
Phenotypic Variability of Andersen–Tawil Syndrome Due to Allelic Mutation c.652C>T in the KCNJ2 Gene—A New Family Case Report
 ,
, 

Abstract
:1. Introduction
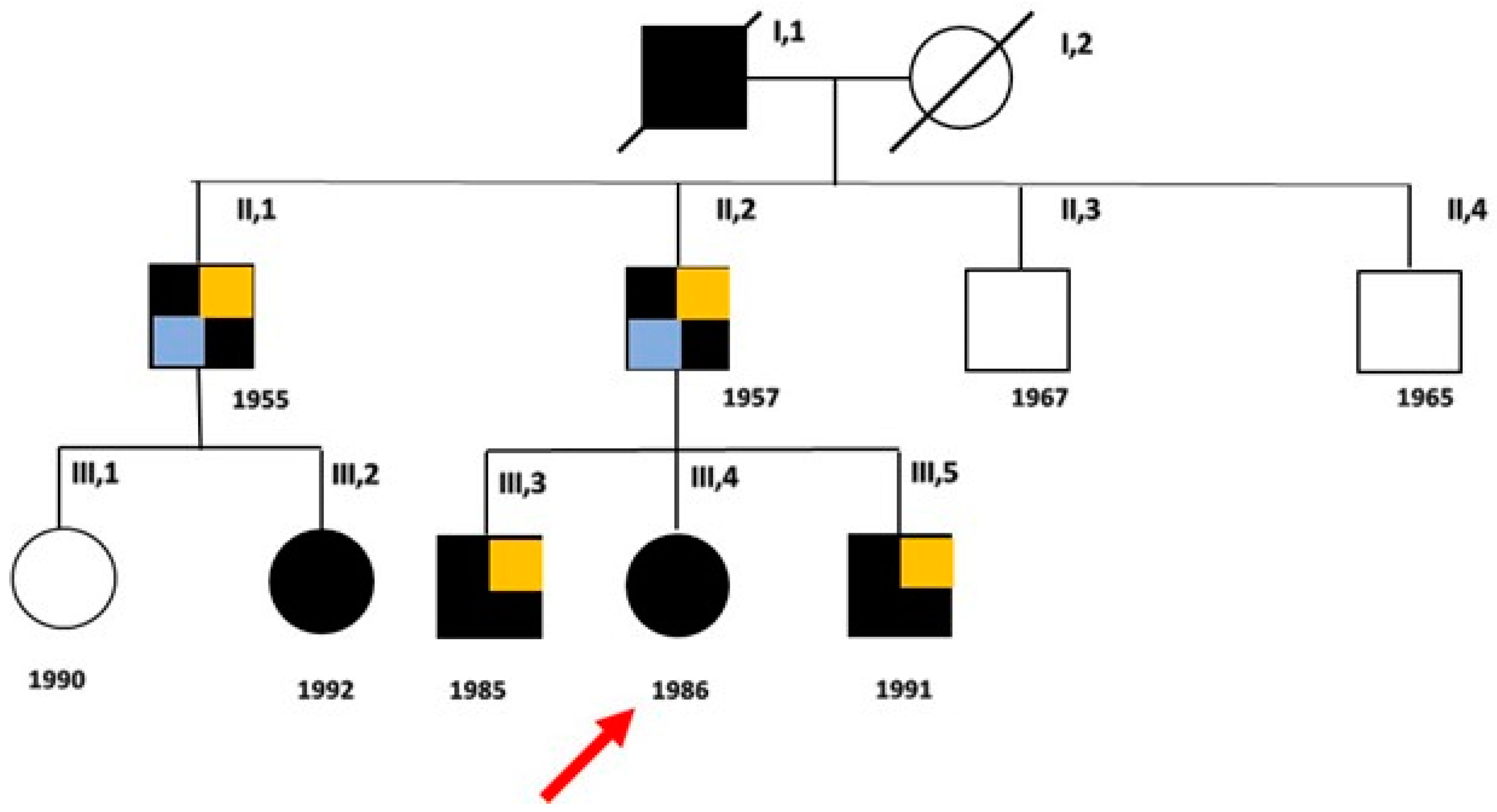
2. Detailed Family Case Description
2.1. Clinical Evaluation of Patients
2.2. Genetic Analysis
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matthews, E.; Holmes, S.; Fialho, D. Skeletal muscle channelopathies: A guide to diagnosis and management. Pract. Neurol. 2021, 21, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Morales, F.; Pusch, M. An Up-to-Date Overview of the Complexity of Genotype-Phenotype Relationships in Myotonic Chan-nelopathies. Front. Neurol. 2020, 10, 1404. [Google Scholar] [CrossRef] [PubMed]
- Dunø, M.; Vissing, J. Myotonia Congenita. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; 3 August 2005 [updated 25 February 2021]; University of Washington: Seattle, WA, USA, 1993–2024. [Google Scholar]
- Cannon, S.C. Sodium Channelopathies of Skeletal Muscle. Handb. Exp. Pharmacol. 2018, 246, 309–330. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Lehmann-Horn, F. Hypokalemic Periodic Paralysis. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; 30 April 2002 [updated 26 July 2018]; University of Washington: Seattle, WA, USA, 1993–2024. [Google Scholar]
- Weber, F. Hyperkalemic Periodic Paralysis. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; 18 July 2003 [updated 1 July 2021]; University of Washington: Seattle, WA, USA, 1993–2024. [Google Scholar]
- Andersen, E.D.; Krasilnikoff, P.A.; Overvad, H. Intermittent muscular weakness, extra-systoles, and multiple developmental anomalies. A new syndrome? Acta Paediatr. Scand. 1971, 60, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Tawil, R.; Ptacek, L.J.; Pavlakis, S.G.; DeVivo, D.C.; Penn, A.S.; Ozdemir, C.; Griggs, R.C. Andersen’s syndrome: Potassium-sensitive pe-riodic paralysis, ventricular ectopy, and dysmorphic features. Ann. Neurol. 1994, 35, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Plaster, N.M.; Tawil, R.; Tristani-Firouzi, M.; Canún, S.; Bendahhou, S.; Tsunoda, A.; Donaldson, M.R.; Iannaccone, S.T.; Brunt, E.; Barohn, R.; et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell 2001, 105, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Manuel, A.I.; Gutiérrez, L.K.; Vera-Pedrosa, M.L.; Cruz, F.M.; Bermúdez-Jiménez, F.J.; Martínez-Carrascoso, I.; Sánchez-Pérez, P.; Macías, Á.; Jalife, J. Molecular stratification of arrhythmogenic mechanisms in the Andersen Tawil syndrome. Cardiovasc. Res. 2023, 119, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Tristani-Firouzi, M.; Jensen, J.L.; Donaldson, M.R.; Sansone, V.; Meola, G.; Hahn, A.; Bendahhou, S.; Kwiecinski, H.; Fidzianska, A.; Plas-ter, N.; et al. