evaluation. Int. J. Pharm. 2019, 560, 27–34. [Google Scholar] [CrossRef]
- Freeling, J.P.; Koehn, J.; Shu, C.; Sun, J.; Ho, R.J.; Al-Jabri, A.A.; Euler, Z.; Alter, G.; Wang, X.; Wang, P.; et al. Anti-HIV Drug-Combination Nanoparticles Enhance Plasma Drug Exposure Duration as Well as Triple-Drug Combination Levels in Cells Within Lymph Nodes and Blood in Primates. AIDS Res. Hum. Retroviruses 2015, 31, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Kraft, J.C.; McConnachie, L.A.; Koehn, J.; Kinman, L.; Collins, C.; Shen, D.D.; Collier, A.C.; Ho, R.J. Long-acting combination anti-HIV drug suspension enhances and sustains higher drug levels in lymph node cells than in blood cells and plasma. Aids 2017, 31, 765–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlesinger, E.; Johengen, D.; Luecke, E.; Rothrock, G.; McGowan, I.; van der Straten, A.; Desai, T. A Tunable, Biodegradable, Thin-Film Polymer Device as a Long-Acting Implant Delivering Tenofovir Alafenamide Fumarate for HIV Pre-exposure Prophylaxis. Pharm. Res. 2016, 33, 1649–1656. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.M.; Krovi, S.A.; Li, L.; Girouard, N.; Demkovich, Z.R.; Myers, D.; Creelman, B.; van der Straten, A. Characterization of a Reservoir-Style Implant for Sustained Release of Tenofovir Alafenamide (TAF) for HIV Pre-Exposure Prophylaxis (PrEP). Pharmaceutics 2019, 11, 315. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Johnson, L.M.; Krovi, S.A.; Demkovich, Z.R.; van der Straten, A. Performance and Stability of Tenofovir Alafenamide Formulations within Subcutaneous Biodegradable Implants for HIV Pre-Exposure Prophylaxis (PrEP). Pharmaceutics 2020, 12, 1057. [Google Scholar] [CrossRef]
- Chua, C.Y.X.; Jain, P.; Ballerini, A.; Bruno, G.; Hood, R.L.; Gupte, M.; Gao, S.; Di Trani, N.; Susnjar, A.; Shelton, K.; et al. Transcutaneously refillable nanofluidic implant achieves sustained level of tenofovir diphosphate for HIV pre-exposure prophylaxis. J. Control. Release 2018, 286, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Pons-Faudoa, F.P.; Sizovs, A.; Shelton, K.A.; Momin, Z.; Niles, J.A.; Bushman, L.R.; Xu, J.; Chua, C.Y.X.; Nichols, J.E.; Demaria, S.; et al. Preventive Efficacy of a Tenofovir Alafenamide Fumarate Nanofluidic Implant in SHIV-Challenged Nonhuman Primates. Adv. Ther. 2020, 4, 2000163. [Google Scholar] [CrossRef]
- Simpson, S.M.; Widanapathirana, L.; Su, J.T.; Sung, S.; Watrous, D.; Qiu, J.; Pearson, E.; Evanoff, A.; Karunakaran, D.; Chacon, J.E.; et al. Design of a Drug-Eluting Subcutaneous Implant of the Antiretroviral Tenofovir Alafenamide Fumarate. Pharm. Res. 2020, 37, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Su, J.T.; Simpson, S.M.; Sung, S.; Tfaily, E.B.; Veazey, R.; Marzinke, M.; Qiu, J.; Watrous, D.; Widanapathirana, L.; Pearson, E.; et al. A Subcutaneous Implant of Tenofovir Alafenamide Fumarate Causes Local Inflammation and Tissue Necrosis in Rabbits and Macaques. Antimicrob. Agents Chemother. 2020, 64, e01893-19. [Google Scholar] [CrossRef] [Green Version]
