
Mitochondrial Dysfunction as the Major Basis of Brain Aging


Abstract
:1. Introduction
2. The Decline of Immune System Function with Age
3. The Excessive Presence of Oxidant Free Radicals with Aging
4. The Emergence of a Chronic State of Low-Level Excitatory Activity
5. The Mitochondrial Basis for Age-Related Deviation of Essential Processes toward Injurious Configurations
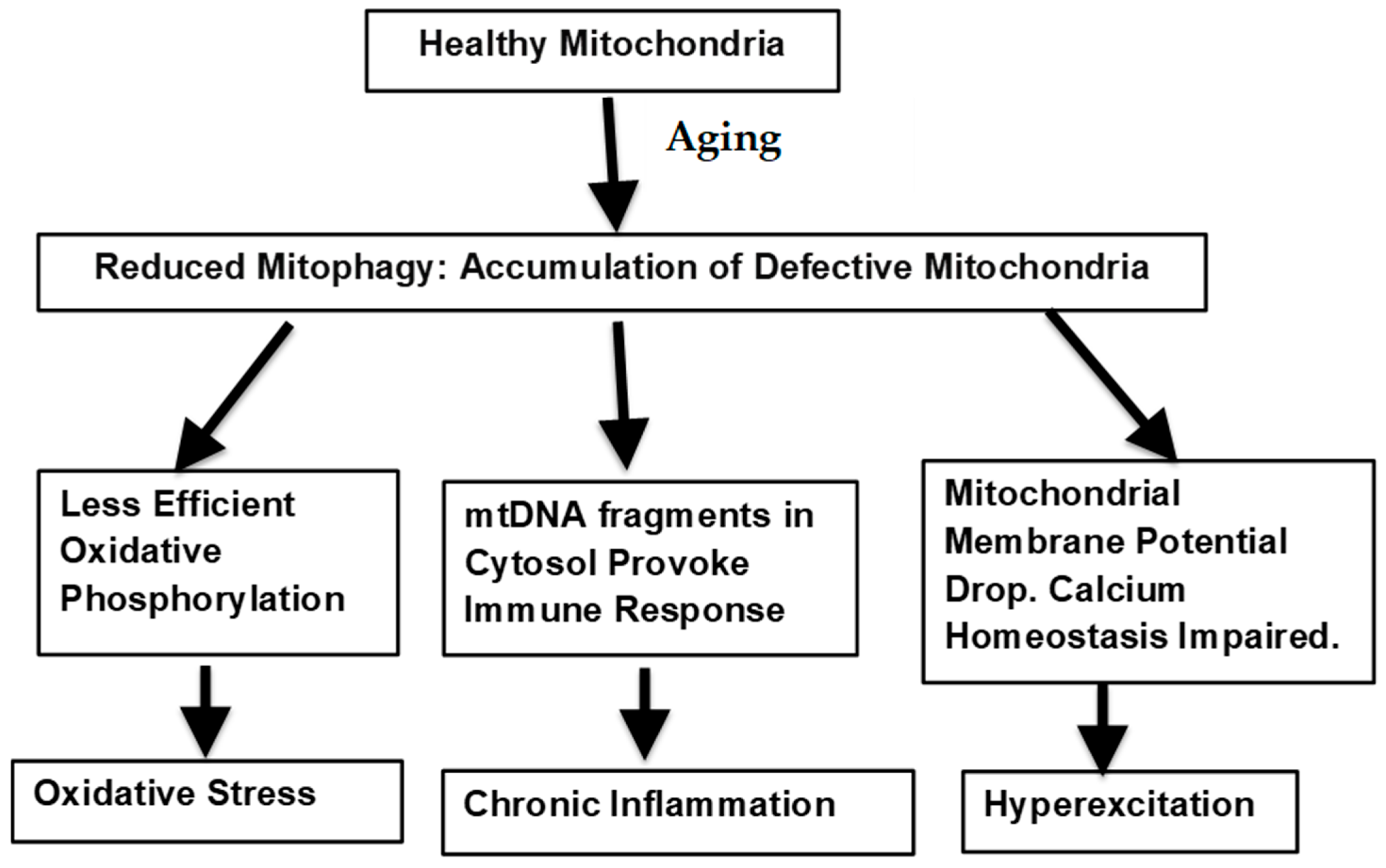
5.1. Free Radical Production
5.2. Undesirable Autoimmune Responses
5.3. Persistent Hyperexcitation
6. Importance of Mitophagy
7. Therapeutic Moderation of the Rate of Aging
8. Conclusions
Funding
Conflicts of Interest
References
- Luo, Y.; Zhang, Y.; Yang, Y.; Wu, S.; Zhao, J.; Li, Y.; Kang, X.; Li, Z.; Chen, J.; Shen, X.; et al. Bifidobacterium infantis and 2′-fucosyllactose supplementation in early life may have potential long-term benefits on gut microbiota, intestinal development, and immune function in mice. J. Dairy Sci. 2023, 106, 7461–7476. [Google Scholar] [CrossRef] [PubMed]
- Amador-Patarroyo, M.J.; Rodriguez-Rodriguez, A.; Montoya-Ortiz, G. How does age at onset influence the outcome of autoimmune diseases? Autoimmune Dis. 2012, 2012, 251730. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Bondy, S.C. Aspects of the immune system that impact brain function. J. Neuroimmunol. 2020, 340, 577167. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Pascoal, T.A.; Benedet, A.L.; Ashton, N.J.; Kang, M.S.; Therriault, J.; Chamoun, M.; Savard, M.; Lussier, F.Z.; Tissot, C.; Karikari, T.K.; et al. Microglial activation and tau propagate jointly across Braak stages. Nat. Med. 2021, 27, 1592–1599, Erratum in Nat. Med. 2021, 27, 2048–2049. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Laws, S.M.; Miles, L.A.; Wiley, J.S.; Huang, X.; Masters, C.L.; Gu, B.J. Genomics of Alzheimer’s disease implicates the innate and adaptive immune systems. Cell. Mol. Life Sci. 2021, 78, 7397–7426. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Sancheti, H.; Liu, Z.; Cadenas, E. Mitochondrial function in ageing: Coordination with signalling and transcriptional pathways. J. Physiol. 2016, 594, 2025–2042. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Jian, Z.; Jin, T.; Li, Y.; Zeng, Z.; Zhang, X.; Xiong, X.; Gu, L. NOX2-mediated reactive oxygen species are double-edged swords in focal cerebral ischemia in mice. J. Neuroinflamm. 2022, 19, 184. [Google Scholar] [CrossRef]
- Goldsteins, G.; Hakosalo, V.; Jaronen, M.; Keuters, M.H.; Lehtonen, Š.; Koistinaho, J. CNS Redox Homeostasis and Dysfunction in Neurodegenerative Diseases. Antioxidants 2022, 11, 405. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: Chronic diseases and aging. Arch. Toxicol. 2023, 97, 2499–2574. [Google Scholar] [CrossRef] [PubMed]
- Sanghai, N.; Tranmer, G.K. Biochemical and Molecular Pathways in Neurodegenerative Diseases: An Integrated View. Cells 2023, 12, 2318. [Google Scholar] [CrossRef] [PubMed]