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syn-drome). J. Clin. Investig. 2002, 110, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.L.; Pieper, G.H.; Wilders, R. Andersen-Tawil syndrome: Clinical and molecular aspects. Int. J. Cardiol. 2013, 170, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.P.; Imbrici, P.; Fialho, D.; Herd, C.; Bilsland, L.G.; Weber, A.; Mueller, R.; Hilton-Jones, D.; Ealing, J.; Boothman, B.R.; et al. Andersen-Tawil syndrome: New potassium channel mutations and possible phenotypic variation. Neurology 2005, 65, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Preisig-Müller, R.; Schlichthörl, G.; Goerge, T.; Heinen, S.; Brüggemann, A.; Rajan, S.; Derst, C.; Veh, R.W.; Daut, J. Heteromerization of Kir2.x potassium channels contributes to the phenotype of Andersen’s syndrome. Proc. Natl. Acad. Sci. USA 2002, 99, 7774–7779. [Google Scholar] [CrossRef] [PubMed]
- Reilly, L.; Eckhardt, L.L. Cardiac potassium inward rectifier Kir2: Review of structure, regulation, pharmacology, and ar-rhythmogenesis. Heart Rhythm. 2021, 18, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Hager, N.A.; McAtee, C.K.; Lesko, M.A.; O’Donnell, A.F. Inwardly Rectifying Potassium Channel Kir2.1 and its “Kirious” Regulation by Protein Trafficking and Roles in Development and Disease. Front. Cell Dev. Biol. 2022, 9, 796136. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.P.; da Silva, M.R.; Soong, T.W.; Fontaine, B.; Donaldson, M.R.; Kung, A.W.; Jongjaroenprasert, W.; Liang, M.C.; Khoo, D.H.; Cheah, J.S.; et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyro-toxic hypokalemic periodic paralysis. Cell 2010, 140, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Dassau, L.; Conti, L.R.; Radeke, C.M.; Ptáček, L.J.; Vandenberg, C.A. Kir2.6 regulates the sur-face expression of Kir2.x inward recti-fier potassium channels. J. Biol. Chem. 2011, 286, 9526–9541. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, M.R.; Jensen, J.L.; Tristani-Firouzi, M.; Tawil, R.; Bendahhou, S.; Suarez, W.A.; Cobo, A.M.; Poza, J.J.; Behr, E.; Wagstaff, J.; et al. PIP2 binding residues of Kir2.1 are common targets of muta-tions causing Andersen syndrome. Neurology 2003, 60, 1811–1816. [Google Scholar] [CrossRef]
- Pouget, J.; Philip, N.; Faugere, G.; Pellissier, J.F. Le syndrome d’Andersen: Une forme particulière de paralysie périodique avec dysrythmie cardiaque [Andersen syndrome: A particular form of paralysis with cardiac dysrhythmia]. Rev. Neurol. 2004, 160 Pt 2, S38–S42. [Google Scholar] [CrossRef] [PubMed]
- Schoonderwoerd, B.A.; Wiesfeld, A.C.; Wilde, A.A.; van den Heuvel, F.; Van Tintelen, J.P.; van den Berg, M.P.; Van Veldhuisen, D.J.; Van Gelder, I.C. A family with Andersen-Tawil syndrome and dilated cardiomyopathy. Heart Rhythm. 2006, 3, 1346–1350. [Google Scholar] [CrossRef] [PubMed]
