

Predictive Modeling of Proteins Encoded by a Plant Virus Sheds a New Light on Their Structure and Inherent Multifunctionality


Abstract
:1. Introduction

2. Materials and Methods
2.1. GFLV Sequence Retrieval and Curation
2.2. Disorder and Secondary Structure Prediction
2.3. In Silico Protein Modeling

2.4. Domain, Motif, and Structural Site Predictive Modeling
2.5. Statistical Considerations, Graphic Generation, and Visualization
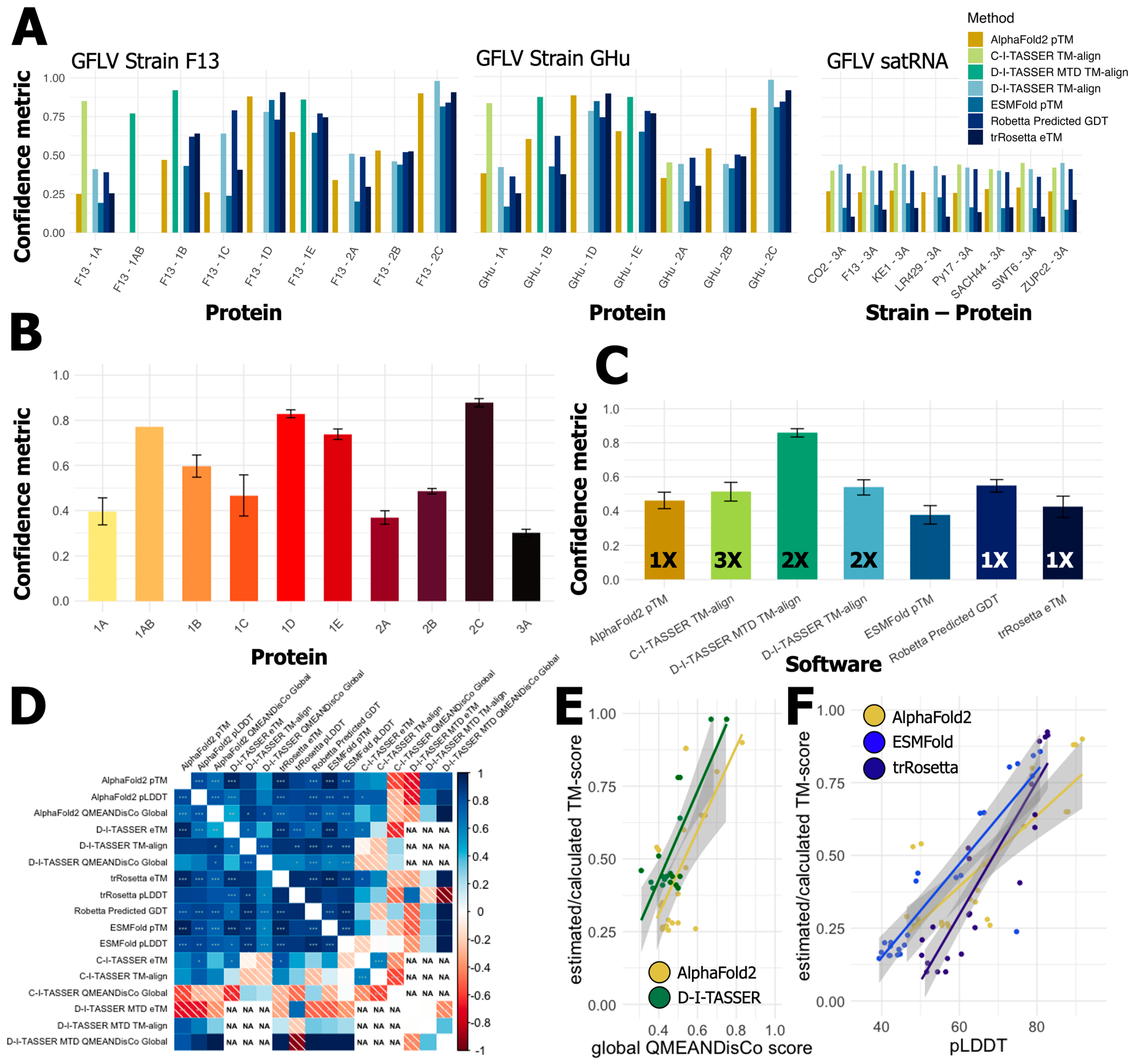
3. Results
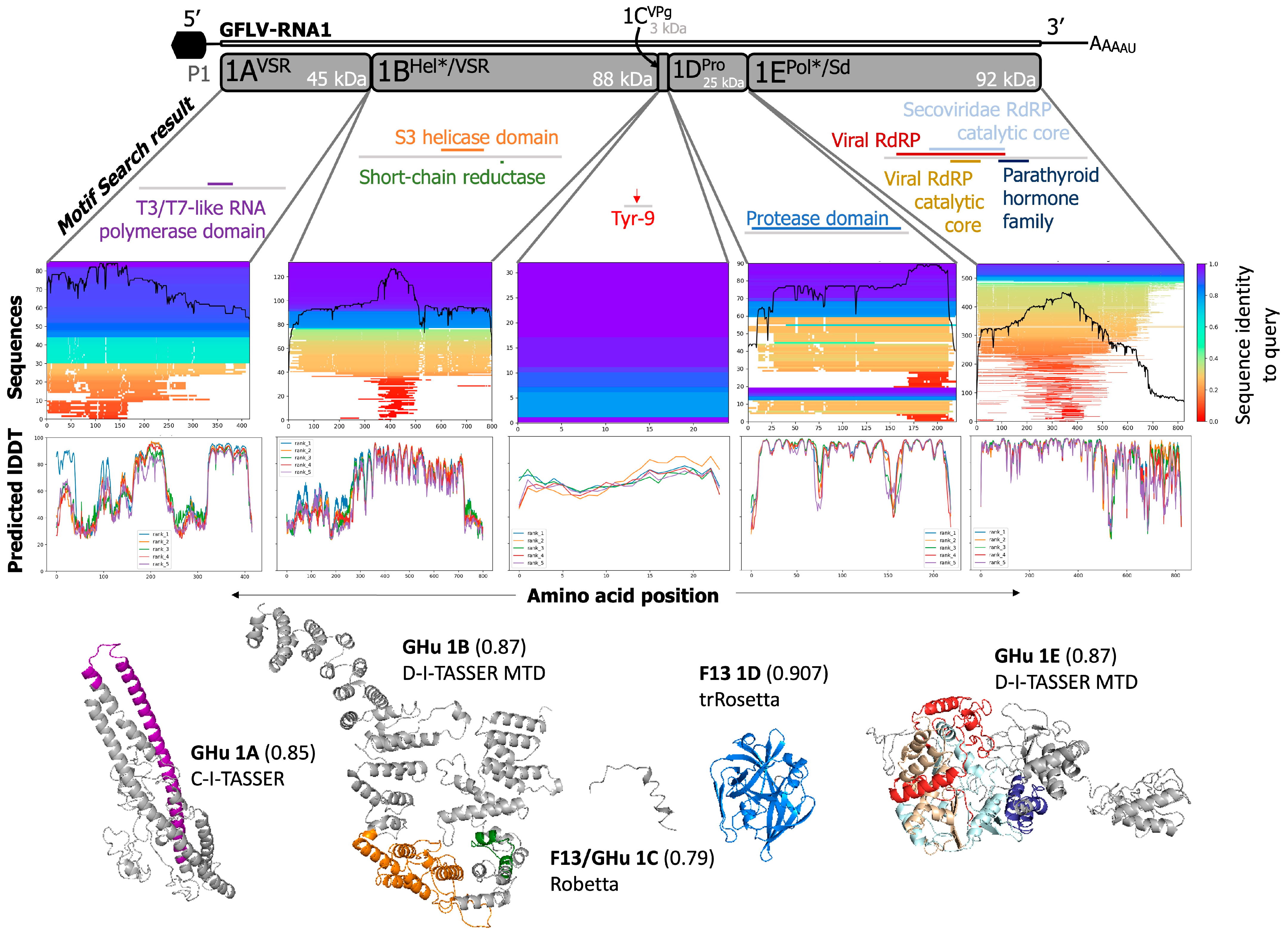
3.1. Current Documented Features of GFLV Proteins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GFLV RNA | Protein a | Experimentally Validated Function b | Putative Function c | Confirmed Localization | Reference(s) |
|---|---|---|---|---|---|
| RNA1 | 1AVSR | Viral silencing suppressor | - | - | [19] |
| 1BHel*/VSR | Viral silencing suppressor | Helicase | Endoplasmic reticulum | [14,16,19,36,38,102] | |
| 1CVPg | Viral genome-linked protein | - | - | [93] | |
| 1DPro | Viral protease | - | - | [22,94,95,96,97] | |
| 1EPol*/Sd | Symptom determinant | RNA-dependent RNA polymerase | - | [20,21,36,38,98,103] | |
| RNA2 | 2AHP/Sd | Homing protein Symptom determinant | - | Perinuclear space | [23,24,99] |
| 2BMP | Movement protein | - | Plasmodesmata | [25,26,99,100] | |
| 2CCP/Td | Coat protein Transmission determinant | - | - | [27,28,29,30,95,101] | |
| satRNA | 3A? | - | - | - | [31,32,33,34,35] |
3.2. Predictions of Functions and Structures for GFLV Proteins

3.3. GFLV RNA1 Proteins
| Molecular Function | Biological Process | Cellular Component | |||||||
|---|---|---|---|---|---|---|---|---|---|
| GFLV Protein | GO | C-Score a | Name | GO | C-Score | Name | GO | C-Score | Name |
| 1AVSR | 0015462 | 0.92 | protein-transmembrane transporting ATPase activity | 0050658 | 0.57 | RNA transport | 0005737 | 0.94 | cytoplasm |
| 0005524 | 0.92 | ATP binding | 0045184 | 0.57 | establishment of protein localization | 0043227 | 0.57 | membrane-bounded organelle | |
| 0005634 | 0.50 | nucleus | |||||||
| 1BHel*/VSR | 0003723 | 0.50 | RNA binding | 0039503 | 0.97 | suppression by virus of host innate immune response | 0019028 | 0.85 | viral capsid |
| 0015075 | 0.49 | ion transmembrane transporter activity | 0009968 | 0.96 | negative regulation of signal transduction | 0033655 | 0.76 | host cell cytoplasm part | |
| 0004386 | 0.37 | helicase activity | 0039537 | 0.95 | suppression by virus of host viral-induced cytoplasmic pattern recognition receptor signaling pathway | 0033648 | 0.72 | host intracellular membrane-bounded organelle | |
| 1CVPg | 0016798 | 0.59 | hydrolase activity, acting on glycosyl bonds | 0044238 | 0.51 | primary metabolic process | 0044444 | 0.67 | cytoplasmic part |
