


Morphological Changes Induced by TKS4 Deficiency Can Be Reversed by EZH2 Inhibition in Colorectal Carcinoma Cells




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and TKS4 Knockout
2.2. EZH2 Inhibitor Treatment
2.3. Total RNA Extraction and Real-Time Quantitative PCR
2.4. Whole Transcriptome Sequencing (RNA-seq)
2.5. RNA Sequencing Data Analysis
2.6. Protein Extraction and Western Blot Analysis
2.7. Immunocytochemistry and Fluorescence Microscopy
2.8. Chromatin Immunoprecipitation Sequencing (ChIP-Seq)
2.9. ChIP-Seq Data Analysis
2.10. Cell Migration Assay
2.11. Transwell Invasion Assay
2.12. Cell Proliferation Assay
2.13. Viability Assay
2.14. Statistical Analysis
3. Results
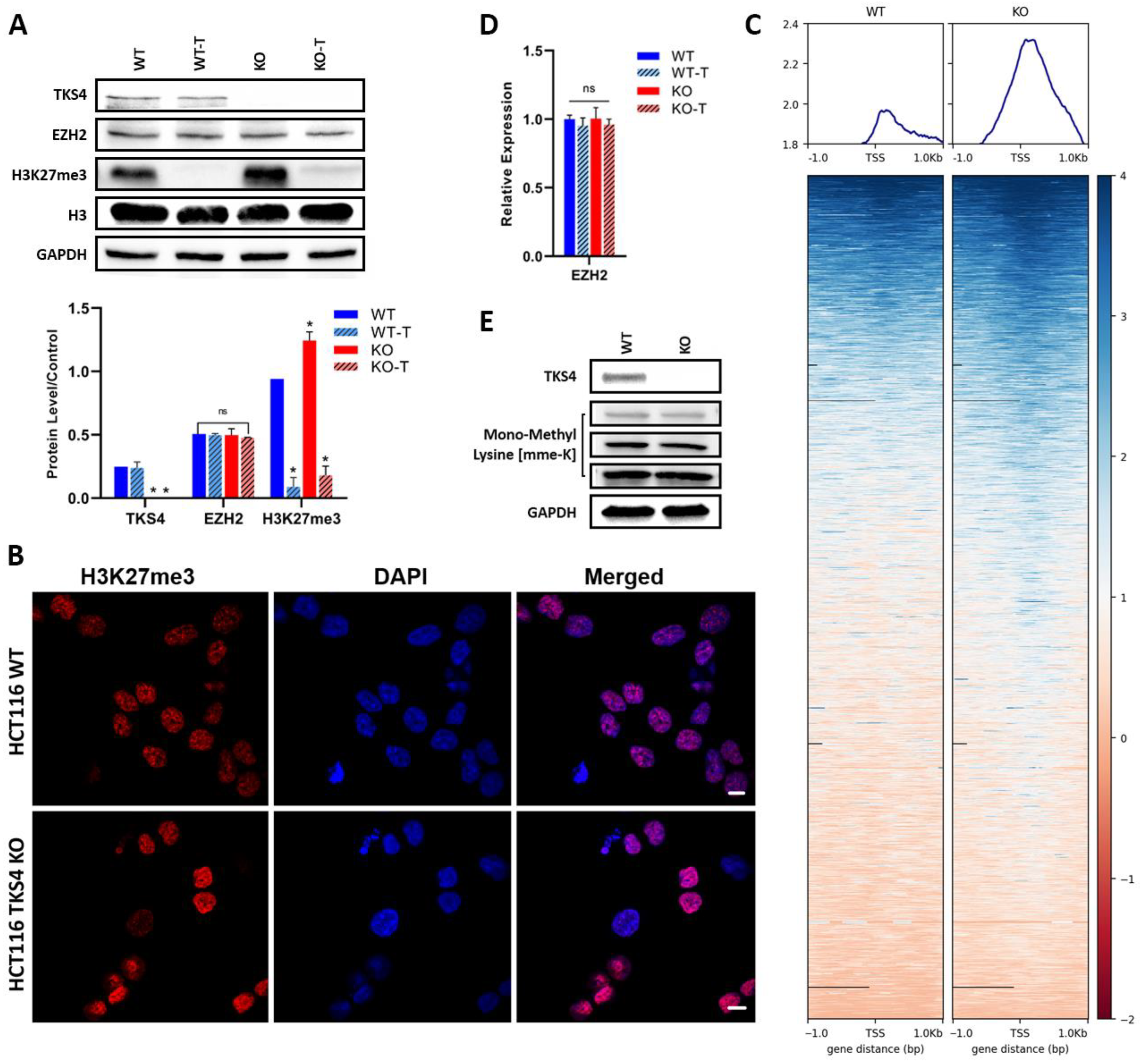
3.1. Elevation of H3K27me3 in TKS4 KO Cells

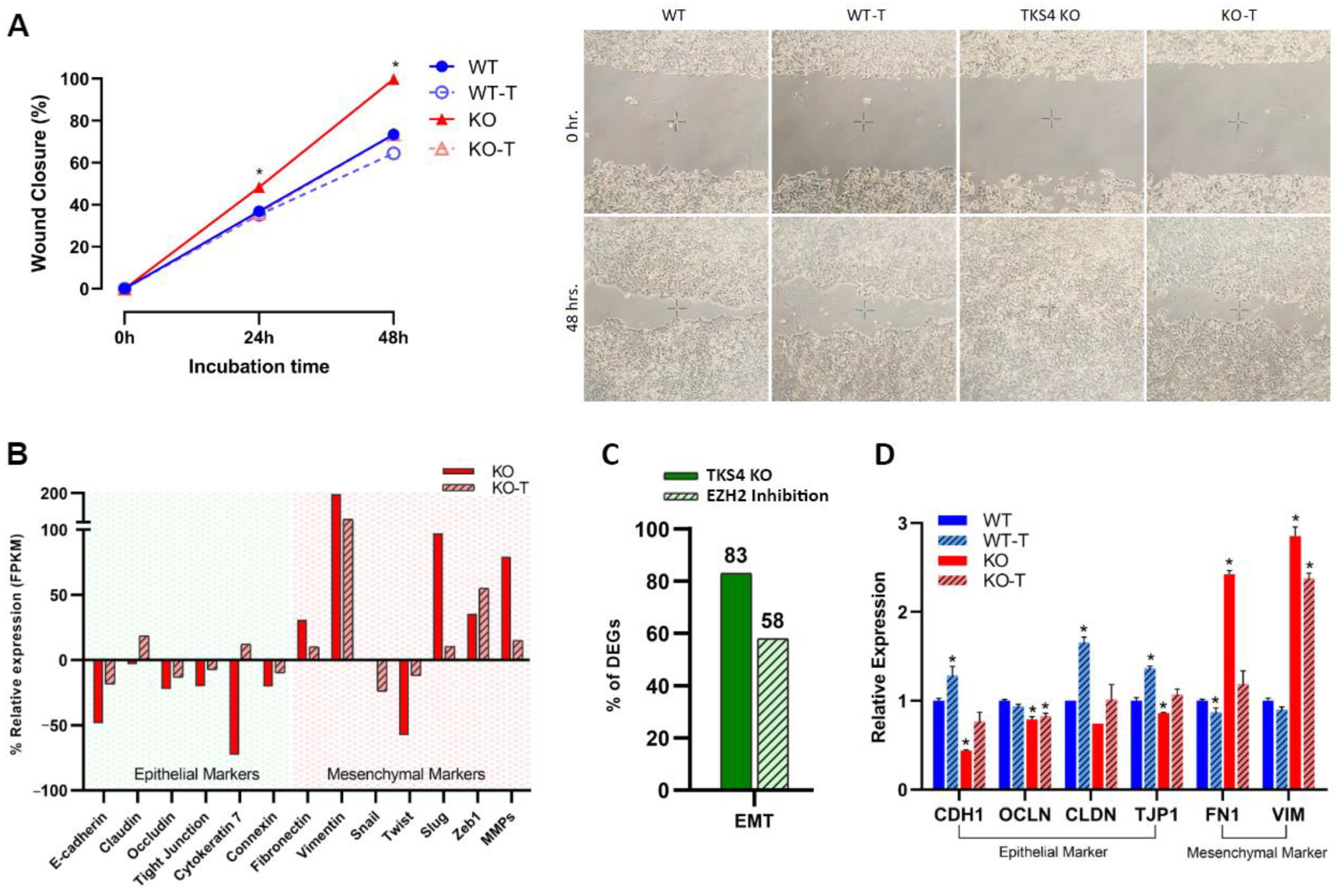
3.2. The Absence of TKS4 Induces Migration and EMT-Like Changes through EZH2 Hyperactivity
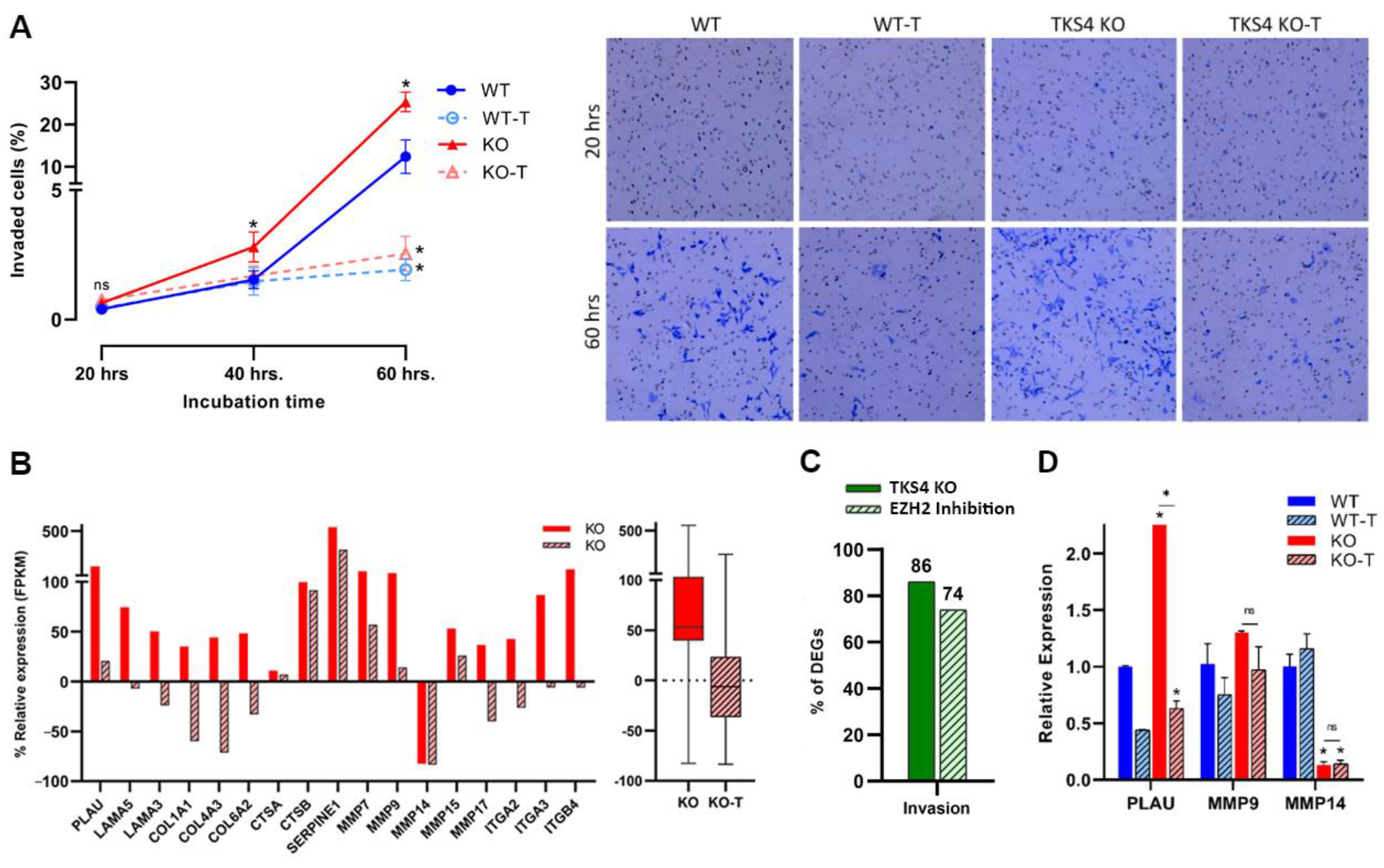
3.3. Absence of TKS4 Induces Invasion through EZH2 Hyperactivity
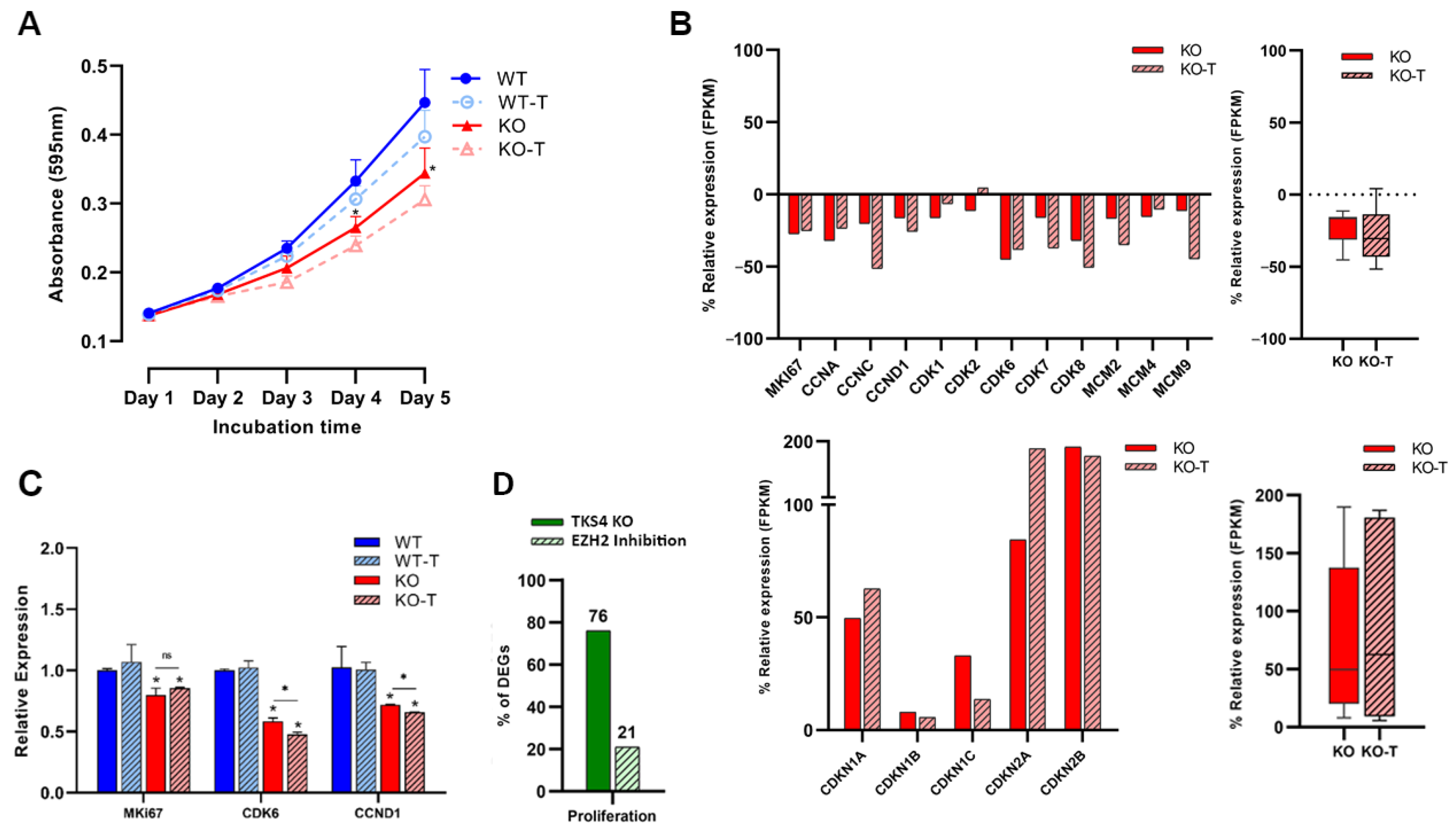
3.4. EZH2 Inhibition Decreases Proliferation Rate
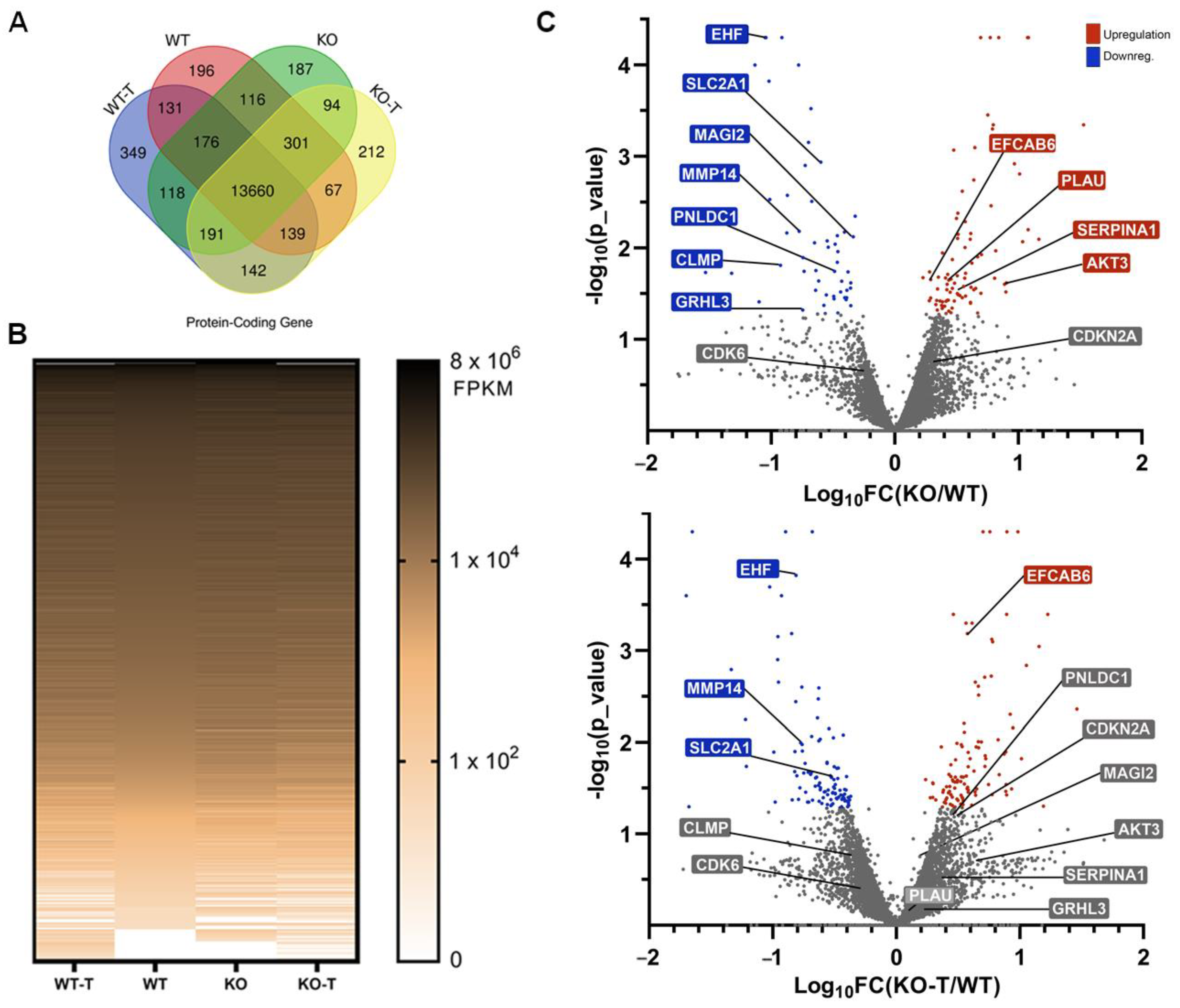
3.5. Effects of TKS4 and EZH2 Inhibition on Global Gene Expression
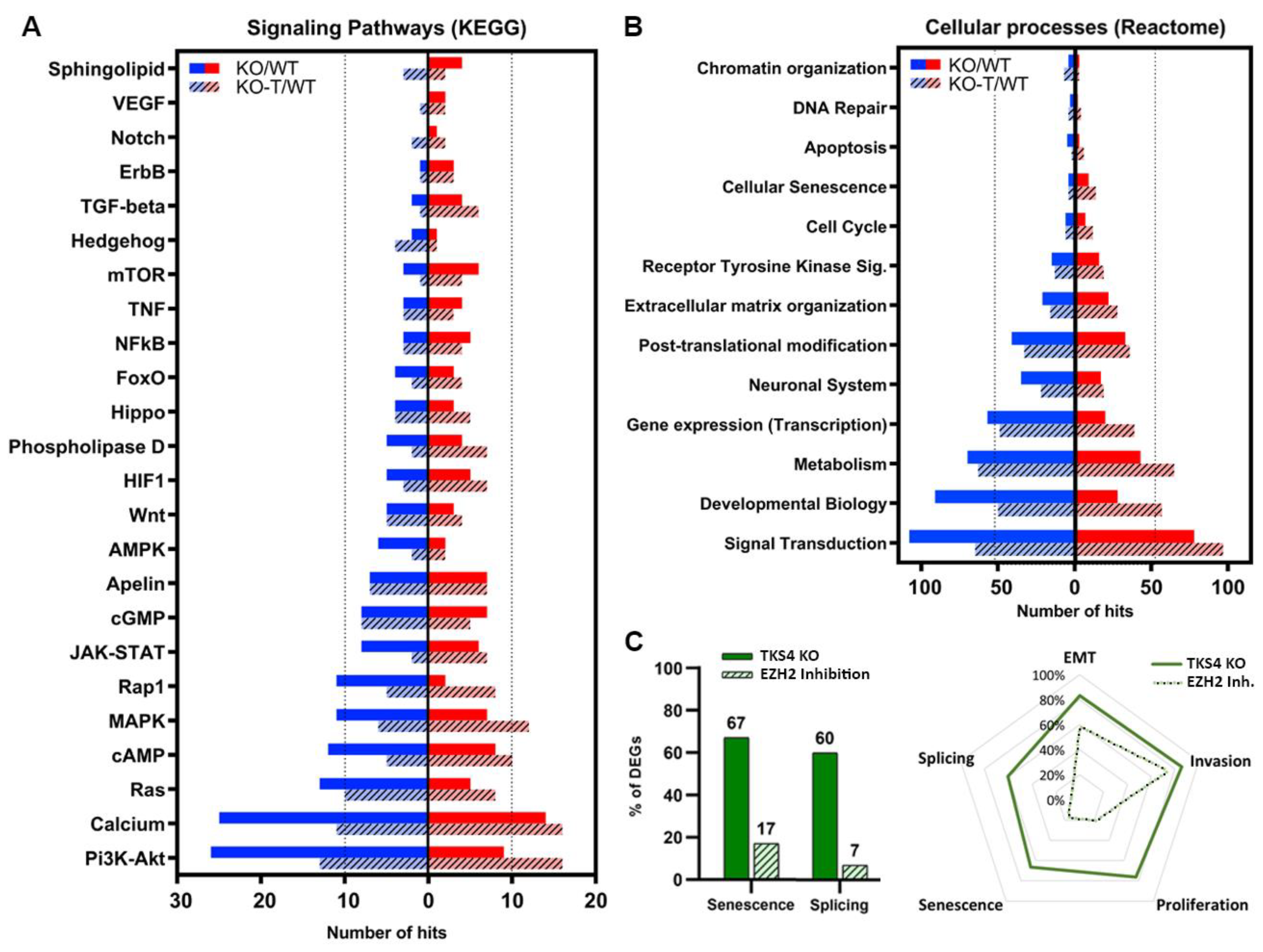
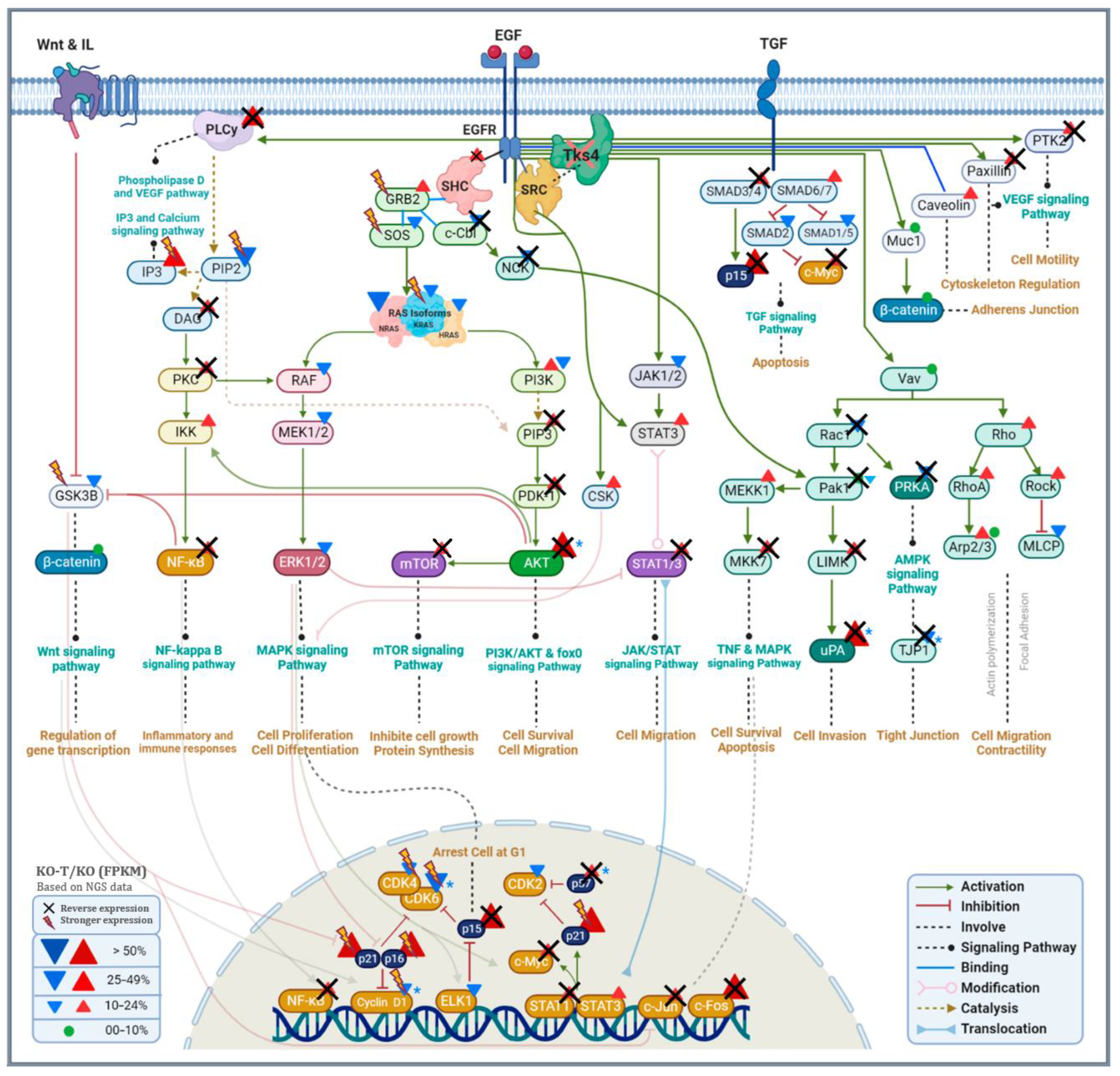
3.6. Alterations in Signaling Pathways
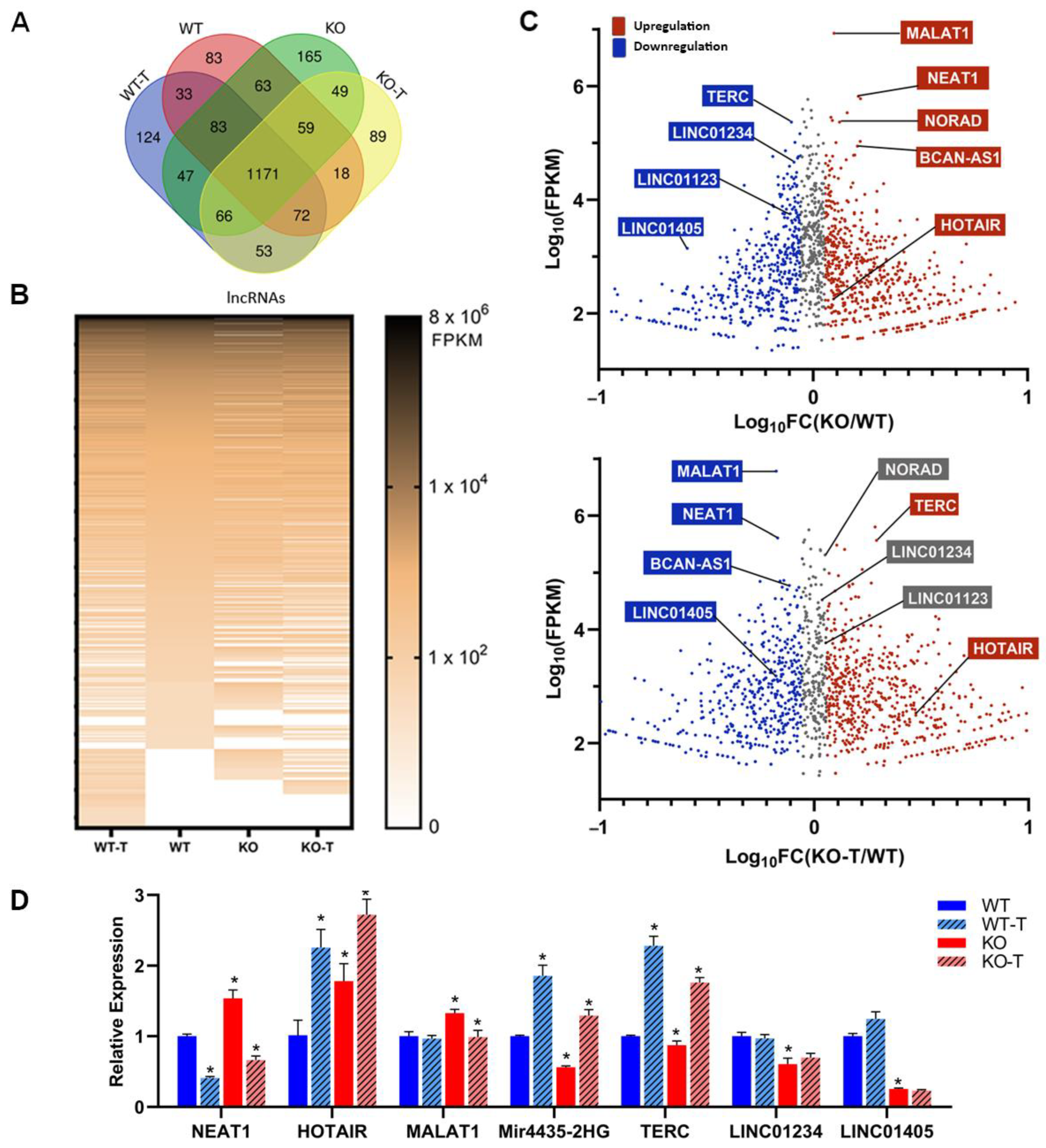
3.7. Changes in Long Non-Coding RNAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buday, L.; Tompa, P. Functional Classification of Scaffold Proteins and Related Molecules. FEBS J. 2010, 277, 4348–4355. [Google Scholar] [CrossRef] [PubMed]
- Kudlik, G.; Takács, T.; Radnai, L.; Kurilla, A.; Szeder, B.; Koprivanacz, K.; Merő, B.L.; Buday, L.; Vas, V. Advances in Understanding TKS4 and TKS5: Molecular Scaffolds Regulating Cellular Processes from Podosome and Invadopodium Formation to Differentiation and Tissue Homeostasis. Int. J. Mol. Sci. 2020, 21, 8117. [Google Scholar] [CrossRef] [PubMed]
- Courtneidge, S.A. Cell Migration and Incion in Human Disease: The Tks Adaptor Proteins. Biochem. Soc. Trans. 2012, 40, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.M.; Kayserili, H.; Lam, J.; Apak, M.Y.; Hennekam, R.C.M. Further Delineation of Frank-Ter Haar Syndrome. Am. J. Med. Genet. A 2004, 131, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Bögel, G.; Gujdár, A.; Geiszt, M.; Lányi, Á.; Fekete, A.; Sipeki, S.; Downward, J.; Buday, L. Frank-Ter Haar Syndrome Protein Tks4 Regulates Epidermal Growth Factor-Dependent Cell Migration. J. Biol. Chem. 2012, 287, 31321–31329. [Google Scholar] [CrossRef] [PubMed]
- Durand, B.; Stoetzel, C.; Schaefer, E.; Calmels, N.; Scheidecker, S.; Kempf, N.; De Melo, C.; Guilbert, A.-S.; Timbolschi, D.; Donato, L.; et al. A Severe Case of Frank-Ter Haar Syndrome and Literature Review: Further Delineation of the Phenotypical Spectrum. Eur. J. Med. Genet. 2020, 63, 103857. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, Z.; Cejudo-Martin, P.; de Brouwer, A.; van der Zwaag, B.; Ruiz-Lozano, P.; Scimia, M.C.; Lindsey, J.D.; Weinreb, R.; Albrecht, B.; Megarbane, A.; et al. Disruption of the Podosome Adaptor Protein TKS4 (SH3PXD2B) Causes the Skeletal Dysplasia, Eye, and Cardiac Abnormalities of Frank-Ter Haar Syndrome. Am. J. Hum. Genet. 2010, 86, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Mao, M.; Thedens, D.R.; Chang, B.; Harris, B.S.; Zheng, Q.Y.; Johnson, K.R.; Donahue, L.R.; Anderson, M.G. The Podosomal-Adaptor Protein SH3PXD2B Is Essential for Normal Postnatal Development. Mamm. Genome 2009, 20, 462–475. [Google Scholar] [CrossRef] [PubMed]
- Szeder, B.; Tárnoki-Zách, J.; Lakatos, D.; Vas, V.; Kudlik, G.; Merő, B.; Koprivanacz, K.; Bányai, L.; Hámori, L.; Róna, G.; et al. Absence of the Tks4 Scaffold Protein Induces Epithelial-Mesenchymal Transition-Like Changes in Human Colon Cancer Cells. Cells 2019, 8, 1343. [Google Scholar] [CrossRef] [PubMed]
- Kurilla, A.; László, L.; Takács, T.; Tilajka, Á.; Lukács, L.; Novák, J.; Pancsa, R.; Buday, L.; Vas, V. Studying the Association of TKS4 and CD2AP Scaffold Proteins and Their Implications in the Partial Epithelial-Mesenchymal Transition (EMT) Process. Int. J. Mol. Sci. 2023, 24, 15136. [Google Scholar] [CrossRef] [PubMed]
- László, L.; Maczelka, H.; Takács, T.; Kurilla, A.; Tilajka, Á.; Buday, L.; Vas, V.; Apáti, Á. A Novel Cell-Based Model for a Rare Disease: The Tks4-KO Human Embryonic Stem Cell Line as a Frank-Ter Haar Syndrome Model System. Int. J. Mol. Sci. 2022, 23, 8803. [Google Scholar] [CrossRef] [PubMed]