- Tengan, C.H.; Antunes, A.C.; Bauab, J.R.; Prado, G.F.; Manzano, G.M.; Gabbai, A.A. Andersen syndrome: An association of periodic paralysis, cardiac arrhythmia and dysmorphic abnormalities. Arq. Neuropsiquiatr. 2006, 64, 582–584. [Google Scholar] [CrossRef] [PubMed]
- Bökenkamp, R.; Wilde, A.A.; Schalij, M.J.; Blom, N.A. Flecainide for recurrent malignant ventricular arrhythmias in two siblings with Andersen-Tawil syndrome. Heart Rhythm. 2007, 4, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Haruna, Y.; Kobori, A.; Makiyama, T.; Yoshida, H.; Akao, M.; Doi, T.; Tsuji, K.; Ono, S.; Nishio, Y.; Shimizu, W.; et al. Geno-type-phenotype correlations of KCNJ2 mutations in Japanese patients with Andersen-Tawil syndrome. Hum. Mutat. 2007, 28, 208. [Google Scholar] [CrossRef] [PubMed]
- Janson, C.M.; Poelzing, S.; Shah, M.J. Combined inhibition of Na⁺ and Ca²⁺ channels: A novel paradigm for the treatment of in-cessant ventricular arrhythmias in Andersen-Tawil syndrome. Heart Rhythm. 2014, 11, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Ardissone, A.; Sansone, V.; Colleoni, L.; Bernasconi, P.; Moroni, I. Intrafamilial phenotypic variability in Andersen-Tawil syn-drome: A diagnostic challenge in a potentially treatable condition. Neuromuscul. Disord. 2017, 27, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Barrón-Díaz, D.R.; Totomoch-Serra, A.; Escobar-Cedillo, R.E.; García-Gutierrez, A.; Reyes-Quintero, Á.E.; Villegas Davirán, S.E.; Ibar-ra-Miramón, C.B.; Márquez, M.F. Andersen-Tawil Syndrome with High Risk of Sudden Cardiac Death in Four Mexican Patients. Car-diac and Extra-Cardiac Phenotypes. Rev. Investig. Clin. 2020, 73, 145–153. [Google Scholar] [CrossRef]
- Takahashi, T.; Tandai, S.; Toki, T.; Sato, T.; Eto, S.; Sato, A.; Ueda, T.; Sato, S.; Ichinose, K.; Ito, E.; et al. KCNJ2 mutation in intrac-table ventricular arrhythmia with Andersen’s syndrome. Pediatr. Int. 2005, 47, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, R.N.; Gula, L.J.; Skanes, A.C.; Krahn, A.D. Andersen-Tawil syndrome: Management challenges during pregnancy, labor, and delivery. J. Cardiovasc. Electrophysiol. 2008, 19, 987–989. [Google Scholar] [CrossRef] [PubMed]
- Rajakulendran, S.; Tan, S.V.; Hanna, M.G. Muscle weakness, palpitations and a small chin: The Andersen-Tawil syndrome. Pract. Neurol. 2010, 10, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Zhou, J.; Kawamura, M.; Itoh, H.; Mizusawa, Y.; Ding, W.G.; Wu, J.; Ohno, S.; Makiyama, T.; Miyamoto, A.; et al. Phenotype variability in patients carrying KCNJ2 mutations. Circ. Cardiovasc. Genet. 2012, 5, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Lefter, S.; Hardiman, O.; Costigan, D.; Lynch, B.; McConville, J.; Hand, C.K.; Ryan, A.M. Andersen-Tawil syndrome with early fixed myopathy. J. Clin. Neuromuscul. Dis. 2014, 16, 79–82. [Google Scholar] [CrossRef]