- Ameen, S.S.; Griem-Krey, N.; Dufour, A.; Hossain, M.I.; Hoque, A.; Sturgeon, S.; Nandurkar, H.; Draxler, D.F.; Medcalf, R.L.; Kamaruddin, M.A.; et al. N-Terminomic Changes in Neurons During Excitotoxicity Reveal Proteolytic Events Associated with Synaptic Dysfunctions and Potential Targets for Neuroprotection. Mol. Cell. Proteom. 2023, 22, 100543. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Su, L.; Liu, Z. Critical role of calpain in inflammation. Biomed. Rep. 2016, 5, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Liu, W. Hypoxia-Induced Mitogenic Factor: A Multifunctional Protein Involved in Health and Disease. Front. Cell Dev. Biol. 2021, 9, 691774. [Google Scholar] [CrossRef] [PubMed]
- Mahaman, Y.A.R.; Huang, F.; Kessete Afewerky, H.; Maibouge, T.M.S.; Ghose, B.; Wang, X. Involvement of calpain in the neuropathogenesis of Alzheimer’s disease. Med. Res. Rev. 2019, 39, 608–630. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, R.; Kitazawa, M.; Chabrier, M.A.; Cheng, D.; Baglietto-Vargas, D.; Kling, A.; Moeller, A.; Green, K.N.; LaFerla, F.M. Calpain inhibitor A-705253 mitigates Alzheimer’s disease-like pathology and cognitive decline in aged 3xTgAD mice. Am. J. Pathol. 2012, 181, 616–625. [Google Scholar] [CrossRef]
- Metwally, E.; Al-Abbadi, H.A.; Hussain, T.; Murtaza, G.; Abdellatif, A.M.; Ahmed, M.F. Calpain signaling: From biology to therapeutic opportunities in neurodegenerative disorders. Front. Vet. Sci. 2023, 10, 1235163. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.; McCoy, H.M.; Zaman, V.; Shields, D.C.; Banik, N.L.; Haque, A. Calpain activation and progression of inflammatory cycles in Parkinson’s disease. Front. Biosci. 2022, 27, 20. [Google Scholar] [CrossRef] [PubMed]
- Morales, I.; Sanchez, A.; Puertas-Avendaño, R.; Rodriguez-Sabate, C.; Perez-Barreto, A.; Rodriguez, M. Neuroglial transmitophagy and Parkinson’s disease. Glia 2020, 68, 2277–2299. [Google Scholar] [CrossRef] [PubMed]
- Bayón-Cordero, L.; Ochoa-Bueno, B.I.; Ruiz, A.; Ozalla, M.; Matute, C.; Sánchez-Gómez, M.V. GABA Receptor Agonists Protect from Excitotoxic Damage Induced by AMPA in Oligodendrocytes. Front. Pharmacol. 2022, 13, 897056. [Google Scholar] [CrossRef] [PubMed]
- Reyes, R.C.; Brennan, A.M.; Shen, Y.; Baldwin, Y.; Swanson, R.A. Activation of neuronal NMDA receptors induces superoxide-mediated oxidative stress in neighboring neurons and astrocytes. J. Neurosci. 2012, 32, 12973–12978. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Companys-Alemany, J.; Turcu, A.L.; Schneider, M.; Müller, C.E.; Vázquez, S.; Griñán-Ferré, C.; Pallàs, M. NMDA receptor antagonists reduce amyloid-β deposition by modulating calpain-1 signaling and autophagy, rescuing cognitive impairment in 5XFAD mice. Cell. Mol. Life Sci. 2022, 79, 408. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Sun, P.; Zhang, H.; Yang, H. Mitochondrial quality control in the brain: The physiological and pathological roles. Front. Neurosci. 2022, 16, 1075141. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Wei, Y.H. Mitochondria and aging. Adv. Exp. Med. Biol. 2012, 942, 311–327. [Google Scholar] [CrossRef]
- Lin, M.T.; Simon, D.K.; Ahn, C.H.; Kim, L.M.; Beal, M.F. High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brain. Hum. Mol. Genet. 2002, 11, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Fontana, G.A.; Gahlon, H.L. Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res. 2020, 48, 11244–11258. [Google Scholar] [CrossRef]