| 0009507 | 0.33 | chloroplast | |||||||
| 0005576 | 0.33 | extracellular region | |||||||
| 1DPro | 0003824 | 0.97 | catalytic activity | 0044003 | 0.94 | modification by symbiont of host morphology or physiology | 0016020 | 0.86 | membrane |
| 0004197 | 0.78 | cysteine-type endopeptidase activity | 0039520 | 0.89 | induction by virus of host autophagy | ||||
| 0003723 | 0.71 | RNA binding | 0039544 | 0.84 | suppression by virus of host RIG-I activity by RIG-I proteolysis | ||||
| 1EPol*/Sd | 0034062 | 0.68 | RNA polymerase activity | 0039507 | 0.97 | suppression by virus of host molecular function | 0019028 | 0.85 | viral capsid |
| 0003676 | 0.63 | nucleic acid binding | 0039503 | 0.97 | suppression by virus of host innate immune response | 0033655 | 0.76 | host cell cytoplasm part | |
| 0035639 | 0.54 | purine ribonucleoside triphosphate binding | 0039694 | 0.77 | viral RNA genome replication | 0016020 | 0.63 | membrane | |
| 2AHP/Sd | 0046872 | 0.47 | metal ion binding | 0044710 | 0.47 | single-organism metabolic process | 0016020 | 0.93 | membrane |
| 0052933 | 0.37 | alcohol dehydrogenase (cytochrome c(L)) activity | 0042597 | 0.64 | periplasmic space | ||||
| 0030288 | 0.57 | outer membrane-bounded periplasmic space | |||||||
| 2BMP | 0046914 | 0.36 | transition metal ion binding | 0044710 | 0.36 | single-organism metabolic process | 0005576 | 0.75 | extracellular region |
| 0044464 | 0.50 | cell part | |||||||
| 0031988 | 0.50 | membrane-bounded vesicle | |||||||
| 2CCP/Td | 0005198 | 0.72 | structural molecule activity | 0046740 | 0.65 | transport of virus in host, cell to cell | 0043231 | 0.38 | intracellular membrane-bounded organelle |
| 0009341 | 0.38 | beta-galactosidase complex | |||||||
| 0019028 | 0.31 | viral capsid | |||||||
| 3A? | 0046914 | 0.36 | transition metal ion binding | 0098662 | 0.36 | inorganic cation transmembrane transport | 0016020 | 0.94 | membrane |
| 0016676 | 0.36 | oxidoreductase activity, acting on a heme group of donors, oxygen as acceptor | 0045333 | 0.36 | cellular respiration | 0044464 | 0.89 | cell part | |
| 0015078 | 0.36 | hydrogen ion transmembrane transporter activity | 0015992 | 0.36 | proton transport | 0005886 | 0.78 | plasma membrane | |
| Molecular Function | Biological Process | Cellular Component | |||||||
|---|---|---|---|---|---|---|---|---|---|
| GFLV Protein | GO | C-Score a | Name | GO | C-Score | Name | GO | C-Score | Name |
| 1AVSR | 0097493 | 0.92 | structural molecule activity conferring elasticity | 0044763 | 0.94 | single-organism cellular process | 0044444 | 0.97 | cytoplasmic part |
| 005101 | 0.92 | actin filament binding | 0045944 | 0.34 | positive regulation of transcription from RNA polymerase II promoter | 0044446 | 0.97 | intracellular organelle part | |
