




The Radical SAM Heme Synthase AhbD from Methanosarcina barkeri Contains Two Auxiliary [4Fe-4S] Clusters


Abstract
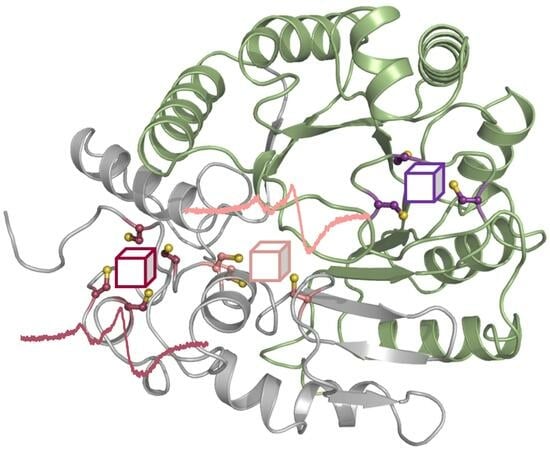
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Site-Directed Mutagenesis of the ahbD Gene
2.3. Production, Purification, and Iron–Sulfur Cluster Reconstitution of Recombinant AhbD
2.4. Determination of Protein Concentration
2.5. Determination of Iron and Sulfide Contents
2.6. Enzyme Activity and Substrate Binding Assay
2.7. Tetrapyrrole Extraction
2.8. High-Performance Liquid Chromatography (HPLC)
2.9. EPR Spectroscopy
3. Results
3.1. A Subset of AhbD Sequences Displays a Full SPASM Motif with Eight Conserved Cysteines
3.2. The AlphaFold2 Model of AhbD from M. barkeri Suggests the Presence of Two Auxiliary Iron–Sulfur Clusters
3.3. AhbD Variants Support the Presence of Two Auxiliary [4Fe-4S] Clusters
3.3.1. Production and Purification of AhbD Cluster Variants Carrying a Single Cluster
3.3.2. EPR Spectroscopy Demonstrates the Presence of Two Auxiliary [4Fe-4S] Clusters
3.4. Role of the Auxiliary Clusters for AhbD Activity
3.4.1. Production and Purification of AhbD Cluster Variants Lacking AuxI or AuxII
3.4.2. The Auxiliary Clusters Are Not Required for FeCopro Binding
3.4.3. AhbD Variant RS/AuxII Accumulates the Monovinyl Intermediate
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paoli, M.; Marles-Wright, J.; Smith, A. Structure-function relationships in heme-proteins. DNA Cell Biol. 2002, 21, 271–280. [Google Scholar] [CrossRef]
- Dailey, H.A.; Dailey, T.A.; Gerdes, S.; Jahn, D.; Jahn, M.; O’Brian, M.R.; Warren, M.J. Prokaryotic heme biosynthesis: Multiple pathways to a common essential product. Microbiol. Mol. Biol. Rev. 2017, 81, e00048-16. [Google Scholar] [CrossRef]
- Dailey, H.A.; Gerdes, S.; Dailey, T.A.; Burch, J.S.; Phillips, J.D. Noncanonical coproporphyrin-dependent bacterial heme biosynthesis pathway that does not use protoporphyrin. Proc. Natl. Acad. Sci. USA 2015, 112, 2210–2215. [Google Scholar] [CrossRef]
- Bali, S.; Lawrence, A.D.; Lobo, S.A.; Saraiva, L.M.; Golding, B.T.; Palmer, D.J.; Howard, M.J.; Ferguson, S.J.; Warren, M.J. Molecular hijacking of siroheme for the synthesis of heme and d1 heme. Proc. Natl. Acad. Sci. USA 2011, 108, 18260–18265. [Google Scholar] [CrossRef]
- Lobo, S.A.L.; Lawrence, A.D.; Romão, C.V.; Warren, M.J.; Teixeira, M.; Saraiva, L.M. Characterisation of Desulfovibrio vulgaris haem b synthase, a radical SAM family member. Biochim. Biophys. Acta 2014, 1844, 1238–1247. [Google Scholar] [CrossRef]
- Kühner, M.; Schweyen, P.; Hoffmann, M.; Ramos, J.V.; Reijerse, E.J.; Lubitz, W.; Bröring, M.; Layer, G. The auxiliary [4Fe-4S] cluster of the Radical SAM heme synthase from Methanosarcina barkeri is involved in electron transfer. Chem. Sci. 2016, 7, 4633–4643. [Google Scholar] [CrossRef]
- Sofia, H.J.; Chen, G.; Hetzler, B.G.; Reyes-Spindola, J.F.; Miller, N.E. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: Functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001, 29, 1097–1106. [Google Scholar] [CrossRef]
- Walsby, C.J.; Ortillo, D.; Broderick, W.E.; Broderick, J.B.; Hoffman, B.M. An anchoring role for FeS clusters: Chelation of the amino acid moiety of S-adenosylmethionine to the unique iron site of the [4Fe-4S] cluster of pyruvate formate-lyase activating enzyme. J. Am. Chem. Soc. 2002, 124, 11270–11271. [Google Scholar] [CrossRef]