- Vizioli, M.G.; Liu, T.; Miller, K.N.; Robertson, N.A.; Gilroy, K.; Lagnado, A.B.; Perez-Garcia, A.; Kiourtis, C.; Dasgupta, N.; Lei, X.; et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020, 4, 428–445. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Contreras, M.; Kennedy, S.R. The Complicated Nature of Somatic mtDNA Mutations in Aging. Front. Aging 2022, 2, 805126. [Google Scholar] [CrossRef] [PubMed]
- Rosa, F.L.L.; de Souza, I.I.A.; Monnerat, G.; Campos de Carvalho, A.C.; Maciel, L. Aging Triggers Mitochondrial Dysfunction in Mice. Int. J. Mol. Sci. 2023, 24, 10591. [Google Scholar] [CrossRef] [PubMed]
- Godoy, J.A.; Rios, J.A.; Picón-Pagès, P.; Herrera-Fernández, V.; Swaby, B.; Crepin, G.; Vicente, R.; Fernández-Fernández, J.M.; Muñoz, F.J. Mitostasis, Calcium and Free Radicals in Health, Aging and Neurodegeneration. Biomolecules 2021, 11, 1012. [Google Scholar] [CrossRef]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef]
- Dutta, N.; Garcia, G.; Higuchi-Sanabria, R. Hijacking Cellular Stress Responses to Promote Lifespan. Front. Aging 2022, 3, 860404. [Google Scholar] [CrossRef] [PubMed]
- Stenberg, S.; Li, J.; Gjuvsland, A.B.; Persson, K.; Demitz-Helin, E.; González Peña, C.; Yue, J.X.; Gilchrist, C.; Ärengård, T.; Ghiaci, P.; et al. Genetically controlled mtDNA deletions prevent ROS damage by arresting oxidative phosphorylation. eLife 2022, 11, e76095. [Google Scholar] [CrossRef] [PubMed]
- Schosserer, M.; Banks, G.; Dogan, S.; Dungel, P.; Fernandes, A.; Marolt Presen, D.; Matheu, A.; Osuchowski, M.; Potter, P.; Sanfeliu, C.; et al. Modelling physical resilience in ageing mice. Mech. Ageing Dev. 2019, 177, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Zanini, G.; Selleri, V.; Lopez Domenech, S.; Malerba, M.; Nasi, M.; Mattioli, A.V.; Pinti, M. Mitochondrial DNA as inflammatory DAMP: A warning of an aging immune system? Biochem. Soc. Trans. 2023, 51, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Marzetti, E. Cell Death and Inflammation: The Role of Mitochondria in Health and Disease. Cells 2021, 10, 537. [Google Scholar] [CrossRef]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef] [PubMed]
- Gulen, M.F.; Samson, N.; Keller, A.; Schwabenland, M.; Liu, C.; Glück, S.; Thacker, V.V.; Favre, L.; Mangeat, B.; Kroese, L.J.; et al. cGAS-STING drives ageing-related inflammation and neurodegeneration. Nature 2023, 620, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Homolak, J. Targeting the microbiota-mitochondria crosstalk in neurodegeneration with senotherapeutics. Adv. Protein Chem. Struct. Biol. 2023, 136, 339–383. [Google Scholar] [CrossRef]
- Victorelli, S.; Salmonowicz, H.; Chapman, J.; Martini, H.; Vizioli, M.G.; Riley, J.S.; Cloix, C.; Hall-Younger, E.; Machado Espindola-Netto, J.; Jurk, D.; et al. Apoptotic stress causes mtDNA release during senescence and drives the SASP. Nature 2023, 622, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Depp, C.; Bas-Orth, C.; Schroeder, L.; Hellwig, A.; Bading, H. Synaptic Activity Protects Neurons Against Calcium-Mediated Oxidation and Contraction of Mitochondria During Excitotoxicity. Antioxid. Redox Signal. 2018, 29, 1109–1124. [Google Scholar] [CrossRef] [PubMed]