| 0005524 | 0.41 | ATP binding | 0043123 | 0.34 | positive regulation of I-kappaB kinase/NF-kappaB signaling | 00043234 | 0.94 | protein complex | |
| 1BHel*/VSR | 0003824 | 0.49 | catalytic activity | 0080134 | 0.93 | regulation of response to stress | 0043231 | 0.85 | intracellular membrane-bounded organelle |
| 0070182 | 0.36 | DNA polymerase binding | 0019054 | 0.93 | modulation by virus of host process | 0005634 | 0.81 | nucleus | |
| 0039537 | 0.92 | suppression by virus of host viral-induced cytoplasmic pattern recognition receptor signaling pathway | 0044444 | 0.41 | cytoplasmic part | ||||
| 1CVPg | 0016798 | 0.59 | hydrolase activity, acting on glycosyl bonds | 0044238 | 0.51 | primary metabolic process | 0044444 | 0.67 | cytoplasmic part |
| 0009507 | 0.33 | chloroplast | |||||||
| 0005576 | 0.33 | extracellular region | |||||||
| 1DPro | 0003824 | 0.98 | catalytic activity | 0044003 | 0.94 | modification by symbiont of host morphology or physiology | 0019028 | 0.97 | viral capsid |
| 0004197 | 0.81 | cysteine-type endopeptidase activity | 0039520 | 0.89 | induction by virus of host autophagy | 0016020 | 0.80 | membrane | |
| 0005524 | 0.74 | ATP binding | 0039544 | 0.85 | suppression by virus of host RIG-I activity by RIG-I proteolysis | ||||
| 1EPol*/Sd | 0003968 | 0.63 | RNA-directed RNA polymerase activity | 0019054 | 0.99 | modulation by virus of host process | 0019028 | 0.87 | viral capsid |
| 0003723 | 0.52 | RNA binding | 0039503 | 0.97 | suppression by virus of host innate immune response | 0030430 | 0.76 | host cell cytoplasm | |
| 0022838 | 0.43 | substrate-specific channel activity | 0039694 | 0.78 | viral RNA genome replication | 0033648 | 0.75 | host intracellular membrane-bounded organelle | |
| 2AHP/Sd | 0005088 | 0.46 | Ras guanyl-nucleotide exchange factor activity | 0051345 | 0.56 | positive regulation of hydrolase activity | 0044424 | 0.75 | intracellular part |
| 0043087 | 0.56 | regulation of GTPase activity | 0016020 | 0.75 | membrane | ||||
| 0035556 | 0.56 | intracellular signal transduction | 0005829 | 0.62 | cytosol | ||||
| 2BMP | 0005576 | 0.78 | extracellular region | ||||||
| 0043227 | 0.72 | membrane-bounded organelle | |||||||
| 0043234 | 0.50 | protein complex | |||||||
| 2CCP/Td | 0005198 | 0.72 | structural molecule activity | 0046740 | 0.64 | transport of virus in host, cell to cell | 0043231 | 0.38 | intracellular membrane-bounded organelle |
| 0009341 | 0.38 | beta-galactosidase complex | |||||||
| 0019028 | 0.31 | viral capsid | |||||||
3.3.1. GFLV 1AVSR
3.3.2. GFLV 1BHel*/VSR
3.3.3. Fusion Protein GFLV-1AVSRBHel*/VSR
3.3.4. GFLV 1CVPg
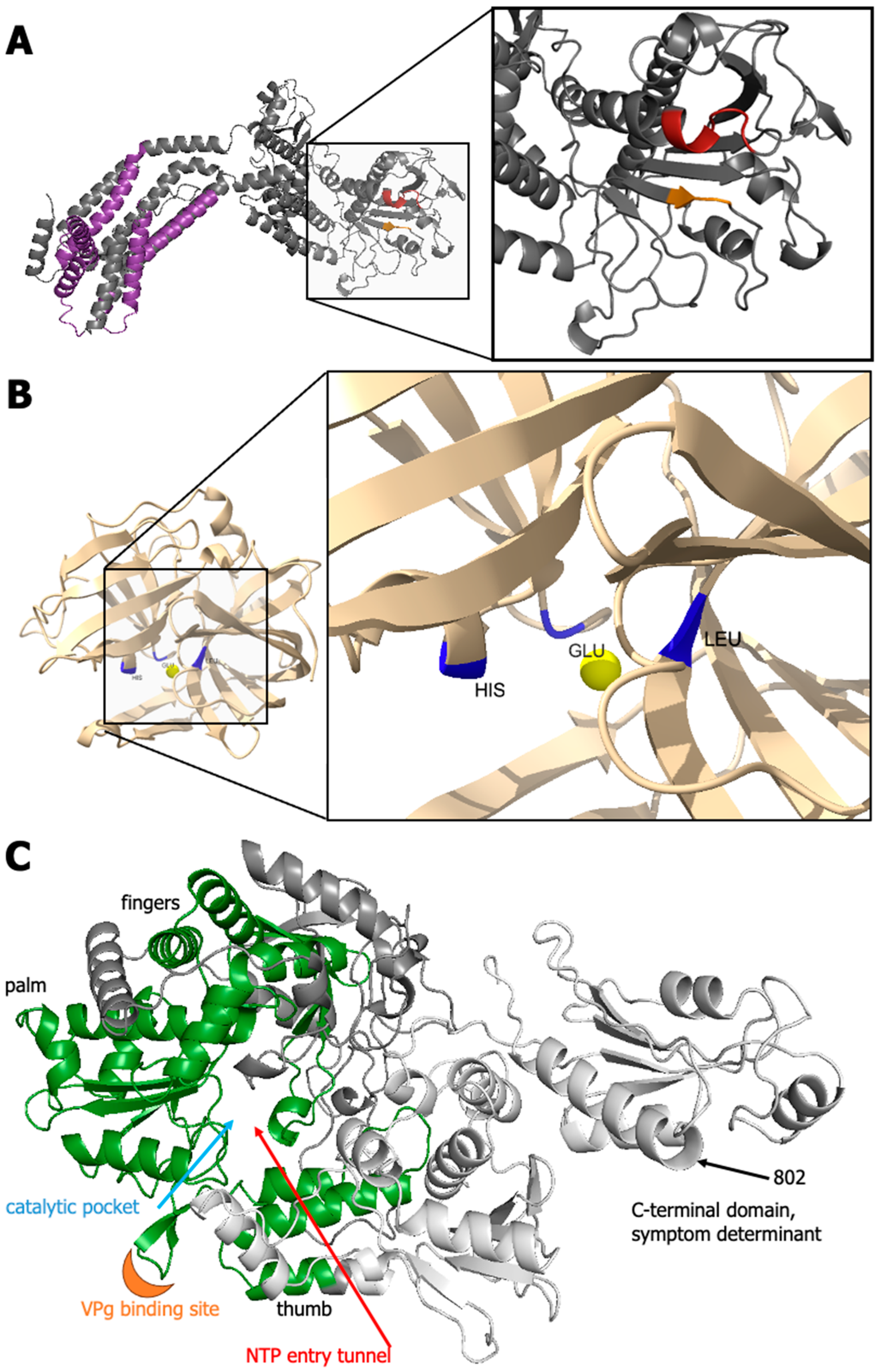
3.3.5. GFLV 1DPro
3.3.6. GFLV 1EPol*/Sd
3.4. GFLV RNA2 Proteins