- Dülk, M.; Szeder, B.; Glatz, G.; Merő, B.L.; Koprivanacz, K.; Kudlik, G.; Vas, V.; Sipeki, S.; Cserkaszky, A.; Radnai, L.; et al. EGF Regulates the Interaction of Tks4 with Src through Its SH2 and SH3 Domains. Biochemistry 2018, 57, 4186–4196. [Google Scholar] [CrossRef] [PubMed]
- Müller, J. Transcriptional Silencing by the Polycomb Protein in Drosophila Embryos. EMBO J. 1995, 14, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb Complex PRC2 and Its Mark in Life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Lavelle, C.; Victor, J.-M. Nuclear Architecture and Dynamics; Academic Press: Cambridge, MA, USA, 2017; ISBN 9780128035030. [Google Scholar]
- Schoenfelder, S.; Sugar, R.; Dimond, A.; Javierre, B.-M.; Armstrong, H.; Mifsud, B.; Dimitrova, E.; Matheson, L.; Tavares-Cadete, F.; Furlan-Magaril, M.; et al. Polycomb Repressive Complex PRC1 Spatially Constrains the Mouse Embryonic Stem Cell Genome. Nat. Genet. 2015, 47, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Ji, F.; Sunwoo, H.; Jain, G.; Lee, J.T.; Sadreyev, R.I.; Dekker, J.; Kingston, R.E. Polycomb Repressive Complex 1 Generates Discrete Compacted Domains That Change during Differentiation. Mol. Cell 2017, 65, 432–446.e5. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Cavalli, G. Recruitment of Polycomb Group Complexes and Their Role in the Dynamic Regulation of Cell Fate Choice. Development 2009, 136, 3531–3542. [Google Scholar] [CrossRef] [PubMed]
- Mazziotta, C.; Lanzillotti, C.; Gafà, R.; Touzé, A.; Durand, M.-A.; Martini, F.; Rotondo, J.C. The Role of Histone Post-Translational Modifications in Merkel Cell Carcinoma. Front. Oncol. 2022, 12, 832047. [Google Scholar] [CrossRef] [PubMed]
- Kanaoka, S.; Okabe, A.; Kanesaka, M.; Rahmutulla, B.; Fukuyo, M.; Seki, M.; Hoshii, T.; Sato, H.; Imamura, Y.; Sakamoto, S.; et al. Chromatin Activation with H3K36me2 and Compartment Shift in Metastatic Castration-Resistant Prostate Cancer. Cancer Lett. 2024, 588, 216815. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Li, G.; Son, J.; Xu, C.-F.; Margueron, R.; Neubert, T.A.; Reinberg, D. Phosphorylation of the PRC2 Component Ezh2 Is Cell Cycle-Regulated and up-Regulates Its Binding to ncRNA. Genes Dev. 2010, 24, 2615–2620. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Kitabayashi, I. Oncogenic Roles of Enhancer of Zeste Homolog 1/2 in Hematological Malignancies. Cancer Sci. 2018, 109, 2342–2348. [Google Scholar] [CrossRef] [PubMed]
- Hosogane, M.; Funayama, R.; Shirota, M.; Nakayama, K. Lack of Transcription Triggers H3K27me3 Accumulation in the Gene Body. Cell Rep. 2016, 16, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, X.; Kim, M.; He, D.; Wang, C.; Fong, K.W.; Liu, X. Downregulation of EZH2 Inhibits Epithelial-Mesenchymal Transition in Enzalutamide-Resistant Prostate Cancer. Prostate 2023, 83, 1458–1469. [Google Scholar] [CrossRef] [PubMed]
- Battistelli, C.; Cicchini, C.; Santangelo, L.; Tramontano, A.; Grassi, L.; Gonzalez, F.J.; de Nonno, V.; Grassi, G.; Amicone, L.; Tripodi, M. The Snail Repressor Recruits EZH2 to Specific Genomic Sites through the Enrollment of the lncRNA HOTAIR in Epithelial-to-Mesenchymal Transition. Oncogene 2016, 36, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, H.; Hu, Y.; Ma, X.; Li, J.; Shi, Y.; Tao, M.; Wang, Y.; Zhong, Q.; Yan, D.; et al. Enhancer of Zeste Homolog 2 Promotes Renal Fibrosis after Acute Kidney Injury by Inducing Epithelial-Mesenchymal Transition and Activation of M2 Macrophage Polarization. Cell Death Dis. 2023, 14, 253. [Google Scholar] [CrossRef] [PubMed]
- Ghobashi, A.H.; Vuong, T.T.; Kimani, J.W.; Ladaika, C.A.; Hollenhorst, P.C.; O’Hagan, H.M. Activation of AKT Induces EZH2-Mediated β-Catenin Trimethylation in Colorectal Cancer. iScience 2023, 26, 107630. [Google Scholar] [CrossRef] [PubMed]
- Ardalan Khales, S.; Forghanifard, M.M.; Abbaszadegan, M.R.; Hosseini, S.E. EZH2 Deregulates BMP, Hedgehog, and Hippo Cell Signaling Pathways in Esophageal Squamous Cell Carcinoma. Adv. Med. Sci. 2023, 68, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Sreeshma, B.; Devi, A. JARID2 and EZH2, the Eminent Epigenetic Drivers in Human Cancer. Gene 2023, 879, 147584. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fan, H.; Liang, X.; Chen, Y. Polycomb Repressor Complex: Its Function in Human Cancer and Therapeutic Target Strategy. Biomed. Pharmacother. 2023, 169, 115897. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Q. The Roles of EZH2 in Cancer and Its Inhibitors. Med. Oncol. 2023, 40, 167. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xu, L.; Yang, L. Recent Advances in EZH2-Based Dual Inhibitors in the Treatment of Cancers. Eur. J. Med. Chem. 2023, 256, 115461. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, M.; Nazdari, N.; Gholamiyan, G.; Paskeh, M.D.A.; Jafari, A.M.; Nemati, F.; Khodaei, E.; Abyari, G.; Behdadfar, N.; Raei, B.; et al. EZH2 as a Potential Therapeutic Target for Gastrointestinal Cancers. Pathol. Res. Pract. 2024, 253, 154988. [Google Scholar] [CrossRef] [PubMed]