- Gunawardana, M.; Remedios-Chan, M.; Miller, C.S.; Fanter, R.; Yang, F.; Marzinke, M.A.; Hendrix, C.W.; Beliveau, M.; Moss, J.A.; Smith, T.J.; et al. Pharmacokinetics of Long-Acting Tenofovir Alafenamide (GS-7340) Subdermal Implant for HIV Prophylaxis. Antimicrob. Agents Chemother. 2015, 59, 3913–3919. [Google Scholar] [CrossRef] [Green Version]
- Gunawardana, M.; Remedios-Chan, M.; Sanchez, D.; Webster, S.; Galvan, P.; Fanter, R.; Castonguay, A.E.; Webster, P.; Moss, J.A.; Kuo, J.; et al. Multispecies Evaluation of a Long-Acting Tenofovir Alafenamide Subdermal Implant for HIV Prophylaxis. Front. Pharmacol. 2020, 11, 569373. [Google Scholar] [CrossRef] [PubMed]
- Gengiah, T.N.; Karim, Q.A.; Harkoo, I.; Mansoor, L.; Zuma, N.Y.; Radebe, P.; Samsunder, N.; Baxter, C.; Maharaj, B.; Baum, M.M.; et al. CAPRISA 018: A phase I/II clinical trial study protocol to assess the safety, acceptability, tolerability and pharmacokinetics of a sustained-release tenofovir alafenamide subdermal implant for HIV prevention in women. BMJ Open 2022, 12, e052880. [Google Scholar] [CrossRef]
- Henry, S.J.; Barrett, S.E.; Forster, S.P.; Teller, R.S.; Yang, Z.; Li, L.; Mackey, M.A.; Doto, G.J.; Ruth, M.P.; Tsuchiya, T.; et al. Exploration of long-acting implant formulations of hepatitis B drug entecavir. Eur. J. Pharm. Sci. 2019, 136, 104958. [Google Scholar] [CrossRef] [PubMed]
- Higashi-Kuwata, N.; Hayashi, S.; Kumamoto, H.; Ogata-Aoki, H.; Das, D.; Venzon, D.; Hattori, S.-I.; Bulut, H.; Hashimoto, M.; Otagiri, M.; et al. Identification of a novel long-acting 4′-modified nucleoside reverse transcriptase inhibitor against HBV. J. Hepatol. 2021, 74, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Puri, A.; Bhattaccharjee, S.A.; Zhang, W.; Clark, M.; Singh, O.N.; Doncel, G.F.; Banga, A.K. Development of a Transdermal Delivery System for Tenofovir Alafenamide, a Prodrug of Tenofovir with Potent Antiviral Activity Against HIV and HBV. Pharmaceutics 2019, 11, 173. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Gao, X.; Singh, O.N.; Zhang, W.; Agrahari, V.; Peet, M.M.; Clark, M.R.; Doncel, G.F.; Banga, A.K. Pharmacokinetics of a weekly transdermal delivery system of tenofovir alafenamide in hairless rats. Int. J. Pharm. 2020, 582, 119342. [Google Scholar] [CrossRef]
- Johnson, T.J.; Clark, M.R.; Albright, T.H.; Nebeker, J.S.; Tuitupou, A.L.; Clark, J.T.; Fabian, J.; McCabe, R.T.; Chandra, N.; Doncel, G.F.; et al. A 90-Day Tenofovir Reservoir Intravaginal Ring for Mucosal HIV Prophylaxis. Antimicrob. Agents Chemother. 2012, 56, 6272–6283. [Google Scholar] [CrossRef] [Green Version]
- Clark, J.T.; Clark, M.R.; Shelke, N.B.; Johnson, T.J.; Smith, E.M.; Andreasen, A.K.; Nebeker, J.S.; Fabian, J.; Friend, D.R.; Kiser, P.F. Engineering a Segmented Dual-Reservoir Polyurethane Intravaginal Ring for Simultaneous Prevention of HIV Transmission and Unwanted Pregnancy. PLOS ONE 2014, 9, e88509. [Google Scholar] [CrossRef]