3.4.1. GFLV 2AHP/Sd
3.4.2. GFLV 2BMP
3.4.3. GFLV 2CCP/Td
3.5. GFLV satRNA Protein 3A?

4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, X.; Park, B.; Choi, D.; Han, K. A Generalized Approach to Predicting Protein-Protein Interactions between Virus and Host. BMC Genom. 2018, 19, 568. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Chakraborty, S.; Kodamana, H.; Chakraborty, S. Application of Machine Learning in Understanding Plant Virus Pathogenesis: Trends and Perspectives on Emergence, Diagnosis, Host-Virus Interplay and Management. Virol. J. 2022, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Gutnik, D.; Evseev, P.; Miroshnikov, K.; Shneider, M. Using AlphaFold Predictions in Viral Research. Curr. Issues Mol. Biol. 2023, 45, 3705–3732. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Young, F.; Robertson, D.L.; Yuan, K. Prediction of Virus-Host Association Using Protein Language Models and Multiple Instance Learning. bioRxiv 2023. [Google Scholar] [CrossRef]
- Andret-Link, P.; Laporte, C.; Valat, L.; Ritzenthaler, C.; Demangeat, G.; Vigne, E.; Laval, V.; Pfeiffer, P.; Stussi-Garaud, C.; Fuchs, M. Grapevine Fanleaf Virus: Still a Major Threat to the Grapevine Industry. J. Plant Pathol. 2004, 86, 183–195. [Google Scholar]
- Digiaro, M.; Elbeaino, T.; Martelli, G.P. Grapevine Fanleaf Virus and Other Old World Nepoviruses. In Grapevine Viruses: Molecular Biology, Diagnostics and Management; Meng, B., Martelli, G.P., Golino, D.A., Fuchs, M., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 47–82. [Google Scholar]
- Martelli, G.P. Virus Diseases of Grapevine. In Encyclopedia of Life Sciences; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2019; pp. 1–13. [Google Scholar]
- Cazalis-Allut, L. Œuvres Agricoles; Victor Masson et fils: Paris, France, 1865; pp. 57–61. [Google Scholar]
- Cadman, C.H.; Dias, H.F.; Harrison, B.D. Sap-Transmissible Viruses Associated with Diseases of Grape Vines in Europe and North America. Nature 1960, 187, 577–579. [Google Scholar] [CrossRef]
- Hewitt, W.B.; Raski, D.J.; Goheen, A.C. Nematode vector of soil-borne fanleaf virus of grapevines. Phytopathology 1958, 48, 586–595. [Google Scholar]
- Taylor, C.E.; Brown, D.J.F. Nematode Vectors of Plant Viruses; Cab International: Wallingford, UK, 1997. [Google Scholar]
- Fuchs, M.; Hily, J.-M.; Petrzik, K.; Sanfaçon, H.; Thompson, J.R.; van der Vlugt, R.; Wetzel, T.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Secoviridae 2022. J. Gen. Virol. 2022, 103, 001807. [Google Scholar] [CrossRef]
- Stothard, P. The Sequence Manipulation Suite: JavaScript Programs for Analyzing and Formatting Protein and DNA Sequences. Biotechniques 2000, 28, 1102–1104. [Google Scholar] [CrossRef]
- Viry, M.; Serghini, M.A.; Hans, F.; Ritzenthaler, C.; Pinck, M.; Pinck, L. Biologically Active Transcripts from Cloned cDNA of Genomic Grapevine Fanleaf Nepovirus RNAs. J. Gen. Virol. 1993, 74, 169–174. [Google Scholar] [CrossRef]
- Fuchs, M.; Schmitt-Keichinger, C.; Sanfaçon, H. A Renaissance in Nepovirus Research Provides New Insights into Their Molecular Interface with Hosts and Vectors. In Advances in Virus Research; Kielian, M., Mettenleiter, T.C., Roossinck, M.J., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 97, pp. 61–105. [Google Scholar]
- Schmitt-Keichinger, C.; Hemmer, C.; Berthold, F.; Ritzenthaler, C. Molecular, Cellular, and Structural Biology of Grapevine Fanleaf Virus. In Grapevine Viruses: Molecular Biology, Diagnostics and Management; Meng, B., Martelli, G.P., Golino, D.A., Fuchs, M., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 83–107. [Google Scholar]
- Joly, A.C.; Garcia, S.; Hily, J.-M.; Koechler, S.; Demangeat, G.; Garcia, D.; Vigne, E.; Lemaire, O.; Zuber, H.; Gagliardi, D. An Extensive Survey of Phytoviral RNA 3′ Uridylation identifies Extreme Variations and Virus-Specific Patterns. Plant Physiol. 2023, 193, 271–290. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Snijder, E.J. Viral Cysteine Proteinases. Perspect. Drug Disc. Des. 1996, 6, 64–86. [Google Scholar] [CrossRef]
- Choi, J.; Pakbaz, S.; Yepes, L.M.; Cieniewicz, E.J.; Schmitt-Keichinger, C.; Labarile, R.; Minutillo, S.A.; Heck, M.; Hua, J.; Fuchs, M. Grapevine Fanleaf Virus RNA1-Encoded Proteins 1A and 1BHel Suppress RNA Silencing. Mol. Plant-Microbe Interact. 2023, 36, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Osterbaan, L.J.; Choi, J.; Kenney, J.; Flasco, M.; Vigne, E.; Schmitt-Keichinger, C.; Rebelo, A.R.; Heck, M.; Fuchs, M. The Identity of a Single Residue of the RNA-Dependent RNA Polymerase of Grapevine Fanleaf Virus Modulates Vein Clearing in Nicotiana benthamiana. Mol. Plant-Microbe Interact. 2019, 32, 790–801. [Google Scholar] [CrossRef] [PubMed]
- Vigne, E.; Gottula, J.W.; Schmitt-Keichinger, C.; Komar, V.; Ackerer, L.; Belval, L.; Rakotomalala, L.; Lemaire, O.; Ritzenthaler, C.; Fuchs, M. A Strain-Specific Segment of the RNA-Dependent RNA Polymerase of Grapevine Fanleaf Virus Determines Symptoms in Nicotiana Species. J. Gen. Virol. 2013, 94, 2803–2813. [Google Scholar] [CrossRef] [PubMed]
- Morris-Krsinich, B.A.M.; Forster, R.L.S.; Mossop, D.W. The Synthesis and Processing of the Nepovirus Grapevine Fanleaf Virus Proteins in Rabbit Reticulocyte Lysate. Virology 1983, 130, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Gaire, F.; Schmitt, C.; Stussi-Garaud, C.; Pinck, L.; Ritzenthaler, C. Protein 2A of Grapevine Fanleaf Nepovirus Is Implicated in RNA2 Replication and Colocalizes to the Replication Site. Virology 1999, 264, 25–36. [Google Scholar] [CrossRef]
- Martin, I.R.; Vigne, E.; Berthold, F.; Komar, V.; Lemaire, O.; Fuchs, M.; Schmitt-Keichinger, C. The 50 Distal Amino Acids of the 2AHP Homing Protein of Grapevine Fanleaf Virus Elicit a Hypersensitive Reaction on Nicotiana Occidentalis. Mol. Plant Pathol. 2018, 19, 731–743. [Google Scholar] [CrossRef]
- Belin, C.; Schmitt, C.; Gaire, F.; Walter, B.; Demangeat, G.; Pinck, L. The Nine C-Terminal Residues of the Grapevine Fanleaf Nepovirus Movement Protein Are Critical for Systemic Virus Spread. J. Gen. Virol. 1999, 80, 1347–1356. [Google Scholar] [CrossRef]
- Ritzenthaler, C.; Pinck, M.; Pinck, L. Grapevine Fanleaf Nepovirus P38 Putative Movement Protein Is Not Transiently Expressed and Is a Stable Final Maturation Product In Vivo. J. Gen. Virol. 1995, 76, 907–915. [Google Scholar] [CrossRef]