- Layer, G.; Moser, J.; Heinz, D.W.; Jahn, D.; Schubert, W.-D. Crystal structure of coproporphyrinogen III oxidase reveals cofactor geometry of Radical SAM enzymes. EMBO J. 2003, 22, 6214–6224. [Google Scholar] [CrossRef]
- Broderick, W.E.; Hoffman, B.M.; Broderick, J.B. Mechanism of radical initiation in the radical S-adenosyl-L-methionine superfamily. Acc. Chem. Res. 2018, 51, 2611–2619. [Google Scholar] [CrossRef]
- Frey, P.A.; Hegeman, A.D.; Ruzicka, F.J. The Radical SAM superfamily. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 63–88. [Google Scholar] [CrossRef]
- Holliday, G.L.; Akiva, E.; Meng, E.C.; Brown, S.D.; Calhoun, S.; Pieper, U.; Sali, A.; Booker, S.J.; Babbitt, P.C. Atlas of the radical SAM superfamily: Divergent evolution of function using a “plug and play” domain. Methods Enzymol. 2018, 606, 1–71. [Google Scholar]
- Bauerle, M.R.; Schwalm, E.L.; Booker, S.J. Mechanistic diversity of radical S-adenosylmethionine (SAM)-dependent methylation. J. Biol. Chem. 2015, 290, 3995–4002. [Google Scholar] [CrossRef]
- Bridwell-Rabb, J.; Li, B.; Drennan, C.L. Cobalamin-dependent radical S-adenosylmethionine enzymes: Capitalizing on old motifs for new functions. ACS Bio Med. Chem. Au 2022, 2, 173–186. [Google Scholar] [CrossRef]
- Lanz, N.D.; Booker, S.J. Auxiliary iron-sulfur cofactors in radical SAM enzymes. Biochim. Biophys. Acta 2015, 1853, 1316–1334. [Google Scholar] [CrossRef]
- Grell, T.A.J.; Goldman, P.J.; Drennan, C.L. SPASM and twitch domains in S-adenosylmethionine (SAM) radical enzymes. J. Biol. Chem. 2015, 290, 3964–3971. [Google Scholar] [CrossRef]
- Haft, D.H.; Basu, M.K. Biological systems discovery in silico: Radical S-adenosylmethionine protein families and their target peptides for posttranslational modification. J. Bacteriol. 2011, 193, 2745–2755. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods. 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Kühner, M.; Haufschildt, K.; Neumann, A.; Storbeck, S.; Streif, J.; Layer, G. The alternative route to heme in the methanogenic archaeon Methanosarcina barkeri. Archaea 2014, 2014, 327637. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef] [PubMed]
- Storbeck, S.; Rolfes, S.; Raux-Deery, E.; Warren, M.J.; Jahn, D.; Layer, G. A novel pathway for the biosynthesis of heme in Archaea: Genome-based bioinformatic predictions and experimental evidence. Archaea 2010, 2010, 175050. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Benjdia, A.; Subramanian, S.; Leprince, J.; Vaudry, H.; Johnson, M.K.; Berteau, O. Anaerobic sulfatase-maturating enzyme—A mechanistic link with glycyl radical-activating enzymes? FEBS J. 2010, 277, 1906–1920. [Google Scholar] [CrossRef] [PubMed]
- Grove, T.L.; Lee, K.-H.; St Clair, J.; Krebs, C.; Booker, S.J. In vitro characterization of AtsB, a radical SAM formylglycine-generating enzyme that contains three [4Fe-4S] clusters. Biochemistry 2008, 47, 7523–7538. [Google Scholar] [CrossRef] [PubMed]
- Barr, I.; Stich, T.A.; Gizzi, A.S.; Grove, T.L.; Bonanno, J.B.; Latham, J.A.; Chung, T.; Wilmot, C.M.; Britt, R.D.; Almo, S.C.; et al. X-ray and EPR characterization of the auxiliary Fe-S clusters in the radical SAM enzyme PqqE. Biochemistry 2018, 57, 1306–1315. [Google Scholar] [CrossRef]
- Ayikpoe, R.; Ngendahimana, T.; Langton, M.; Bonitatibus, S.; Walker, L.M.; Eaton, S.S.; Eaton, G.R.; Pandelia, M.-E.; Elliott, S.J.; Latham, J.A. Spectroscopic and electrochemical characterization of the mycofactocin biosynthetic protein, MftC, provides insight into its redox flipping mechanism. Biochemistry 2019, 58, 940–950. [Google Scholar] [CrossRef]