- Neves, D.; Salazar, I.L.; Almeida, R.D.; Silva, R.M. Molecular mechanisms of ischemia and glutamate excitotoxicity. Life Sci. 2023, 328, 121814. [Google Scholar] [CrossRef]
- Baltan, S. Excitotoxicity and mitochondrial dysfunction underlie age-dependent ischemic white matter injury. Adv. Neurobiol. 2014, 11, 151–170. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, calcium and mitochondria: A triad in synaptic neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Walters, G.C.; Usachev, Y.M. Mitochondrial calcium cycling in neuronal function and neurodegeneration. Front. Cell Dev. Biol. 2023, 11, 1094356. [Google Scholar] [CrossRef]
- Gaziev, A.I.; Abdullaev, S.; Podlutsky, A. Mitochondrial function and mitochondrial DNA maintenance with advancing age. Biogerontology 2014, 15, 417–438. [Google Scholar] [CrossRef] [PubMed]
- Fairley, L.H.; Grimm, A.; Eckert, A. Mitochondria Transfer in Brain Injury and Disease. Cells 2022, 11, 3603. [Google Scholar] [CrossRef]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., 2nd; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648, Erratum in Nat. Neurosci. 2021, 24, 289. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Amadoro, G.; Latina, V.; Valenti, D. Therapeutic Potential of Targeting Mitochondria for Alzheimer’s Disease Treatment. J. Clin. Med. 2022, 11, 6742. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, N.S.; Wilkins, H.M. Mitophagy and the Brain. Int. J. Mol. Sci. 2020, 21, 9661. [Google Scholar] [CrossRef] [PubMed]
- Schmid, E.T.; Pyo, J.H.; Walker, D.W. Neuronal induction of BNIP3-mediated mitophagy slows systemic aging in Drosophila. Nat. Aging 2022, 2, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Caponio, D.; Veverová, K.; Zhang, S.Q.; Shi, L.; Wong, G.; Vyhnalek, M.; Fang, E.F. Compromised autophagy and mitophagy in brain ageing and Alzheimer’s diseases. Aging Brain 2022, 2, 100056. [Google Scholar] [CrossRef] [PubMed]
- Rappe, A.; McWilliams, T.G. Mitophagy in the aging nervous system. Front. Cell Dev. Biol. 2022, 10, 978142. [Google Scholar] [CrossRef] [PubMed]
- Onyango, I.G.; Bennett, J.P.; Stokin, G.B. Mitochondrially-Targeted Therapeutic Strategies for Alzheimer’s Disease. Curr. Alzheimer Res. 2021, 18, 753–771. [Google Scholar] [CrossRef] [PubMed]
- Varghese, N.; Werner, S.; Grimm, A.; Eckert, A. Dietary Mitophagy Enhancer: A Strategy for Healthy Brain Aging? Antioxidants 2020, 9, 932. [Google Scholar] [CrossRef]
- Gebert, M.; Sławski, J.; Kalinowski, L.; Collawn, J.F.; Bartoszewski, R. The Unfolded Protein Response: A Double-Edged Sword for Brain Health. Antioxidants 2023, 12, 1648. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Yin, J.; Chen, J.; Ma, X.; Wu, M.; Liu, G.; Yao, K.; Tan, B.; Yin, Y. Mitochondria-Targeted Antioxidants: A Step towards Disease Treatment. Oxidative Med. Cell. Longev. 2020, 2020, 8837893. [Google Scholar] [CrossRef] [PubMed]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, H.S.; Chung, J.H. Molecular mechanisms of mitochondrial DNA release and activation of the cGAS-STING pathway. Exp. Mol. Med. 2023, 55, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Hua, F.; Fang, P.; Li, C.; Deng, F.; Chen, S.; Ying, J.; Wang, X. Regulation of Mitophagy by Sirtuin Family Proteins: A Vital Role in Aging and Age-Related Diseases. Front. Aging Neurosci. 2022, 14, 845330. [Google Scholar] [CrossRef] [PubMed]