- Andret-Link, P.; Schmitt-Keichinger, C.; Demangeat, G.; Komar, V.; Fuchs, M. The Specific Transmission of Grapevine Fanleaf Virus by Its Nematode Vector Xiphinema Index Is Solely Determined by the Viral Coat Protein. Virology 2004, 320, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Schellenberger, P.; Andret-Link, P.; Schmitt-Keichinger, C.; Bergdoll, M.; Marmonier, A.; Vigne, E.; Lemaire, O.; Fuchs, M.; Demangeat, G.; Ritzenthaler, C. A Stretch of 11 Amino Acids in the βB- βC Loop of the Coat Protein of Grapevine Fanleaf Virus Is Essential for Transmission by the Nematode Xiphinema Index. J. Virol. 2010, 84, 7924–7933. [Google Scholar] [CrossRef] [PubMed]
- Schellenberger, P.; Sauter, C.; Lorber, B.; Bron, P.; Trapani, S.; Bergdoll, M.; Marmonier, A.; Schmitt-Keichinger, C.; Lemaire, O.; Demangeat, G.; et al. Structural Insights into Viral Determinants of Nematode Mediated Grapevine Fanleaf Virus Transmission. PLoS Pathog. 2011, 7, e1002034. [Google Scholar] [CrossRef] [PubMed]
- Serghini, M.A.; Fuchs, M.; Pinck, M.; Reinbolt, J.; Walter, B.; Pinck, L. RNA2 of Grapevine Fanleaf Virus: Sequence Analysis and Coat Protein Cistron Location. J. Gen. Virol. 1990, 71, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Čepin, U.; Gutiérrez-Aguirre, I.; Ravnikar, M.; Pompe-Novak, M. Frequency of Occurrence and Genetic Variability of Grapevine Fanleaf Virus Satellite RNA. Plant Pathol. 2016, 65, 510–520. [Google Scholar] [CrossRef]
- Fuchs, M.; Pinck, M.; Serghini, M.A.; Ravelonandro, M.; Walter, B.; Pinck, L. The Nucleotide Sequence of Satellite RNA in Grapevine Fanleaf Virus, Strain F13. J. Gen. Virol. 1989, 70, 955–962. [Google Scholar] [CrossRef]
- Pinck, L.; Fuchs, M.; Pinck, M.; Ravelonandro, M.; Walter, B. A Satellite RNA in Grapevine Fanleaf Virus Strain F13. J. Gen. Virol. 1988, 69, 233–239. [Google Scholar] [CrossRef]
- Saldarelli, P.; Minafra, A.; Walter, B. A Survey of Grapevine Fanleaf Nepovirus Isolates for the Presence of Satellite RNA. Vitis 1993, 32, 99–102. [Google Scholar]
- Gottula, J.; Lapato, D.; Cantilina, K.; Saito, S.; Bartlett, B.; Fuchs, M. Genetic Variability, Evolution, and Biological Effects of Grapevine Fanleaf Virus Satellite RNAs. Phytopathology 2013, 103, 1180–1187. [Google Scholar] [CrossRef]
- Ritzenthaler, C.; Viry, M.; Pinck, M.; Margis, R.; Fuchs, M.; Pinck, L. Complete Nucleotide Sequence and Genetic Organization of Grapevine Fanleaf Nepovirus RNA1. J. Gen. Virol. 1991, 72, 2357–2365. [Google Scholar] [CrossRef]
- Pompe-Novak, M.; Gutiérrez-Aguirre, I.; Vojvoda, J.; Blas, M.; Tomažič, I.; Vigne, E.; Fuchs, M.; Ravnikar, M.; Petrovič, N. Genetic Variability within RNA2 of Grapevine Fanleaf Virus. Eur. J. Plant Pathol. 2007, 117, 307–312. [Google Scholar] [CrossRef]
- Eichmeier, A.; Baránek, M.; Pidra, M. Genetic Variability of Grapevine Fanleaf Virus Isolates within Genes 1BHel and 1EPol. J. Plant Pathol. 2011, 93, 511–515. [Google Scholar]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Orlov, I.; Hemmer, C.; Ackerer, L.; Lorber, B.; Ghannam, A.; Poignavent, V.; Hleibieh, K.; Sauter, C.; Schmitt-Keichinger, C.; Belval, L.; et al. Structural Basis of Nanobody Recognition of Grapevine Fanleaf Virus and of Virus Resistance Loss. Proc. Natl. Acad. Sci. USA 2020, 117, 10848–10855. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Zheng, W.; Wuyun, Q.; Freddolino, P.L.; Zhang, Y. Integrating Deep Learning, Threading Alignments, and a Multi-MSA Strategy for High-Quality Protein Monomer and Complex Structure Prediction in CASP15. Proteins Struct. Funct. Bioinform. 2023, 12, 1684–1703. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A Unified Platform for Automated Protein Structure and Function Prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein Structure and Function Prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER Server for Protein 3D Structure Prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Roy, A.; Zhang, Y. BioLiP: A Semi-Manually Curated Database for Biologically Relevant Ligand–Protein Interactions. Nucleic Acids Res. 2013, 41, D1096–D1103. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Hu, J.; Zhang, C.; Zhang, G.; Zhang, Y. Assembling Multidomain Protein Structures through Analogous Global Structural Alignments. Natl. Acad. Sci. Lett. 2019, 116, 15930–15938. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zheng, W.; Li, Y.; Pearce, R.; Zhang, C.; Bell, E.W.; Zhang, G.; Zhang, Y. I-TASSER-MTD: A Deep-Learning-Based Platform for Multi-Domain Protein Structure and Function Prediction. Nat. Protoc. 2022, 17, 2326–2353. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Bell, E.W.; Zhang, Y. Folding Non-Homologous Proteins by Coupling Deep-Learning Contact Maps with I-TASSER Assembly Simulations. Cell Rep. Methods 2021, 3, 100014. [Google Scholar] [CrossRef] [PubMed]
- Holm, L. DALI and the Persistence of Protein Shape. Protein Sci. 2020, 29, 128–140. [Google Scholar] [CrossRef]
- Holm, L. Dali Server: Structural Unification of Protein Families. Nucleic Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef]
- Zhang, C.; Freddolino, P.L.; Zhang, Y. COFACTOR: Improved Protein Function Prediction by Combining Structure, Sequence and Protein-Protein Interaction Information. Nucleic Acids Res. 2017, 45, W291–W299. [Google Scholar] [CrossRef]
- Roy, A.; Yang, J.; Zhang, Y. COFACTOR: An Accurate Comparative Algorithm for Structure-Based Protein Function Annotation. Nucleic Acids Res. 2012, 40, W471–W477. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-Scale Prediction of Atomic-Level Protein Structure with a Language Model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef]
- Chowdhury, R.; Bouatta, N.; Biswas, S.; Floristean, C.; Kharkar, A.; Roy, K.; Rochereau, C.; Ahdritz, G.; Zhang, J.; Church, G.M.; et al. Single-Sequence Protein Structure Prediction Using a Language Model and Deep Learning. Nat. Biotechnol. 2022, 40, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Ferruz, N.; Schmidt, S.; Höcker, B. ProtGPT2 Is a Deep Unsupervised Language Model for Protein Design. Nat. Commun. 2022, 13, 4348. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The trRosetta Server for Fast and Accurate Protein Structure Prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved Protein Structure Prediction Using Predicted Interresidue Orientations. Proc. Natl. Acad. Sci. USA 2020, 3, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Mortuza, S.M.; Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Zhang, Y. Improving Fragment-Based Ab Initio Protein Structure Assembly Using Low-Accuracy Contact-Map Predictions. Nat. Commun. 2021, 12, 5011. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 6557, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; DiMaio, F.; Wang, R.Y.R.; Kim, D.; Miles, C.; Brunette, T.J.; Thompson, J.; Baker, D. High Resolution Comparative Modeling with RosettaCM. Structure 2013, 10, 1735–1742. [Google Scholar] [CrossRef]