- Thurman, A.R.; Schwartz, J.L.; Brache, V.; Clark, M.R.; McCormick, T.; Chandra, N.; Marzinke, M.A.; Stanczyk, F.Z.; Dezzutti, C.S.; Hillier, S.L.; et al. Randomized, placebo controlled phase I trial of safety, pharmacokinetics, pharmacodynamics and acceptability of tenofovir and tenofovir plus levonorgestrel vaginal rings in women. PLOS ONE 2018, 13, e0199778. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Q.-L.; Yu, Z.-J.; Ji, F.; Li, G.-M.; Zhang, G.-F.; Xu, J.-H.; Chen, Z.-M.; Cui, G.-L.; Li, W.; Zhang, D.-W.; et al. Tenofovir Alafenamide to Prevent Perinatal Hepatitis B Transmission: A Multicenter, Prospective, Observational Study. Clin. Infect. Dis. 2021, 73, e3324–e3332. [Google Scholar] [CrossRef]
- Pan, C.Q.; Cao, L.; Huang, Y. Editorial: Tenofovir alafenamide fumarate-a new bullet to prevent mother-to-child transmission of hepatitis B virus. Authors’ reply. Aliment. Pharmacol. Ther. 2020, 52, 1746–1747. [Google Scholar]
- Wang, M.; Bian, Q.; Zhu, Y.; Pang, Q.; Chang, L.; Li, R.; Tiongson, B.C.; Zhang, H.; Pan, C.Q. Real-world study of tenofovir disoproxil fumarate to prevent hepatitis B transmission in mothers with high viral load. Aliment. Pharmacol. Ther. 2018, 49, 211–217. [Google Scholar] [CrossRef]
- Pan, C.Q.; Duan, Z.; Dai, E.; Zhang, S.; Han, G.; Wang, Y.; Zhang, H.; Zou, H.; Zhu, B.; Zhao, W.; et al. Tenofovir to Prevent Hepatitis B Transmission in Mothers with High Viral Load. N. Engl. J. Med. 2016, 374, 2324–2334. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Lucifora, J.; Mason, W.S.; Sureau, C.; Beck, J.; Levrero, M.; Kann, M.; Knolle, P.A.; Benkirane, M.; Durantel, D.; et al. Towards an HBV cure: State-of-the-art and unresolved questions—report of the ANRS workshop on HBV cure. Gut 2015, 64, 1314–1326. [Google Scholar] [CrossRef]
- Durantel, D.; Zoulim, F. New antiviral targets for innovative treatment concepts for hepatitis B virus and hepatitis delta virus. J. Hepatol. 2016, 64, S117–S131. [Google Scholar] [CrossRef]
- Stone, D.; Niyonzima, N.; Jerome, K.R. Genome editing and the next generation of antiviral therapy. Hum. Genet. 2016, 135, 1071–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, M.G.; Combe, E.; Inchauspe, A.; Mangeot, P.E.; Delberghe, E.; Chapus, F.; Neveu, G.; Alam, A.; Carter, K.; Testoni, B.; et al. CRISPR-Cas9 Targeting of Hepatitis B Virus Covalently Closed Circular DNA Generates Transcriptionally Active Episomal Variants. mBio 2022, 13, e0288821. [Google Scholar] [CrossRef]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef]
- Fanning, G.C.; Zoulim, F.; Hou, J.; Bertoletti, A. Therapeutic strategies for hepatitis B virus infection: Towards a cure. Nat. Rev. Drug Discov. 2019, 18, 827–844. [Google Scholar] [CrossRef]
- Lim, S.G.; Baumert, T.F.; Boni, C.; Gane, E.; Levrero, M.; Lok, A.S.; Maini, M.K.; Terrault, N.A.; Zoulim, F. The scientific basis of combination therapy for chronic hepatitis B functional cure. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 238–253. [Google Scholar] [CrossRef]
- Maepa, M.B.; Roelofse, I.; Ely, A.; Arbuthnot, P. Progress and Prospects of Anti-HBV Gene Therapy Development. Int. J. Mol. Sci. 2015, 16, 17589–17610. [Google Scholar] [CrossRef] [Green Version]