- Jung, J.H.; Chae, J.H.; Song, J.H.; Ahn, S.H. The Case|An unusual case of recurrent hypokalemic periodic paralysis. Kidney Int. 2017, 91, 1523–1524. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Xu, M.; Sun, J.; Qiao, K.; Song, J.; Cai, S.; Zhu, W.; Zhou, L.; Xi, J.; Lu, J.; et al. Identification of gene mutations in patients with primary periodic paralysis using targeted next-generation sequencing. BMC Neurol. 2019, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Horigome, H.; Ishikawa, Y.; Kokubun, N.; Yoshinaga, M.; Sumitomo, N.; Lin, L.; Kato, Y.; Tanabe-Kameda, Y.; Ohno, S.; Nagashima, M.; et al. Multivariate analysis of TU wave complex on electrocardiogram in Andersen-Tawil syndrome with KCNJ2 mutations. Ann. Noninvasive Electrocardiol. 2020, 25, e12721. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, K.; Lv, T.; Xie, Y.; Liu, F.; Zhang, P. Case report: Mexiletine suppresses ventricular arrhythmias in Andersen-Tawil syn-drome. Front. Cardiovasc. Med. 2022, 9, 992185. [Google Scholar] [CrossRef] [PubMed]
- Sarova-Pinhas, I.; Braham, J.; Shalev, A. Premenstrual periodic paralysis. J. Neurol. Neurosurg. Psychiatry 1981, 44, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Grob, D.; Johns Rj Liljestrand, A. Potassium movement in patients with familial periodic paralysis: Relationship to the defect in muscle function. Am. J. Med. 1957, 23, 356–375. [Google Scholar] [CrossRef] [PubMed]
- Resnick, J.S.; Engel, W.K.; Griggs, R.C.; Stam, A.C. Acetazolamide prophylaxis in hypokalemic periodic paralysis. N. Engl. J. Med. 1968, 278, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Griggs, R.C.; Engel, W.K.; Resnick, J.S. Acetazolamide treatment of hypokalemic periodic paralysis. Prevention of attacks and improvement of persistent weakness. Ann. Intern. Med. 1970, 73, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.D.; Griggs, R.C.; Moxley, R.T., 3rd. Regulation of plasma potassium in hyperkalemic periodic paralysis. Neurology 1979, 29, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Poskanzer, D.C.; Kerr, D.N. A third type of periodic paralysis, with normokalemia and favourable response to sodium chloride. Am. J. Med. 1961, 31, 328–342. [Google Scholar] [CrossRef] [PubMed]
- Soom, M.; Schönherr, R.; Kubo, Y.; Kirsch, C.; Klinger, R.; Heinemann, S.H. Multiple PIP2 binding sites in Kir2.1 inwardly rectifying potassium channels. FEBS Lett. 2001, 490, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Lopes, C.M.; Zhang, H.; Rohacs, T.; Jin, T.; Yang, J.; Logothetis, D.E. Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron 2002, 34, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zu, H.; Yao, K. A case report of Andersen-Tawil syndrome misdiagnosed with myodystrophy. Front. Neurol. 2023, 14, 1170693. [Google Scholar] [CrossRef] [PubMed]