- Raman, S.; Vernon, R.; Thompson, J.; Tyka, M.; Sadreyev, R.; Pei, J.; Kim, D.; Kellogg, E.; DiMaio, F.; Lane, O.; et al. Structure Prediction for CASP8 with All-Atom Refinement Using Rosetta. Proteins Struct. Funct. Bioinform. 2009, 77, 89–99. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V.A. Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 15, 2237–2243. [Google Scholar] [CrossRef]
- Gabler, F.; Nam, S.Z.; Till, S.; Mirdita, M.; Steinegger, M.; Söding, J.; Lupas, A.N.; Alva, V. Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Curr. Protoc. Bioinform. 2020, 1, e108. [Google Scholar] [CrossRef]
- Yang, Y.; Faraggi, E.; Zhao, H.; Zhou, Y. Improving Protein Fold Recognition and Template-Based Modeling by Employing Probabilistic-Based Matching between Predicted One-Dimensional Structural Properties of the Query and Corresponding Native Properties of Templates. Bioinformatics 2011, 27, 2076–2082. [Google Scholar] [CrossRef] [PubMed]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-Based Protein Structure Modeling Using the RaptorX Web Server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Bryant, S.H. CD-Search: Protein Domain Annotations on the Fly. Nucleic Acids Res. 2004, 32, W327–W331. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Sigrist, C.J.A.; Cerutti, L.; de Castro, E.; Langendijk-Genevaux, P.S.; Bulliard, V.; Bairoch, A.; Hulo, N. PROSITE, a Protein Domain Database for Functional Characterization and Annotation. Nucleic Acids Res. 2010, 38, D161–D166. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; Gwadz, M.; Lu, S.; Marchler, G.H.; Song, J.S.; Thanki, N.; Yamashita, R.A.; et al. The Conserved Domain Database in 2023. Nucleic Acids Res. 2023, 51, D384–D388. [Google Scholar] [CrossRef] [PubMed]
- Sperschneider, J.; Catanzariti, A.-M.; DeBoer, K.; Petre, B.; Gardiner, D.M.; Singh, K.B.; Dodds, P.N.; Taylor, J.M. LOCALIZER: Subcellular Localization Prediction of Both Plant and Effector Proteins in the Plant Cell. Sci. Rep. 2017, 7, 44598. [Google Scholar] [CrossRef]
- Sahu, S.S.; Loaiza, C.D.; Kaundal, R. Plant-mSubP: A Computational Framework for the Prediction of Single- and Multi-Target Protein Subcellular Localization Using Integrated Machine-Learning Approaches. AoB Plants 2020, 12, plz068. [Google Scholar] [CrossRef]
- Blum, T.; Briesemeister, S.; Kohlbacher, O. MultiLoc2: Integrating Phylogeny and Gene Ontology Terms Improves Subcellular Protein Localization Prediction. BMC Bioinform. 2009, 10, 274. [Google Scholar] [CrossRef]
- Armenteros, J.J.A.; Salvatore, M.; Emanuelsson, O.; Winther, O.; von Heijne, G.; Elofsson, A.; Nielsen, H. Detecting Sequence Signals in Targeting Peptides Using Deep Learning. Life Sci. Alliance 2019, 2, e201900429. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 Improves Signal Peptide Predictions Using Deep Neural Networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- De Castro, E.; Sigrist, C.J.A.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P.S.; Gasteiger, E.; Bairoch, A.; Hulo, N. ScanProsite: Detection of PROSITE Signature Matches and ProRule-Associated Functional and Structural Residues in Proteins. Nucleic Acids Res. 2006, 34, W362–W365. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Taylor, J.; Lin, V.; Altman, T.; Barbera, P.; Meleshko, D.; Lohr, D.; Novakovsky, G.; Buchfink, B.; Al-Shayeb, B.; et al. Petabase-Scale Sequence Alignment Catalyses Viral Discovery. Nature 2022, 602, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.E.; Sillitoe, I.; Dawson, N.; Lam, S.D.; Clarke, T.; Lee, D.; Orengo, C.; Lees, J. Gene3D: Extensive Prediction of Globular Domains in Proteins. Nucleic Acids Res. 2018, 46, D435–D439. [Google Scholar] [CrossRef] [PubMed]
- Sillitoe, I.; Bordin, N.; Dawson, N.; Waman, V.P.; Ashford, P.; Scholes, H.M.; Pang, C.S.M.; Woodridge, L.; Rauer, C.; Sen, N.; et al. CATH: Increased Structural Coverage of Functional Space. Nucleic Acids Res. 2021, 49, D266–D273. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Li, Y.; Zhang, Y.; Yu, D.-J. ATPbind: Accurate Protein–ATP Binding Site Prediction by Combining Sequence-Profiling and Structure-Based Comparisons. J. Chem. Inf. Model. 2018, 58, 501–510. [Google Scholar] [CrossRef]
- Chen, K.; Mizianty, M.J.; Kurgan, L. Prediction and Analysis of Nucleotide-Binding Residues Using Sequence and Sequence-Derived Structural Descriptors. Bioinformatics 2012, 28, 331–341. [Google Scholar] [CrossRef]
- Gutierrez, S.; Tyczynski, W.G.; Boomsma, W.; Teufel, F.; Winther, O. MembraneFold: Visualising Transmembrane Protein Structure and Topology. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Armenteros, J.J.A.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM Predicts Alpha and Beta Transmembrane Proteins Using Deep Neural Networks. bioRxiv 2022. [Google Scholar] [CrossRef]
- Juretić, D.; Zoranić, L.; Zucić, D. Basic Charge Clusters and Predictions of Membrane Protein Topology. J. Chem. Inf. Comput. Sci. 2002, 42, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Käll, L.; Krogh, A.; Sonnhammer, E.L.L. A Combined Transmembrane Topology and Signal Peptide Prediction Method. J. Mol. Biol. 2004, 338, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Nugent, T.; Jones, D.T. Transmembrane Protein Topology Prediction Using Support Vector Machines. BMC Bioinform. 2009, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. lDDT: A Local Superposition-Free Score for Comparing Protein Structures and Models Using Distance Difference Tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef] [PubMed]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. QMEANDisCo-Distance Constraints Applied on Model Quality Estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. Scoring Function for Automated Assessment of Protein Structure Template Quality. Proteins Struct. Funct. Bioinform. 2004, 57, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Pinck, M.; Reinbolt, J.; Loudes, A.M.; Le Ret, M.; Pinck, L. Primary Structure and Location of the Genome-Linked Protein (VPg) of Grapevine Fanleaf Nepovirus. FEBS Lett. 1991, 284, 117–119. [Google Scholar] [CrossRef]