- Jacksi, M.; Schad, E.; Buday, L.; Tantos, A. Absence of Scaffold Protein Tks4 Disrupts Several Signaling Pathways in Colon Cancer Cells. Int. J. Mol. Sci. 2023, 24, 1310. [Google Scholar] [CrossRef]
- Boyd, D.D.; Levine, A.E.; Brattain, D.E.; McKnight, M.K.; Brattain, M.G. Comparison of Growth Requirements of Two Human Intratumoral Colon Carcinoma Cell Lines in Monolayer and Soft Agarose1. Cancer Res. 1988, 48, 2469–2474. [Google Scholar] [PubMed]
- Cadena-Herrera, D.; Esparza-De Lara, J.E.; Ramírez-Ibañez, N.D.; López-Morales, C.A.; Pérez, N.O.; Flores-Ortiz, L.F.; Medina-Rivero, E. Validation of Three Viable-Cell Counting Methods: Manual, Semi-Automated, and Automated. Biotechnol. Rep. 2015, 7, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Girard, N.; Bazille, C.; Lhuissier, E.; Benateau, H.; Llombart-Bosch, A.; Boumediene, K.; Bauge, C. 3-Deazaneplanocin A (DZNep), an Inhibitor of the Histone Methyltransferase EZH2, Induces Apoptosis and Reduces Cell Migration in Chondrosarcoma Cells. PLoS ONE 2014, 9, e98176. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Zhou, X.; Li, R.; Michal, J.J.; Zhang, S.; Dodson, M.V.; Zhang, Z.; Harland, R.M. Whole Transcriptome Analysis with Sequencing: Methods, Challenges and Potential Solutions. Cell. Mol. Life Sci. 2015, 72, 3425–3439. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential Gene and Transcript Expression Analysis of RNA-Seq Experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.G.C.T. cummeRbund; Bioconductor, 2017. Version 3.18, Bioconductor. [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef] [PubMed]
- Pijuan, J.; Barceló, C.; Moreno, D.F.; Maiques, O.; Sisó, P.; Marti, R.M.; Macià, A.; Panosa, A. In vitro Cell Migration, Invasion, and Adhesion Assays: From Cell Imaging to Data Analysis. Front. Cell Dev. Biol. 2019, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Katona, B.W.; Liu, Y.; Ma, A.; Jin, J.; Hua, X. EZH2 Inhibition Enhances the Efficacy of an EGFR Inhibitor in Suppressing Colon Cancer Cells. Cancer Biol. Ther. 2014, 15, 1677–1687. [Google Scholar] [CrossRef] [PubMed]
- Gall, T.M.H.; Frampton, A.E. Gene of the Month: E-Cadherin (CDH1). J. Clin. Pathol. 2013, 66, 928–932. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, C.E.; Mitchell, L.A.; Koval, M. Roles for Claudins in Alveolar Epithelial Barrier Function. Ann. N. Y. Acad. Sci. 2012, 1257, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Conceição, A.L.G.; Da Silva, C.T.; Badial, R.M.; Valsechi, M.C.; Stuqui, B.; Gonçalves, J.D.; Jasiulionis, M.G.; De Freitas Calmon, M.; Rahal, P. Downregulation of OCLN and GAS1 in Clear Cell Renal Cell Carcinoma. Oncol. Rep. 2017, 37, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Riz, I.; Hawley, R.G. Increased Expression of the Tight Junction Protein TJP1/ZO-1 Is Associated with Upregulation of TAZ-TEAD Activity and an Adult Tissue Stem Cell Signature in Carfilzomib-Resistant Multiple Myeloma Cells and High-Risk Multiple Myeloma Patients. Oncoscience 2017, 4, 79. [Google Scholar] [CrossRef] [PubMed]
- Franke, W.W.; Schiller, D.L.; Moll, R.; Winter, S.; Schmid, E.; Engelbrecht, I.; Denk, H.; Krepler, R.; Platzer, B. Diversity of Cytokeratins: Differentiation Specific Expression of Cytokeratin Polypeptides in Epithelial Cells and Tissues. J. Mol. Biol. 1981, 153, 933–959. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, N.; Zhu, J.; Hong, X.-T.; Liu, H.; Ou, Y.-R.; Su, F.; Wang, R.; Li, Y.-M.; Wu, Q. Downregulated connexin32 Promotes EMT through the Wnt/β-Catenin Pathway by Targeting Snail Expression in Hepatocellular Carcinoma. Int. J. Oncol. 2017, 50, 1977–1988. [Google Scholar] [CrossRef] [PubMed]
- Defamie, N.; Chepied, A.; Mesnil, M. Connexins, Gap Junctions and Tissue Invasion. FEBS Lett. 2014, 588, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Sudo, T.; Iwaya, T.; Nishida, N.; Sawada, G.; Takahashi, Y.; Ishibashi, M.; Shibata, K.; Fujita, H.; Shirouzu, K.; Mori, M.; et al. Expression of Mesenchymal Markers Vimentin and Fibronectin: The Clinical Significance in Esophageal Squamous Cell Carcinoma. Ann. Surg. Oncol. 2012, 20, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Leader, M.; Collins, M.; Patel, J.; Henry, K. Vimentin: An Evaluation of Its Role as a Tumour Marker. Histopathology 1987, 11, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Satake, S.; Nakayama, F.; Matsumoto, M.; Ohnuma, K.; Komori, T.; Semba, S.; Ito, A.; Yokozaki, H. Snail-Associated Epithelial–mesenchymal Transition Promotes Oesophageal Squamous Cell Carcinoma Motility and Progression. J. Pathol. 2008, 215, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Steinbichler, T.B.; Dudas, J.; Ingruber, J.; Glueckert, R.; Sprung, S.; Fleischer, F.; Cidlinsky, N.; Dejaco, D.; Kofler, B.; Giotakis, A.I.; et al. Slug Is a Surrogate Marker of Epithelial to Mesenchymal Transition (EMT) in Head and Neck Cancer. J. Clin. Med. Res. 2020, 9, 2061. [Google Scholar] [CrossRef] [PubMed]