- Davis, K.M.; Schramma, K.R.; Hansen, W.A.; Bacik, J.P.; Khare, S.D.; Seyedsayamdost, M.R.; Ando, N. Structures of the peptide-modifying radical SAM enzyme SuiB elucidate the basis of substrate recognition. Proc. Natl. Acad. Sci. USA 2017, 114, 10420–10425. [Google Scholar] [CrossRef]
- Holm, L. Dali server: Structural unification of protein families. Nucleic Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef]
- Goldman, P.J.; Grove, T.L.; Sites, L.A.; McLaughlin, M.I.; Booker, S.J.; Drennan, C.L. X-ray structure of an AdoMet radical activase reveals an anaerobic solution for formylglycine posttranslational modification. Proc. Natl. Acad. Sci. USA 2013, 110, 8519–8524. [Google Scholar] [CrossRef]
- Grove, T.L.; Himes, P.M.; Hwang, S.; Yumerefendi, H.; Bonanno, J.B.; Kuhlman, B.; Almo, S.C.; Bowers, A.A. Structural insights into thioether bond formation in the biosynthesis of sactipeptides. J. Am. Chem. Soc. 2017, 139, 11734–11744. [Google Scholar] [CrossRef] [PubMed]
- Rush, K.W.; Eastman, K.A.S.; Kincannon, W.M.; Blackburn, N.J.; Bandarian, V. Peptide selenocysteine substitutions reveal direct substrate-enzyme interactions at auxiliary clusters in radical S-adenosyl-L-methionine maturases. J. Am. Chem. Soc. 2023, 145, 10167–10177. [Google Scholar] [CrossRef] [PubMed]
- Grove, T.L.; Ahlum, J.H.; Qin, R.M.; Lanz, N.D.; Radle, M.I.; Krebs, C.; Booker, S.J. Further characterization of Cys-type and Ser-type anaerobic sulfatase maturating enzymes suggests a commonality in the mechanism of catalysis. Biochemistry 2013, 52, 2874–2887. [Google Scholar] [CrossRef]
- Balo, A.R.; Caruso, A.; Tao, L.; Tantillo, D.J.; Seyedsayamdost, M.R.; Britt, R.D. Trapping a cross-linked lysine-tryptophan radical in the catalytic cycle of the radical SAM enzyme SuiB. Proc. Natl. Acad. Sci. USA 2021, 118, e2101571118. [Google Scholar] [CrossRef]
- Zhu, W.; Walker, L.M.; Tao, L.; Iavarone, A.T.; Wei, X.; Britt, R.D.; Elliott, S.J.; Klinman, J.P. Structural properties and catalytic implications of the SPASM domain iron-sulfur clusters in Methylorubrum extorquens PqqE. J. Am. Chem. Soc. 2020, 142, 12620–12634. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AhbD Variant | Cys to Ala | Iron (mol/mol AhbD) 1 | Sulfide (mol/mol AhbD) 1 |
|---|---|---|---|
| wt | 11.8 ± 1.5 | 10.1 ± 1.1 | |
| AuxI | 23 + 26 + 312 + 315 | 2.7 ± 1.4 | 3.2 ± 0.7 |
| AuxII | 23 + 26 + 256 + 274 + 324 | 2.4 ± 0.8 | 3.4 2 |
| RS/AuxI | 312 + 315 + 321 | 8.3 ± 1.0 | 5.6 ± 1.0 |
| RS/AuxII | 324 | 7.2 ± 2.7 | 4.5 ± 1.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fix, I.; Heidinger, L.; Friedrich, T.; Layer, G. The Radical SAM Heme Synthase AhbD from Methanosarcina barkeri Contains Two Auxiliary [4Fe-4S] Clusters. Biomolecules 2023, 13, 1268. https://doi.org/10.3390/biom13081268
Fix I, Heidinger L, Friedrich T, Layer G. The Radical SAM Heme Synthase AhbD from Methanosarcina barkeri Contains Two Auxiliary [4Fe-4S] Clusters. Biomolecules. 2023; 13(8):1268. https://doi.org/10.3390/biom13081268
Chicago/Turabian StyleFix, Isabelle, Lorenz Heidinger, Thorsten Friedrich, and Gunhild Layer. 2023. "The Radical SAM Heme Synthase AhbD from Methanosarcina barkeri Contains Two Auxiliary [4Fe-4S] Clusters" Biomolecules 13, no. 8: 1268. https://doi.org/10.3390/biom13081268
APA StyleFix, I., Heidinger, L., Friedrich, T., & Layer, G. (2023). The Radical SAM Heme Synthase AhbD from Methanosarcina barkeri Contains Two Auxiliary [4Fe-4S] Clusters. Biomolecules, 13(8), 1268. https://doi.org/10.3390/biom13081268