- Margis, R.; Viry, M.; Pinck, M.; Pinck, L. Cloning and in Vitro Characterization of the Grapevine Fanleaf Virus Proteinase Cistron. Virology 1991, 185, 779–787. [Google Scholar] [CrossRef]
- Margis, R.; Ritzenthaler, C.; Reinbolt, J.; Pinck, M.; Pinck, L. Genome Organization of Grapevine Fanleaf Nepovirus RNA2 Deduced from the 122K Polyprotein P2 in Vitro Cleavage Products. J. Gen. Virol. 1993, 74, 1919–1926. [Google Scholar] [CrossRef]
- Margis, R.; Viry, M.; Pinck, M.; Bardonnet, N.; Pinck, L. Differential Proteolytic Activities of Precursor and Mature Forms of the 24K Proteinase of Grapevine Fanleaf Nepovirus. Virology 1994, 200, 79–86. [Google Scholar] [CrossRef]
- Margis, R.; Pinck, L. Effects of Site-Directed Mutagenesis on the Presumed Catalytic Triad and Substrate-Binding Pocket of Grapevine Fanleaf Nepovirus 24-kDa Proteinase. Virology 1992, 190, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Osterbaan, L.J.; Hoyle, V.; Curtis, M.; DeBlasio, S.; Rivera, K.D.; Heck, M.; Fuchs, M. Identification of Protein Interactions of Grapevine Fanleaf Virus RNA-Dependent RNA Polymerase during Infection of Nicotiana benthamiana by Affinity Purification and Tandem Mass Spectrometry. J. Gen. Virol. 2021, 102, 001607. [Google Scholar] [CrossRef] [PubMed]
- Laporte, C.; Vetter, G.; Loudes, A.-M.; Robinson, D.G.; Hillmer, S.; Stussi-Garaud, C.; Ritzenthaler, C. Involvement of the Secretory Pathway and the Cytoskeleton in Intracellular Targeting and Tubule Assembly of Grapevine Fanleaf Virus Movement Protein in Tobacco BY-2 Cells. Plant Cell 2003, 15, 2058–2075. [Google Scholar] [CrossRef] [PubMed]
- Amari, K.; Boutant, E.; Hofmann, C.; Schmitt-Keichinger, C.; Fernandez-Calvino, L.; Didier, P.; Lerich, A.; Mutterer, J.; Thomas, C.L.; Heinlein, M.; et al. A Family of Plasmodesmal Proteins with Receptor-Like Properties for Plant Viral Movement Proteins. PLoS Pathog. 2010, 6, e1001119. [Google Scholar] [CrossRef] [PubMed]
- Quacquarelli, A.; Gallitelli, D.; Savino, V.; Martelli, G.P. Properties of Grapevine Fanleaf Virus. J. Gen. Virol. 1976, 32, 349–360. [Google Scholar] [CrossRef]
- Ritzenthaler, C.; Laporte, C.; Gaire, F.; Dunoyer, P.; Schmitt, C.; Duval, S.; Piéquet, A.; Loudes, A.-M.; Rohfritsch, O.; Stussi-Garaud, C.; et al. Grapevine Fanleaf Virus Replication Occurs on Endoplasmic Reticulum-Derived Membranes. J. Virol. 2002, 76, 8808–8819. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.G.; DeBlasio, S.; Yang, Y.; Thannhauser, T.; Heck, M.; Fuchs, M. Profiling Plant Proteome and Transcriptome Changes during Grapevine Fanleaf Virus Infection. J. Proteome Res. 2023, 22, 1997–2017. [Google Scholar] [CrossRef]
- Olechnovič, K.; Monastyrskyy, B.; Kryshtafovych, A.; Venclovas, Č. Comparative Analysis of Methods for Evaluation of Protein Models Against Native Structures. Bioinformatics 2019, 6, 937–944. [Google Scholar] [CrossRef]
- Miller, P.W.; Pokutta, S.; Mitchell, J.M.; Chodaparambil, J.V.; Clarke, D.N.; Nelson, W.J.; Weis, W.I.; Nichols, S.A. Analysis of a Vinculin Homolog in a Sponge (Phylum Porifera) Reveals That Vertebrate-like Cell Adhesions Emerged Early in Animal Evolution. J. Biol. Chem. 2018, 293, 11674–11686. [Google Scholar] [CrossRef]
- Nepal, M.; Che, R.; Ma, C.; Zhang, J.; Fei, P. FANCD2 and DNA Damage. Int. J. Mol. Sci. 2017, 8, 1804. [Google Scholar] [CrossRef]
- Al-Lazikani, B.; Jung, J.; Xiang, Z.; Honig, B. Protein Structure Prediction. Curr. Opin. Chem. Biol. 2001, 5, 51–56. [Google Scholar] [CrossRef]
- Kuhlman, B.; Bradley, P. Advances in Protein Structure Prediction and Design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Li, Y.; Zhang, C.; Zhou, X.; Pearce, R.; Bell, E.W.; Huang, X.; Zhang, Y. Protein Structure Prediction Using Deep Learning Distance and Hydrogen-Bonding Restraints in CASP14. Proteins Struct. Funct. Bioinform. 2021, 89, 1734–1751. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Malone, B.; Llewellyn, E.; Grasso, M.; Shelton, P.M.M.; Olinares, P.D.B.; Maruthi, K.; Eng, E.T.; Vatandaslar, H.; Chait, B.T.; et al. Structural Basis for Helicase-Polymerase Coupling in the SARS-CoV-2 Replication-Transcription Complex. Cell 2020, 182, 1560–1573. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, U.C.; Shrivastava, R. Interaction of Viral Proteins with Metal Ions: Role in Maintaining the Structure and Functions of Viruses. FEMS Microbiol. Immunol. 2005, 43, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Sanfaçon, H.; Skern, T. AlphaFold Modeling of Nepovirus 3C-Like Proteinases Provides New Insights into Their Diverse Substrate Specificities. Virology 2023, 590, 109956. [Google Scholar] [CrossRef]
- Sanfaçon, H. Re-examination of Nepovirus Polyprotein Cleavage Sites Highlights the Diverse Specificities and Evolutionary Relationships of Nepovirus 3C-Like Proteases. Arch. Virol. 2022, 167, 2529–2543. [Google Scholar] [CrossRef]
- Cuthbertson, J.M.; Doyle, D.A.; Sansom, M.S.P. Transmembrane Helix Prediction: A Comparative Evaluation and Analysis. Protein Eng. Des. Sel. 2005, 18, 295–308. [Google Scholar] [CrossRef]
- Ramasarma, T. Transmembrane Domains Participate in Functions of Integral Membrane Proteins. Indian J. Biochem. Biophys. 1996, 33, 20–29. [Google Scholar]
- Borkakoti, N.; Thornton, J.M. AlphaFold2 Protein Structure Prediction: Implications for Drug Discovery. Curr. Opin. Struct. 2023, 78, 102526. [Google Scholar] [CrossRef]
- Ren, F.; Ding, X.; Zheng, M.; Korzinkin, M.; Cai, X.; Zhu, W.; Mantsyzov, A.; Aliper, A.; Aladinskiy, V.; Cao, Z.; et al. AlphaFold Accelerates Artificial Intelligence Powered Drug Discovery: Efficient Discovery of a Novel CDK20 Small Molecule Inhibitor. Chem. Sci. 2023, 14, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Meller, A.; Bhakat, S.; Solieva, S.; Bowman, G.R. Accelerating Cryptic Pocket Discovery Using AlphaFold. J. Chem. Theory Comput. 2023, 19, 4355–4363. [Google Scholar] [CrossRef] [PubMed]
- Sen Gupta, P.S.; Panda, S.K.; Nayak, A.K.; Rana, M.K. Identification and Investigation of a Cryptic Binding Pocket of the P37 Envelope Protein of Monkeypox Virus by Molecular Dynamics Simulations. J. Phys. Chem. Lett. 2023, 14, 3230–3235. [Google Scholar] [CrossRef] [PubMed]