- Porto, E.M.; Komor, A.C.; Slaymaker, I.M.; Yeo, G.W. Base editing: Advances and therapeutic opportunities. Nat. Rev. Drug Discov. 2020, 19, 839–859. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; Choi, V.M.; Xia, D.F.; Vo, T.D.; Gregory, P.D.; Holmes, M.C. Transient cold shock enhances zinc-finger nuclease–mediated gene disruption. Nat. Methods 2010, 7, 459–460. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.L.; Althage, A.; Chung, J.; Chisari, F.V. Hydrodynamic injection of viral DNA: A mouse model of acute hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2002, 99, 13825–13830. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, W.; Lin, J.; Wang, F.; Wu, M.; Chen, C.; Zheng, Y.; Peng, X.; Li, J.; Yuan, Z. An Efficient Antiviral Strategy for Targeting Hepatitis B Virus Genome Using Transcription Activator-Like Effector Nucleases. Mol. Ther. 2014, 22, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Bloom, K.; Maepa, M.B.; Ely, A.; Arbuthnot, P. Gene Therapy for Chronic HBV-Can We Eliminate cccDNA? Genes 2018, 9, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komor, A.C.; Badran, A.H.; Liu, D.R. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 2016, 168, 20–36. [Google Scholar] [CrossRef] [Green Version]
- Stone, D.; Long, K.R.; Loprieno, M.A.; Feelixge, H.S.D.S.; Kenkel, E.J.; Liley, R.M.; Rapp, S.; Roychoudhury, P.; Nguyen, T.; Stensland, L.; et al. CRISPR-Cas9 gene editing of hepatitis B virus in chronically infected humanized mice. Mol. Ther.—Methods Clin. Dev. 2021, 20, 258–275. [Google Scholar] [CrossRef] [PubMed]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- Mason, W.S.; Gill, U.S.; Litwin, S.; Zhou, Y.; Peri, S.; Pop, O.; Hong, M.L.; Naik, S.; Quaglia, A.; Bertoletti, A.; et al. HBV DNA Integration and Clonal Hepatocyte Expansion in Chronic Hepatitis B Patients Considered Immune Tolerant. Gastroenterology 2016, 151, 986–998.e4. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, X.; Steer, C.J.; Song, G.; Niu, J. Efficient silencing of hepatitis B virus S gene through CRISPR-mediated base editing. Hepatol. Commun. 2022, 6, 1652–1663. [Google Scholar] [CrossRef]
- Kim, H.; Kim, J.-S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014, 15, 321–334. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Cao, Y.; Yang, Z. Roles of APOBEC3 in hepatitis B virus (HBV) infection and hepatocarcinogenesis. Bioengineered 2021, 12, 2074–2086. [Google Scholar] [CrossRef]
- Yang, Y.-C.; Chen, Y.-H.; Kao, J.-H.; Ching, C.; Liu, I.-J.; Wang, C.-C.; Tsai, C.-H.; Wu, F.-Y.; Liu, C.-J.; Chen, P.-J.; et al. Permanent Inactivation of HBV Genomes by CRISPR/Cas9-Mediated Non-cleavage Base Editing. Mol. Ther.—Nucleic Acids 2020, 20, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Smekalova, E.; Martinez, M.G.; Combe, E.; Dejene, S.; Packer, M.; LeBoeuf, D.; Kumar, A.; Barrera, L.; Dorkin, R.; Chen, R.; et al. Cytosine Base Editing Inhibits Hepatitis B Virus Replication and Reduces HBsAg Expression In Vitro and In Vivo. In Proceedings of the HBV 2022 International Meeting, Paris, France, 18–22 September 2022; Available online: https://beamtx.com/media/1rrjxj2o/hbvposter_2022_final.pdf (accessed on 1 January 2023).