- Pozharskaya, V.; Torres-González, E.; Rojas, M.; Gal, A.; Amin, M.; Dollard, S.; Roman, J.; Stecenko, A.A.; Mora, A.L. Twist: A Regulator of Epithelial-Mesenchymal Transition in Lung Fibrosis. PLoS ONE 2009, 4, e7559. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.E.; Nathan, V.; Osborne, J.K.; Farrow, R.K.; Deb, D.; Sullivan, J.P.; Dospoy, P.D.; Augustyn, A.; Hight, S.K.; Sato, M.; et al. ZEB1 Drives Epithelial-to-Mesenchymal Transition in Lung Cancer. J. Clin. Investig. 2016, 126, 3219–3235. [Google Scholar] [CrossRef] [PubMed]
- Vilorio-Marqués, L.; Martín, V.; Diez-Tascón, C.; González-Sevilla, M.F.; Fernández-Villa, T.; Honrado, E.; Davila-Batista, V.; Molina, A.J. The Role of EZH2 in Overall Survival of Colorectal Cancer: A Meta-Analysis. Sci. Rep. 2017, 7, 13806. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Mihalcioiu, C.; Rabbani, S.A. Multifaceted Role of the Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front. Oncol. 2018, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K. Laminin-5 (laminin-332): Unique Biological Activity and Role in Tumor Growth and Invasion. Cancer Sci. 2006, 97, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Caley, M.P.; Martins, V.L.; Moore, K.; Lashari, M.; Nissinen, L.; Kähäri, V.-M.; Alexander, S.; Jones, E.; Harwood, C.A.; Jones, J.; et al. Loss of the Laminin Subunit Alpha-3 Induces Cell Invasion and Macrophage Infiltration in Cutaneous Squamous Cell Carcinoma. Br. J. Dermatol. 2021, 184, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Patarroyo, M.; Tryggvason, K.; Virtanen, I. Laminin Isoforms in Tumor Invasion, Angiogenesis and Metastasis. Semin. Cancer Biol. 2002, 12, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Wang, Y.; Wu, Y.; Gao, Y.; Li, Q.; Abdulrahman, A.A.; Liu, X.-F.; Ji, G.-Q.; Gao, J.; Li, L.; et al. Identification of COL1A1 as an Invasion-related Gene in Malignant Astrocytoma. Int. J. Oncol. 2018, 53, 2542–2554. [Google Scholar] [CrossRef] [PubMed]
- Pasco, S.; Brassart, B.; Ramont, L.; Maquart, F.-X.; Monboisse, J.-C. Control of Melanoma Cell Invasion by Type IV Collagen. Cancer Detect. Prev. 2005, 29, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Sun, J.; Li, C.; Zhang, K. COL4A1 Promotes the Proliferation and Migration of Oral Squamous Cell Carcinoma Cells by Binding to NID1. Exp. Ther. Med. 2023, 25, 176. [Google Scholar] [CrossRef] [PubMed]
- Owusu-Ansah, K.G.; Song, G.; Chen, R.; Edoo, M.I.A.; Li, J.; Chen, B.; Wu, J.; Zhou, L.; Xie, H.; Jiang, D.; et al. COL6A1 Promotes Metastasis and Predicts Poor Prognosis in Patients with Pancreatic Cancer. Int. J. Oncol. 2019, 55, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Weng, W.; Xu, M.; Wang, Q.; Tan, C.; Sun, H.; Wang, L.; Huang, D.; Du, X.; Sheng, W. miR-106b-5p Inhibits the Invasion and Metastasis of Colorectal Cancer by Targeting CTSA. Onco. Targets. Ther. 2018, 11, 3835–3845. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kwon, W.; Park, J.-K.; Baek, S.-M.; Lee, S.-W.; Cho, G.-J.; Ha, Y.-S.; Lee, J.N.; Kwon, T.G.; Kim, M.O.; et al. Suppression of Cathepsin a Inhibits Growth, Migration, and Invasion by Inhibiting the p38 MAPK Signaling Pathway in Prostate Cancer. Arch. Biochem. Biophys. 2020, 688, 108407. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Zheng, H.; Rong, X.; Rong, X.; Zhang, J.; Fang, W.; Zhao, P.; Luo, R. Over-Expression of Cathepsin B in Hepatocellular Carcinomas Predicts Poor Prognosis of HCC Patients. Mol. Cancer 2016, 15, 17. [Google Scholar] [CrossRef]
- Harbeck, N.; Thomssen, C.; Berger, U.; Ulm, K.; Kates, R.E.; Höfler, H.; Jänicke, F.; Graeff, H.; Schmitt, M. Invasion Marker PAI-1 Remains a Strong Prognostic Factor after Long-term Follow-up Both for Primary Breast Cancer and Following First Relapse. Breast Cancer Res. Treat. 1999, 54, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Westermarck, J.; Kähäri, V.M. Regulation of Matrix Metalloproteinase Expression in Tumor Invasion. FASEB J. 1999, 13, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, T.; Okada, Y. MT1-MMP and MMP-7 in Invasion and Metastasis of Human Cancers. Cancer Metastasis Rev. 2003, 22, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, H.; Urvalek, A.M.; Li, T.; Yu, L.; Lamar, J.; DiPersio, C.M.; Feustel, P.J.; Zhao, J. KLF8 Promotes Human Breast Cancer Cell Invasion and Metastasis by Transcriptional Activation of MMP9. Oncogene 2011, 30, 1901–1911. [Google Scholar] [CrossRef] [PubMed]