- van Breugel, M.; Rosa e Silva, I.; Andreeva, A. Structural Validation and Assessment of AlphaFold2 Predictions for Centrosomal and Centriolar Proteins and Their Complexes. Commun. Biol. 2022, 5, 312. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Salmen, W.; Sankaran, B.; Lasanajak, Y.; Smith, D.F.; Crawford, S.E.; Estes, M.K.; Prasad, B.V.V. Novel Fold of Rotavirus Glycan-Binding Domain Predicted by AlphaFold2 and Determined by X-Ray Crystallography. Commun. Biol. 2022, 5, 419. [Google Scholar] [CrossRef] [PubMed]
- Gomes, P.S.F.C.; Gomes, D.E.B.; Bernardi, R.C. Protein Structure Prediction in the Era of AI: Challenges and Limitations When Applying to in silico Force Spectroscopy. Front. Bioinform. 2022, 2, 983306. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Feng, B.Y.; Varshney, A.; Pierce, B.G. Benchmarking AlphaFold for Protein Complex Modeling Reveals Accuracy Determinants. Protein Sci. 2022, 31, e4379. [Google Scholar] [CrossRef]
- Lee, S.; Kim, S.; Lee, G.R.; Kwon, S.; Woo, H.; Seok, C.; Park, H. Evaluating GPCR Modeling and Docking Strategies in the Era of Deep Learning-Based Protein Structure Prediction. Comput. Struct. Biotechnol. J. 2023, 21, 158–167. [Google Scholar] [CrossRef]
- Azzaz, F.; Yahi, N.; Chahinian, H.; Fantini, J. The Epigenetic Dimension of Protein Structure Is an Intrinsic Weakness of the AlphaFold Program. Biomolecules 2022, 12, 1527. [Google Scholar] [CrossRef]



| Program | Algorithm | Modeling Method | Output | Confidence Metric | Confidence Range | Reference | |
|---|---|---|---|---|---|---|---|
| 1 | AlphaFold2 (ColabFold) | Neural network | Template based | Tertiary protein model | eTM and pLDDT | 0.00–1.00 and 0.00–100 | [41,42,43] |
| 2 | C-Quark | Contact-assisted ab initio | Free modeling | Tertiary protein model | eTM | 0.00–1.00 | [61] |
| 3 | C-I-TASSER | Neural network | Free modeling | Tertiary protein model | pTM/TM-align | 0.00–1.00 | [48,51] |
| 4 | D-I-TASSER | Neural network | Template based | Tertiary protein model | pTM/TM-align | 0.00–1.00 | [44,45,46,47,48] |
| 5 | D-I-TASSER MTD | Neural network | Template based | Tertiary protein model | pTM/TM-align | 0.00–1.00 | [48,49,50] |
| 6 | ESMFold | Neural network | Single input sequence | Tertiary protein model | pTM/pLDDT | 0.00–1.00 and 0.00–100 | [56] |
| 7 | Robetta | Extension of RoseTTAFold and trRosetta | Free modeling | Tertiary protein model | GDT | 0.00–1.00 | [62,63,64] |
| 8 | trRosetta | Neural network | Free modeling | Tertiary protein model | eTM/pLDDT | 0.00–1.00 and 0.00–100 | [59,60] |
| 9 | BioLiP | Semi-manually curated database | Guided | Functional predictions | C-Score | 0.00–1.00 | [48] |
| 10 | RGN2 | Neural network | Free modeling | Tertiary protein model | GDT | 0.00–1.00 | [57] |
| 11 | ProtGPT2 | Neural network | Free modeling | Tertiary protein model | pLDDT | 0.00–100 | [58] |
| 12 | QMEANDisCo | Distance based model quality estimation | NA | Confidence metric | QMEANDisCo | 0.00–100 | [92] |
| 13 | Phyre2 | Multiple sequence alignment | Multiple sequence alignment | Secondary/tertiary protein model | Coverage and Identity | 0.00–100 | [79] |
| 14 | palmID | Sequence conservation | NA | Confidence for RdRP | RdRP score | 0.00–100 | [80] |
| 15 | MOTIF Search | Sequence conservation | Multiple sequence alignment | Primary sequence domain detection | E-value or p-value | Lower p-value is better | [69,70,71,72] |
| 16 | CATH/Gene3D | Semi-manually curated database | Multiple sequence alignment | Primary sequence domain detection | Extensive metrics | Multiple | [81,82] |
| 17 | LOCALIZER | Machine learning | Feature detection based on known sequences | Primary sequence domain detection | Ranking | Priority ranking | [73] |
| 18 | Plant mSubP | Machine learning | Feature detection based on known sequences | Primary sequence domain detection | Percentage probability | 0.00–1.00 (Percentage) | [74] |
| 19 | MultiLoc2 | Machine learning | Feature detection based on known sequences | Primary sequence domain detection | Percentage probability | 0.00–1.00 (Percentage) | [75] |
| 20 | TargetP a | Neural network | Feature detection based on known sequences | Primary sequence domain detection | Ranking | Percentage | [76] |
| 21 | SignalP a | Neural network | Feature detection based on known sequences | Primary sequence domain detection | Ranking | Percentage | [77] |
| 22 | MembraneFold a | AlphaFold/OmegaFold based | Feature detection based on known sequences | Primary sequence domain detection | pLDDT for membrane proteins | 0.00–100 | [85] |
| 23 | DeepTMHMM a | Neural network | Feature detection based on known sequences | Primary sequence domain detection | Probability | 0.00–1.00 | [86] |
| 24 | Split 4.0 | Consensus hidden Markov model | Feature detection based on known sequences | Primary sequence domain detection | Binary | Threshold | [87] |
| 25 | Phobius | Hidden Markov model | Feature detection based on known sequences | Primary sequence domain detection | Probability | Threshold | [88] |
| 26 | MEMESAT3 (PSIPRED) | Hidden Markov model (HMM) and model recognition | Feature detection based on known sequences | Primary sequence domain detection | Log likelihood ratio | Threshold | [89] |
| 27 | ATPbind | Machine learning | Feature detection based on structural configuation | Tertiary sequence domain detection | Identity of residues | Threshold | [83] |
| 28 | NsitePred | Machine learning | Feature detection based on known sequences | Primary sequence domain detection | Probability | 0.00–1.00 | [84] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, B.G.; Choi, J.; Fuchs, M.F. Predictive Modeling of Proteins Encoded by a Plant Virus Sheds a New Light on Their Structure and Inherent Multifunctionality. Biomolecules 2024, 14, 62. https://doi.org/10.3390/biom14010062
Roy BG, Choi J, Fuchs MF. Predictive Modeling of Proteins Encoded by a Plant Virus Sheds a New Light on Their Structure and Inherent Multifunctionality. Biomolecules. 2024; 14(1):62. https://doi.org/10.3390/biom14010062
Chicago/Turabian StyleRoy, Brandon G., Jiyeong Choi, and Marc F. Fuchs. 2024. "Predictive Modeling of Proteins Encoded by a Plant Virus Sheds a New Light on Their Structure and Inherent Multifunctionality" Biomolecules 14, no. 1: 62. https://doi.org/10.3390/biom14010062
APA StyleRoy, B. G., Choi, J., & Fuchs, M. F. (2024). Predictive Modeling of Proteins Encoded by a Plant Virus Sheds a New Light on Their Structure and Inherent Multifunctionality. Biomolecules, 14(1), 62. https://doi.org/10.3390/biom14010062