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Schoenmaker, L.; Witzigmann, D.; Kulkarni, J.A.; Verbeke, R.; Kersten, G.; Jiskoot, W.; Crommelin, D.J.A. mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int. J. Pharm. 2021, 601, 120586. [Google Scholar] [CrossRef]
- Xu, L.; Wang, X.; Liu, Y.; Yang, G.; Falconer, R.J.; Zhao, C.-X. Lipid Nanoparticles for Drug Delivery. Adv. NanoBiomed Res. 2022, 2, 2100109. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, L. Lipid Nanoparticles for Gene Delivery. Adv. Genet. 2014, 88, 13–36. [Google Scholar] [CrossRef] [Green Version]
- Rathbone, T.; Ates, I.; Fernando, L.; Addlestone, E.; Lee, C.M.; Richards, V.P.; Cottle, R.N. Electroporation-Mediated Delivery of Cas9 Ribonucleoproteins Results in High Levels of Gene Editing in Primary Hepatocytes. CRISPR J. 2022, 5, 397–409. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chen, R.; Lu, F.M. Exosome-mediated CRISPR/Cas9 system targets to cut the intercellular transmission function of hepatitis B virus genome. Zhonghua Gan Zang Bing Za Zhi 2019, 27, 610–614. [Google Scholar]
- Jansen, L.; Vaillant, A.; Stelma, F.; Kootstra, N.; Bazinet, M.; Al-Mahtab, M.; Reesink, H. O114: Serum HBV-RNA levels decline significantly in chronic hepatitis B patients dosed with the nucleic-acid polymer REP2139-CA. J. Hepatol. 2015, 62, S250. [Google Scholar] [CrossRef]
- Sepp-Lorenzino, L.; Sprague, A.; Mayo, T. Parallel 4: Hepatitis B: Novel treatments and treatment targets: 36—GalNAc-siRNA conjugate ALN-HBV targets a highly conserved, pan-genotypic X-orf viral site and mediates profound and durable HBsAg silencing in vitro and in vivo. Hepatology 2015, 62, 222A–225A. [Google Scholar]
- Dusheiko, G. Will we need novel combinations to cure HBV infection? Liver Int. 2020, 40, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano, V.; Barreiro, P.; Cachay, E.; Kottilil, S.; Fernandez-Montero, J.V.; de Mendoza, C. Advances in hepatitis B therapeutics. Ther. Adv. Infect. Dis. 2020, 7, 2049936120965027. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogunnaike, M.; Das, S.; Raut, S.S.; Sultana, A.; Nayan, M.U.; Ganesan, M.; Edagwa, B.J.; Osna, N.A.; Poluektova, L.Y. Chronic Hepatitis B Infection: New Approaches towards Cure. Biomolecules 2023, 13, 1208. https://doi.org/10.3390/biom13081208
Ogunnaike M, Das S, Raut SS, Sultana A, Nayan MU, Ganesan M, Edagwa BJ, Osna NA, Poluektova LY. Chronic Hepatitis B Infection: New Approaches towards Cure. Biomolecules. 2023; 13(8):1208. https://doi.org/10.3390/biom13081208
Chicago/Turabian StyleOgunnaike, Mojisola, Srijanee Das, Samiksha S. Raut, Ashrafi Sultana, Mohammad Ullah Nayan, Murali Ganesan, Benson J. Edagwa, Natalia A. Osna, and Larisa Y. Poluektova. 2023. "Chronic Hepatitis B Infection: New Approaches towards Cure" Biomolecules 13, no. 8: 1208. https://doi.org/10.3390/biom13081208
APA StyleOgunnaike, M., Das, S., Raut, S. S., Sultana, A., Nayan, M. U., Ganesan, M., Edagwa, B. J., Osna, N. A., & Poluektova, L. Y. (2023). Chronic Hepatitis B Infection: New Approaches towards Cure. Biomolecules, 13(8), 1208. https://doi.org/10.3390/biom13081208