- Ala-Aho, R.; Johansson, N.; Baker, A.H.; Kähäri, V.-M. Expression of Collagenase-3 (MMP-13) Enhances Invasion of Human Fibrosarcoma HT-1080 Cells. Int. J. Cancer 2002, 97, 283–289. [Google Scholar] [CrossRef]
- Yan, T.; Lin, Z.; Jiang, J.; Lu, S.; Chen, M.; Que, H.; He, X.; Que, G.; Mao, J.; Xiao, J.; et al. MMP14 Regulates Cell Migration and Invasion through Epithelial-Mesenchymal Transition in Nasopharyngeal Carcinoma. Am. J. Transl. Res. 2015, 7, 950–958. [Google Scholar] [PubMed]
- Huang, W.; Zhu, J.; Shi, H.; Wu, Q.; Zhang, C. ITGA2 Overexpression Promotes Esophageal Squamous Cell Carcinoma Aggression via FAK/AKT Signaling Pathway. Onco. Targets. Ther. 2021, 14, 3583–3596. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Chen, M.; He, Q.; Yan, Q.; Zhai, C. MicroRNA-199a-5p Suppresses Cell Proliferation, Migration and Invasion by Targeting ITGA3 in Colorectal Cancer. Mol. Med. Rep. 2020, 22, 2307–2317. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-L.; Liu, L.; Li, D.-D.; He, Y.-P.; Guo, L.-H.; Sun, L.-P.; Liu, L.-N.; Xu, H.-X.; Zhang, X.-P. Integrin β4 Promotes Cell Invasion and Epithelial-Mesenchymal Transition through the Modulation of Slug Expression in Hepatocellular Carcinoma. Sci. Rep. 2017, 7, 40464. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Kaufman, P.D. Ki-67: More than a Proliferation Marker. Chromosoma 2018, 127, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Kao, T.P.; Huang, H. CDK1 Promotes Cell Proliferation and Survival via Phosphorylation and Inhibition of FOXO1 Transcription Factor. Oncogene 2008, 27, 4733–4744. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi-Taesch, N.M.; Salim, F.; Kleinberger, J.; Troxell, R.; Cozar-Castellano, I.; Selk, K.; Cherok, E.; Takane, K.K.; Scott, D.K.; Stewart, A.F. Induction of Human β-Cell Proliferation and Engraftment Using a Single G1/S Regulatory Molecule, cdk6. Diabetes 2010, 59, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, M.; Zhang, X.; Huang, H.; Huang, J.; Ke, J.; Ding, H.; Xiao, J.; Shan, X.; Liu, Q.; et al. Upregulation of CDK7 in Gastric Cancer Cell Promotes Tumor Cell Proliferation and Predicts Poor Prognosis. Exp. Mol. Pathol. 2016, 100, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Y.; Luo, Q.-F.; Wei, C.-K.; Li, D.-F.; Fang, L. siRNA-Mediated Silencing of CDK8 Inhibits Proliferation and Growth in Breast Cancer Cells. Int. J. Clin. Exp. Pathol. 2014, 7, 92. [Google Scholar] [PubMed]
- Porter, L.A.; Dellinger, R.W.; Tynan, J.A.; Barnes, E.A.; Kong, M.; Lenormand, J.-L.; Donoghue, D.J. Human Speedy a Novel Cell Cycle Regulator That Enhances Proliferation through Activation of Cdk2. J. Cell Biol. 2002, 157, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. MiR-219-5p Suppresses Cell Proliferation and Cell Cycle Progression in Esophageal Squamous Cell Carcinoma by Targeting CCNA2. Cell. Mol. Biol. Lett. 2019, 24, 4. [Google Scholar] [CrossRef] [PubMed]
- Lew, D.J.; Reed, S.I. A Proliferation of Cyclins. Trends Cell Biol. 1992, 2, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S. Cyclin D1 Serves as a Cell Cycle Regulatory Switch in Actively Proliferating Cells. Curr. Opin. Cell Biol. 2003, 15, 158–163. [Google Scholar]
- Dai, Y.; Grant, S. Cyclin-Dependent Kinase Inhibitors. Curr. Opin. Pharmacol. 2003, 3, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Manohar, S.M.; Joshi, K.S. Molecular Pharmacology of Multitarget Cyclin-Dependent Kinase Inhibitors in Human Colorectal Carcinoma Cells. Expert Opin. Ther. Targets 2023, 27, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Andrés-Sánchez, N.; Fisher, D.; Krasinska, L. Physiological Functions and Roles in Cancer of the Proliferation Marker Ki-67. J. Cell Sci. 2022, 135, 258932. [Google Scholar] [CrossRef] [PubMed]
- Passardi, A.; Gibbons, D. Molecular Targets for the Treatment of Metastatic Colorectal Cancer; Frontiers Media SA: Lausanne, Switzerland, 2024; ISBN 9782832542064. [Google Scholar]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating Cancer with Selective CDK4/6 Inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, S.; Ryan, J.J.; Kudlicka, K.; Iino, N.; Zhou, H.; Farquhar, M.G. Cell Junction-Associated Proteins IQGAP1, MAGI-2, CASK, Spectrins, and Alpha-Actinin Are Components of the Nephrin Multiprotein Complex. Proc. Natl. Acad. Sci. USA 2005, 102, 9814–9819. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Han, C.-J.; Zhang, J.-Z.; He, W.-Z.; Zhao, G.-J.; Cheng, X.; Zhang, L.; Deng, K.-Q.; Liu, Y.; Fan, H.-F.; et al. Hepatic Interferon Regulatory Factor 6 Alleviates Liver Steatosis and Metabolic Disorder by Transcriptionally Suppressing Peroxisome Proliferator-Activated Receptor γ in Mice. Hepatology 2019, 69, 2471–2488. [Google Scholar] [CrossRef] [PubMed]