- Schmiedel, J.; Jackson, S.; Schäfer, J.; Reichmann, H. Mitochondrial cytopathies. J. Neurol. 2003, 250, 267–277. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial cytopathies. Cell Calcium. 2016, 60, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Viering, D.H.H.M.; Vermeltfoort, L.; Bindels, R.J.M.; Deinum, J.; de Baaij, J.H.F. Electrolyte Disorders in Mitochondrial Cytopathies: A Systematic Review. J. Am. Soc. Nephrol. 2023, 34, 1875–1888. [Google Scholar] [CrossRef] [PubMed]
- Dominic, E.A.; Ramezani, A.; Anker, S.D.; Verma, M.; Mehta, N.; Rao, M. Mitochondrial cytopathies and cardiovascular disease. Heart 2014, 100, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Elorza, A.A.; Soffia, J.P. mtDNA Heteroplasmy at the Core of Aging-Associated Heart Failure. An Integrative View of OXPHOS and Mitochondrial Life Cycle in Cardiac Mitochondrial Physiology. Front. Cell Dev. Biol. 2021, 9, 625020. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ATS Familial Cases | |||||||||||
| Clinical Presentation | Laboratory Investigations | Cardiological Features | Dysmorphisms/Other Features | ||||||||
| Authors [Reference] | Relationship | Age at Onset/Sex | First Symptoms | Muscle Involvement | K+ Levels in mEq/L (n.v. 4.2–5.6) | CK Levels in U/L (n.v. < 90) | ECG and Echo Features at 1st Visit and During FU | QTc in msec | Facial and Other Dysmorphisms | Additional Features | Age at Last FU |
| Davies et al. [13] | Proband | 9/F | VA | PP | n.r. | 120 | VA | n.r. | Abnormal dentition, dental enamel discoloration | Mild Ataxia | n.r. |
| Brother | 16/M | PP | PP | Low | n.r. | -- | n.r. | -- | -- | n.r. | |
| Brother | 14/F | PP | PP | Low | 154 | VA | n.r. | -- | -- | n.r. | |
| Mother | 8/F | VA | PP | n.r. | n.r. | VA | n.r. | -- | -- | n.r. | |
| Davies et al. [13] | Proband | 24/M | Painful PP | PP | n.r. | n.r. | -- | n.r. | -- | -- | n.r. |
| Son | 14/M | PP | PP | n.r. | n.r. | -- | n.r. | -- | -- | n.r. | |
| Son | 13/M | PP | PP | low | 184 | -- | n.r. | -- | -- | n.r. | |
| Donaldson et al. [19] | Proband | 10/F | Weakness of the lower limbs; paralysis | PP; muscle weakness of lower limbs | 2.0 | n.r. | bVT; PVC; prominent U wave; LQTS; EVB | 445 | Micrognathia, scoliosis, clinodactyly, small ears, hypertelorism, short and wide neck, fingers syndactyly | --- | 29 |
| Pouget et al. [20] | Father | 38/M | Muscle pain; EVB | n.r. | n.r. | EVB | n.r. | --- | --- | 53 | |
| Schoonderwoerd et al. [21] | Proband | 18/F | Cardiac arrest | Exercise intolerance | 2.6 | n.r. | Prominent U wave; VF; DCM frequent PVCs; VT | n.r. | Mild facial asymmetry, clinodactyly, syndactyly of second and third toes | -- | n.r. |