- Davinelli, S.; De Stefani, D.; De Vivo, I.; Scapagnini, G. Polyphenols as Caloric Restriction Mimetics Regulating Mitochondrial Biogenesis and Mitophagy. Trends Endocrinol. Metab. 2020, 31, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Cilleros-Holgado, P.; Gómez-Fernández, D.; Piñero-Pérez, R.; Romero-Domínguez, J.M.; Reche-López, D.; López-Cabrera, A.; Álvarez-Córdoba, M.; Munuera-Cabeza, M.; Talaverón-Rey, M.; Suárez-Carrillo, A.; et al. Mitochondrial Quality Control via Mitochondrial Unfolded Protein Response (mtUPR) in Ageing and Neurodegenerative Diseases. Biomolecules 2023, 13, 1789. [Google Scholar] [CrossRef] [PubMed]
- Leandro, G.S.; Sykora, P.; Bohr, V.A. The impact of base excision DNA repair in age-related neurodegenerative diseases. Mutat. Res. 2015, 776, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lin, L.; Zhang, Q.; Yang, J.; Kamili, E.; Chu, J.; Li, X.; Yang, S.; Xu, Y. Heteroplasmy and Individual Mitogene Pools: Characteristics and Potential Roles in Ecological Studies. Biology 2023, 12, 1452. [Google Scholar] [CrossRef]
- Nadalutti, C.A.; Ayala-Peña, S.; Santos, J.H. Mitochondrial DNA damage as driver of cellular outcomes. Am. J. Physiol. Cell Physiol. 2022, 322, C136–C150. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Essential Activity | Ineffective Potentially Harmful Variant | Means by Which Mitochondria can Contribute to This Transition |
|---|---|---|
| Generation of energy by oxidation of substrates | Leakage of harmful free radicals into the cytosol | Decreased efficiency of the mitochondrial oxidative phosphorylation |
| Dispersion of invasive organisms by oxidant free radicals | Balance between antioxidant molecules and reactive oxygen disrupted | Sub-optimal redox status leads to failure of Mitochondrial Unfolded Protein Response (mtUPR) and misfolding of proteins |
| Removal of invasive organisms and unhealthy cells by glial and phagocytic immune attack | Extended untargeted inflammatory activity leading to random cell injury | DNA from impaired mitochondria leaks into cytosol provoking autoinflammatory response |
| Maintenance of neuronal signaling | Persistent unfocussed neuronal excitatory activity | Failure of mitochondria to sequester calcium and activation of calpains leads to increased intrasynaptic glutamate release. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bondy, S.C. Mitochondrial Dysfunction as the Major Basis of Brain Aging. Biomolecules 2024, 14, 402. https://doi.org/10.3390/biom14040402
Bondy SC. Mitochondrial Dysfunction as the Major Basis of Brain Aging. Biomolecules. 2024; 14(4):402. https://doi.org/10.3390/biom14040402
Chicago/Turabian StyleBondy, Stephen C. 2024. "Mitochondrial Dysfunction as the Major Basis of Brain Aging" Biomolecules 14, no. 4: 402. https://doi.org/10.3390/biom14040402
APA StyleBondy, S. C. (2024). Mitochondrial Dysfunction as the Major Basis of Brain Aging. Biomolecules, 14(4), 402. https://doi.org/10.3390/biom14040402