- Raschperger, E. Studies on CAR and CLMP: Two Proteins of Epithelial Tight Junctions; Institutionen för Cell- och Molekylärbiologi (CMB)/Department of Cell and Molecular Biology: Solna, Sweden, 2006. [Google Scholar]
- Kimura-Yoshida, C.; Mochida, K.; Nakaya, M.-A.; Mizutani, T.; Matsuo, I. Cytoplasmic Localization of GRHL3 upon Epidermal Differentiation Triggers Cell Shape Change for Epithelial Morphogenesis. Nat. Commun. 2018, 9, 4059. [Google Scholar] [CrossRef] [PubMed]
- Cech, T.R.; Steitz, J.A. The Noncoding RNA Revolution-Trashing Old Rules to Forge New Ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.-L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A.; et al. Long Non-Coding RNAs: Definitions, Functions, Challenges and Recommendations. Nat. Rev. Mol. Cell Biol. 2023, 24, 430–447. [Google Scholar] [CrossRef] [PubMed]
- Snyder, M.; Iraola-Guzmán, S.; Saus, E.; Gabaldón, T. Discovery and Validation of Clinically Relevant Long Non-Coding RNAs in Colorectal Cancer. Cancers 2022, 14, 3866. [Google Scholar] [CrossRef] [PubMed]
- Aprile, M.; Costa, V.; Cimmino, A.; Calin, G.A. Emerging Role of Oncogenic Long Non-Coding RNA as Cancer Biomarkers. Int. J. Cancer 2022, 152, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long Non-Coding RNA HOTAIR Reprograms Chromatin State to Promote Cancer Metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.N.; Chai, Z.T.; Zhu, X.D.; Zhang, N.; Zhan, D.H.; Ye, B.G.; Wang, C.H.; Qin, C.D.; Zhao, Y.M.; Zhu, W.P.; et al. MicroRNA-26a Suppresses Epithelial-Mesenchymal Transition in Human Hepatocellular Carcinoma by Repressing Enhancer of Zeste Homolog 2. J. Hematol. Oncol. 2016, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Yu, J.; Dhanasekaran, S.M.; Kim, J.H.; Mani, R.S.; Tomlins, S.A.; Mehra, R.; Laxman, B.; Cao, X.; Yu, J.; et al. Repression of E-Cadherin by the Polycomb Group Protein EZH2 in Cancer. Oncogene 2008, 27, 7274–7284. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, X.; Hu, J.; He, G.; Li, X.; Wu, P.; Ren, X.; Wang, F.; Liao, W.; Liang, L.; et al. The Positive Feedback between Snail and DAB2IP Regulates EMT, Invasion and Metastasis in Colorectal Cancer. Oncotarget 2015, 6, 27427. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Guo, J.; Guo, J.; Sun, S.; Yang, P.; Wang, J.; Li, Y.; Xie, L.; Cai, J.; Wang, Z. EZH2-Mediated Epigenetic Silencing of TIMP2 Promotes Ovarian Cancer Migration and Invasion. Sci. Rep. 2017, 7, 3568. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.-C.; Liu, L.-C.; Ye, H.-Y.; Wu, J.-Y.; Yu, Y.-L. EZH2 Promotes Migration and Invasion of Triple-Negative Breast Cancer Cells via Regulating TIMP2-MMP-2/-9 Pathway. Am. J. Cancer Res. 2018, 8, 422. [Google Scholar] [PubMed]
- Rao, Z.-Y.; Cai, M.-Y.; Yang, G.-F.; He, L.-R.; Mai, S.-J.; Hua, W.-F.; Liao, Y.-J.; Deng, H.-X.; Chen, Y.-C.; Guan, X.-Y.; et al. EZH2 Supports Ovarian Carcinoma Cell Invasion And/or Metastasis via Regulation of TGF-β1 and Is a Predictor of Outcome in Ovarian Carcinoma Patients. Carcinogenesis 2010, 31, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Mitre, G.P.; Balbinot, K.M.; Ribeiro, A.L.R.; da Silva Kataoka, M.S.; de Melo Alves Júnior, S.; de Jesus Viana Pinheiro, J. Key Proteins of Invadopodia Are Overexpressed in Oral Squamous Cell Carcinoma Suggesting an Important Role of MT1-MMP in the Tumoral Progression. Diagn. Pathol. 2021, 16, 33. [Google Scholar] [CrossRef] [PubMed]
- Gimona, M.; Buccione, R.; Courtneidge, S.A.; Linder, S. Assembly and Biological Role of Podosomes and Invadopodia. Current Opin. Cell Biol. 2008, 20, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ming, T.; Tang, S.; Ren, S.; Yang, H.; Liu, M.; Tao, Q.; Xu, H. Wnt Signaling in Colorectal Cancer: Pathogenic Role and Therapeutic Target. Mol. Cancer 2022, 21, 144. [Google Scholar] [CrossRef] [PubMed]
- Wilkins-Port, C.E.; Ye, Q.; Mazurkiewicz, J.E.; Higgins, P.J. TGF-beta1 + EGF-Initiated Invasive Potential in Transformed Human Keratinocytes Is Coupled to a plasmin/MMP-10/MMP-1-Dependent Collagen Remodeling Axis: Role for PAI-1. Cancer Res. 2009, 69, 4081–4091. [Google Scholar] [CrossRef] [PubMed]