| Mother | 34/F | Unexpected syncopal event | -- | n.r. | n.r. | Frequent PVCs; VT; DCM | n.r. | Asymmetric face, prognathia, camptodactyly of toes, syndactyly of the second and third toes | -- | n.r. | |
| Brother | 16/M | Muscle weakness | Muscle weakness of limbs | n.r. | n.r. | Prominent U wave; PVC; pVT | n.r. | Low set ears, prominent forehead, bifid uvula, webbed neck, small mandible, clinodactyly, syndactyly of the second and third toes | -- | n.r. | |
| Tengan et al. [22] | Proband | 13/M | Muscle weakness | PP, weakness of proximal limb muscles | Normal | n.r. | Prolonged QT; isolated EVB | 490 | Short stature, micrognathia, retrognathia, clinodactyly of fourth and fifth fingers, arched palate, thoracic scoliosis | Obesity; OSA, daytime sleepiness | n.r. |
| Daughter | 6/F | Muscle weakness | Episodes of weakness | Normal | n.r. | -- | n.r. | Short stature, micrognathia, clinodactyly of fourth and fifth fingers | OSA | n.r. | |
| Bokenkamp et al. [23] | Proband | 3/F | Arrhythmia | -- | -- | n.r. | Polymorphic EVB, prolonged QT, prominent U wave, mild aortic root dilation | 480 | Mild dysmorphic features, including broad forehead, hypertelorism, small mandible and clinodactyly | -- | 6 |
| Father | /M | Polymorphic EVB | -- | -- | n.r. | Polymorphic EVB | -- | Mild dysmorphic features | |||
| Brother | 6/M | Polymorphic EVB; syncope | 1 episode of muscle weakness | -- | n.r. | Polymorphic EVB | -- | Mild dysmorphic features | -- | 7 | |
| Haruna et al. [24] | Proband | 6/F | Syncopal events | PP | n.r. | n.r. | PVC, bVT, monomorphic VT | 483 | -- | -- | n.r. |
| Father | 38/M | Muscle weakness | PP | n.r. | n.r. | PVC | 384 | -- | -- | n.r. | |
| Grandfather | /M | Muscle weakness | PP | n.r. | n.r. | -- | 410 | -- | -- | n.r. | |
| Haruna et al. [24] | Proband | 11/F | Muscle weakness | -- | n.r. | n.r. | bVT, mVT | 365 | Dysmorphic features | -- | n.r. |
| Father | 47/M | Muscle weakness | -- | n.r. | n.r. | -- | 394 | -- | -- | n.r. | |
| Brother | 5/M | Myalgia | -- | n.r. | n.r. | -- | 342 | -- | -- | n.r. | |
| Janson et al. [25] | Proband | 10/M | VEB, bVT | PP, episodes of muscle weakness | n.r. | n.r. | EVB, bVT | -- | Micrognathia, wide-spaced eyes, clinodactyly of the fifth digit | -- | 15 |
| Mother | F | PP, VT | PP | n.r. | n.r. | VT, CM | -- | Micrognathia, wide-spaced eyes, clinodactyly of the fifth digit | -- | n.r. | |
| Ardissone et al. [26] | Proband | 1.4/M | Acute muscle pain | Muscle pain, cramps, generalized weakness | 3.5 | 346 | Sinus Tachycardia | Normal | Broad forehead, hypoplastic mandible, low-set ears, short stature, low weight | -- | 4 |
| Mother | 20/F | Muscle pain post-exercise, muscle weakness | Severe generalized muscle weakness | -- | n.r. | -- | -- | Broad forehead, hypoplastic mandible, low-set ears, short stature, low weight | -- | n.r. | |
| Brother | M | VT, long QT | -- | -- | n.r. | VT, long QT, arrhythmias | -- | Broad forehead, hypoplastic jaw, low-set ears | -- | n.r | |
| Brother | M | VT, long QT | Muscle stiffness | -- | n.r. | VT, long QT, arrhythmias | -- | Broad forehead, hypoplastic jaw, low-set ears | -- | n.r. | |
| Barron-Diaz et al. [27] | Proband | 25/F | VT | Limb weakness | n.r. | n.r. | PVC, VT | 441 | Short stature, broad forehead, jaw hypoplasia, dental alterations, camptodactyly, clinodactyly, feet syndactyly, dental alterations | -- | n.r. |
| Sister | 7/F | VT, weakness, syncopal episodes | Limb weakness | n.r. | n.r. | PVC, VT, arrhythmias, mitral regurgitation | 544 | Triangular face, scoliosis, road forehead, ptosis palpebral, mandibular hypoplasia, dental alterations, camptodactyly in hands, clinodactyly, feet syndactyly | -- | n.r. | |
| Our Family Case | Uncle | 34/M | Cramps, myalgias, muscle weakness | General muscle hypertrophy | 4.2 | 297 | Coronary artery by-pass graftings; IHD; sVT; sEVB; EVB | 360 | --- | Metabolic syndrome | 66 |
| Father | 6/M | Cramps, myalgias post-exercise | Lower limbs | 5.4 | 827 | AMI; eccentric hypertrophy of LV; ICD implant sVT; nsVT | 450 | Dental crowding | Metabolic syndrome; OSA | 68 | |
| Cousin | 24/F | Cramps; muscle weakness; myotonia | Lower limbs | 3.9 | 171 | Normal | 370 | Small ears | 5 | ||
| Older brother | 12/M | Cramps; muscle stiffness at lower limbs | Lower limbs | 4.7 | 150 | Sinus arrhythmia; decreased amplitude of T waves in LPL | 380 | Jaw hypoplasia; dental crowding; clinodactyly fifth toe; syndactyly of II/III fingers of hands | Obesity | 39 | |
| Proband | 16/F | Premenstrual episodes of PP, muscle stiffness | Upper and lower limbs | 4.4 | 97 | Sinus Bradycardia; EVB during FU | 370 | Micrognathia, elusive chin, low set of ears, dental crowding, hypotelorism, mall hands/ feet | ___ | 37 | |
| Young brother | 11/M | Muscle stiffness | Upper limbs | 4.9 | 426 | Sinus Bradycardia; transient WPM | 370 | Scoliosis, micrognathia, small ears, hypertelorism, short neck, fingers clinodactyly and syndactyly | Obesity | 32 | |
| (b) | |||||||||||
| ATS Sporadic Cases | |||||||||||
| Haruna et al. [24] | Proband | 6/M | Muscle weakness | PP | n.r. | n.r. | PVC | 339 | Dysmorphic features | -- | n.r. |
| Takahashi et al. [28] | Proband | 5/F | Syncopal events, muscle weakness | Muscle weakness | Normal | n.r. | Long QT, prominent U wave, PVC, intractable VA | 490 | -- | -- | 18 |
| Subbiah et al. [29] | Proband | 17/F | Muscle weakness, bVT | PP, episodes of weakness | n.r. | n.r. | Symptomatic EVB, bVT, prominent U wave | 460 | -- | -- | n.r. |