- Pavón, M.A.; Arroyo-Solera, I.; Téllez-Gabriel, M.; León, X.; Virós, D.; López, M.; Gallardo, A.; Céspedes, M.V.; Casanova, I.; López-Pousa, A.; et al. Enhanced Cell Migration and Apoptosis Resistance May Underlie the Association between High SERPINE1 Expression and Poor Outcome in Head and Neck Carcinoma Patients. Oncotarget 2015, 6, 29016–29033. [Google Scholar] [CrossRef] [PubMed]
- Buikhuisen, J.Y.; Gomez Barila, P.M.; Torang, A.; Dekker, D.; de Jong, J.H.; Cameron, K.; Vitale, S.; Stassi, G.; van Hooff, S.R.; Castro, M.A.A.; et al. AKT3 Expression in Mesenchymal Colorectal Cancer Cells Drives Growth and Is Associated with Epithelial-Mesenchymal Transition. Cancers 2021, 13, 801. [Google Scholar] [CrossRef]
- He, S.; Liu, Y.; Meng, L.; Sun, H.; Wang, Y.; Ji, Y.; Purushe, J.; Chen, P.; Li, C.; Madzo, J.; et al. Ezh2 Phosphorylation State Determines Its Capacity to Maintain CD8 T Memory Precursors for Antitumor Immunity. Nat. Commun. 2017, 8, 2125. [Google Scholar] [CrossRef] [PubMed]
- Yamada, L.; Saito, M.; Thar Min, A.K.; Saito, K.; Ashizawa, M.; Kase, K.; Nakajima, S.; Onozawa, H.; Okayama, H.; Endo, H.; et al. Selective Sensitivity of EZH2 Inhibitors Based on Synthetic Lethality in ARID1A-Deficient Gastric Cancer. Gastric Cancer 2021, 24, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Li, G.; Wang, W.; Sun, Y.; Zhang, Y.; Zhong, C.; Stovall, D.B.; Li, D.; Shi, J.; Sui, G. Disruption of YY1-EZH2 Interaction Using Synthetic Peptides Inhibits Breast Cancer Development. Cancers 2021, 13, 2402. [Google Scholar] [CrossRef] [PubMed]
- Kosalai, S.T.; Morsy, M.H.A.; Papakonstantinou, N.; Mansouri, L.; Stavroyianni, N.; Kanduri, C.; Stamatopoulos, K.; Rosenquist, R.; Kanduri, M. EZH2 Upregulates the PI3K/AKT Pathway through IGF1R and MYC in Clinically Aggressive Chronic Lymphocytic Leukaemia. Epigenetics 2019, 14, 1125–1140. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT Signaling Pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hao, A.; Li, X.; Du, Z.; Li, H.; Wang, H.; Yang, H.; Fang, Z. Melatonin Inhibits Tumorigenicity of Glioblastoma Stem-like Cells via the AKT-EZH2-STAT3 Signaling Axis. J. Pineal Res. 2016, 61, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Liu, J.; Chang, Q.; Beezhold, K.; Lu, Y.; Chen, F. JNK and STAT3 Signaling Pathways Converge on Akt-Mediated Phosphorylation of EZH2 in Bronchial Epithelial Cells Induced by Arsenic. Cell Cycle 2013, 12, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Biswas, R.; Bagchi, A. NFkB Pathway and Inhibition: An Overview. Comput. Mol. Biol. 2016, 6, 1–20. [Google Scholar] [CrossRef]
- Zinatizadeh, M.R.; Schock, B.; Chalbatani, G.M.; Zarandi, P.K.; Jalali, S.A.; Miri, S.R. The Nuclear Factor Kappa B (NF-kB) Signaling in Cancer Development and Immune Diseases. Genes Dis. 2021, 8, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Zaslavsky, A.; Fedele, G.; McLaughlin, S.K.; Reczek, E.E.; De Raedt, T.; Guney, I.; Strochlic, D.E.; MacConaill, L.E.; Beroukhim, R.; et al. An Oncogene–tumor Suppressor Cascade Drives Metastatic Prostate Cancer by Coordinately Activating Ras and Nuclear Factor-κB. Nat. Med. 2010, 16, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Waldner, M.J.; Neurath, M.F. Targeting the VEGF Signaling Pathway in Cancer Therapy. Expert Opin. Ther. Targets 2012, 16, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.Q.; Zhang, L.; Gao, B.S.; Wan, Y.G.; Zhang, X.H.; Chen, B.; Wang, Y.T.; Sun, N.; Fu, Y.W. EZH2 Promotes Tumor Progression by Increasing VEGF Expression in Clear Cell Renal Cell Carcinoma. Clin. Transl. Oncol. 2014, 17, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-T.; Zhu, F.; Lin, W.-R.; Ying, R.-B.; Yang, Y.-P.; Zeng, L.-H. The Novel EZH2 Inhibitor, GSK126, Suppresses Cell Migration and Angiogenesis via down-Regulating VEGF-A. Cancer Chemother. Pharmacol. 2016, 77, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Hu, X.; Xu, Y.; Wu, C.; Chen, J.; Ren, Y.; Kong, L.; Sun, S.; Zhang, L.; Jin, R.; et al. Targeting of EZH2 Inhibits Epithelial-mesenchymal Transition in Head and Neck Squamous Cell Carcinoma via Regulating the STAT3/VEGFR2 Axis. Int. J. Oncol. 2019, 55, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR Signaling Pathway and mTOR Inhibitors in Cancer: Progress and Challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Wang, M.; Zhang, G.; Bao, Y.; Wu, Y.; Li, X.; Yang, W.; Cui, H. E2F7-EZH2 Axis Regulates PTEN/AKT/mTOR Signalling and Glioblastoma Progression. Br. J. Cancer 2020, 123, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Tsou, P.; Sawalha, A.H. Increased Expression of EZH2 Is Mediated by Higher Glycolysis and mTORC1 Activation in Lupus CD4+ T Cells. Immunometabolism 2020, 2, e200013. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Shen, B.; Tan, M.; Mu, X.; Qin, Y.; Zhang, F.; Liu, Y. TGF-β–Induced Upregulation of malat1 Promotes Bladder Cancer Metastasis by Associating with suz12. Clin. Cancer Res. 2014, 20, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhu, C.; Jin, Y. The Oncogenic and Tumor Suppressive Functions of the Long Noncoding RNA MALAT1: An Emerging Controversy. Front. Genet. 2020, 11, 505991. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sehgal, L.; Jain, N.; Khashab, T.; Mathur, R.; Samaniego, F. LncRNA MALAT1 Promotes Development of Mantle Cell Lymphoma by Associating with EZH2. J. Transl. Med. 2016, 14, 346. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Sun, X.; Zhou, X.; Han, L.; Chen, L.; Shi, Z.; Zhang, A.; Ye, M.; Wang, Q.; Liu, C.; et al. Long Non-Coding RNA HOTAIR Promotes Glioblastoma Cell Cycle Progression in an EZH2 Dependent Manner. Oncotarget 2015, 6, 537. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, Z.; Mei, Q.; Guo, M.; Fu, X.; Han, W. Long Non-Coding RNA HOTAIR, a Driver of Malignancy, Predicts Negative Prognosis and Exhibits Oncogenic Activity in Oesophageal Squamous Cell Carcinoma. Br. J. Cancer 2013, 109, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, J.-Y.; Tian, F.-Z.; Zhao, G.; Hu, H.; Ma, Y.-F.; Yang, Y.-L. Long Noncoding RNA NEAT1 Promotes Growth and Metastasis of Cholangiocarcinoma Cells. Oncol. Res. 2018, 26, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Li, O.; Jiang, B.; Yi, W.-M.; Zhang, Y.; Yang, P.-Z.; Guo, C.; Sun, Z.-P.; Peng, C. LncRNA NEAT1 Promotes Cell Proliferation, Migration, and Invasion via the miR-186-5p/PTP4A1 Axis in Cholangiocarcinoma. Kaohsiung J. Med. Sci. 2021, 37, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Cai, J.; Wang, Q.; Wang, Y.; Liu, M.; Yang, J.; Zhou, J.; Kang, C.; Li, M.; Jiang, C. Long Noncoding RNA, Regulated by the EGFR Pathway, Contributes to Glioblastoma Progression Through the WNT/-Catenin Pathway by Scaffolding EZH2. Clin. Cancer Res. 2018, 24, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zuo, H.; Jin, J.; Lv, W.; Xu, Z.; Fan, Y.; Zhang, J.; Zuo, B. Long Noncoding RNA Neat1 Modulates Myogenesis by Recruiting Ezh2. Cell Death Dis. 2019, 10, 505. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Yin, C.; Li, Y.; Tian, D.; Xiang, Y.; Li, Q.; Tang, Y.; Zhang, Y. Long Noncoding RNA NEAT1 Promotes Cardiac Fibrosis in Heart Failure through Increased Recruitment of EZH2 to the Smad7 Promoter Region. J. Transl. Med. 2022, 20, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, L.; Zhang, S.; Ming, Y.; Liu, S.; Cheng, K.; Zhao, Y. Long Noncoding RNA NEAT1 Suppresses Hepatocyte Proliferation in Fulminant Hepatic Failure through Increased Recruitment of EZH2 to the LATS2 Promoter Region and Promotion of H3K27me3 Methylation. Exp. Mol. Med. 2020, 52, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.-F.; Li, W.; Liu, Y.-G.; Zhang, C.; Gao, W.-N.; Wang, L. Inhibition of MIR4435-2HG on Invasion, Migration, and EMT of Gastric Carcinoma Cells by Mediating MiR-138-5p/Sox4 Axis. Front. Oncol. 2021, 11, 661288. [Google Scholar] [CrossRef] [PubMed]
- Pei, L.; Yan, D.; He, Q.; Kong, J.; Yang, M.; Ruan, H.; Lin, Q.; Huang, L.; Huang, J.; Lin, T.; et al. LncRNA MIR4435-2HG Drives Cancer Progression by Modulating Cell Cycle Regulators and mTOR Signaling in Stroma-Enriched Subtypes of Urothelial Carcinoma of the Bladder. Cell. Oncol. 2023, 46, 1509–1527. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Yang, Z.; Yang, H.; Li, D.; Qiu, X. Long Non-Coding RNA MIR4435-2HG Promotes Colorectal Cancer Proliferation and Metastasis Through miR-206/YAP1 Axis. Front. Oncol. 2020, 10, 160. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacksi, M.; Schad, E.; Tantos, A. Morphological Changes Induced by TKS4 Deficiency Can Be Reversed by EZH2 Inhibition in Colorectal Carcinoma Cells. Biomolecules 2024, 14, 445. https://doi.org/10.3390/biom14040445
Jacksi M, Schad E, Tantos A. Morphological Changes Induced by TKS4 Deficiency Can Be Reversed by EZH2 Inhibition in Colorectal Carcinoma Cells. Biomolecules. 2024; 14(4):445. https://doi.org/10.3390/biom14040445
Chicago/Turabian StyleJacksi, Mevan, Eva Schad, and Agnes Tantos. 2024. "Morphological Changes Induced by TKS4 Deficiency Can Be Reversed by EZH2 Inhibition in Colorectal Carcinoma Cells" Biomolecules 14, no. 4: 445. https://doi.org/10.3390/biom14040445
APA StyleJacksi, M., Schad, E., & Tantos, A. (2024). Morphological Changes Induced by TKS4 Deficiency Can Be Reversed by EZH2 Inhibition in Colorectal Carcinoma Cells. Biomolecules, 14(4), 445. https://doi.org/10.3390/biom14040445