| Rajakulendran et al. [30] | Proband | 9/F | Attacks of muscle weakness | Muscle weakness | Normal | n.r. | -- | 365 | Short stature, micromelia, low set ears, clinodactyly of hands, syndactyly of toes | -- | n.r. |
| Kimura et al. [31] | Proband | 6/F | Syncopal events | PP | n.r. | n.r. | PVC | 508 | -- | -- | n.r. |
| Proband | 11/F | -- | -- | n.r. | n.r. | pVT | n.r. | Dysmorphic features | -- | n.r. | |
| Proband | 6/M | -- | PP | n.r. | n.r. | bVT | 468 | Dysmorphic features | -- | n.r. | |
| Proband | 19/F | Syncopal events | -- | n.r. | n.r. | bVT | 400 | Dysmorphic features | -- | n.r. | |
| Proband | 12/M | -- | -- | n.r. | n.r. | bVT, pVT, PVC | 427 | Dysmorphic features | -- | n.r. | |
| Proband | 6/F | -- | -- | n.r. | n.r. | pVT, PVC | 392 | -- | -- | n.r. | |
| Proband | 5/M | -- | -- | n.r. | n.r. | pVT, PVC | Normal | -- | -- | n.r. | |
| Lefter et al. [32] | Proband | 10/M | Lower limb weakness | PP, myalgia, weakness, fixed proximal myopathy | Normal | 850 | Prominent U wave | n.r. | Short stature, micromelia, micrognatia, low set ears, fifth digit clinodactyly, syndactyly of the left foot digits 2–3 | -- | 19 |
| Jung et al. [33] | Proband | 6/M | pVT | Paralysis of lower limbs | 2.5 | n.r. | pVT | n.r. | Micrognatia, clinodactyly | -- | 25 |
| Luo et al. [34] | Proband | 11/M | PP | PP | Normal | n.r. | -- | n.r. | -- | -- | n.r. |
| Horigome et al. [35] | Proband | 6/M | PP | PP | n.r. | n.r. | bVT | n.r. | Short stature, dysmorphic features | -- | n.r. |
| Proband | 19/M | PP | PP | n.r. | n.r. | PVC | n.r. | Dysmorphic features | -- | n.r. | |
| Proband | 24/F | PP | PP | n.r. | n.r. | PVC | n.r. | Short stature, dysmorphic features | -- | n.r. | |
| Proband | 28/F | PP | PP | n.r. | n.r. | bVT | n.r. | Short stature, dysmorphic features | -- | n.r. | |
| Proband | 54/F | PP | PP | n.r. | n.r. | PVC | n.r. | Dysmorphic features | -- | n.r. | |
| Yang et al. [36] | Proband | 7/M | Syncopal events | Lower limb myotonia | 3.9 | n.r. | PVC, bVT, prominent U wave | 420 | Short stature, mandibular hypoplasia, single palmar crease, long bone, over hyper-extension | -- | n.r. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onore, M.E.; Picillo, E.; D’Ambrosio, P.; Morra, S.; Nigro, V.; Politano, L. Phenotypic Variability of Andersen–Tawil Syndrome Due to Allelic Mutation c.652C>T in the KCNJ2 Gene—A New Family Case Report. Biomolecules 2024, 14, 507. https://doi.org/10.3390/biom14040507
Onore ME, Picillo E, D’Ambrosio P, Morra S, Nigro V, Politano L. Phenotypic Variability of Andersen–Tawil Syndrome Due to Allelic Mutation c.652C>T in the KCNJ2 Gene—A New Family Case Report. Biomolecules. 2024; 14(4):507. https://doi.org/10.3390/biom14040507
Chicago/Turabian StyleOnore, Maria Elena, Esther Picillo, Paola D’Ambrosio, Salvatore Morra, Vincenzo Nigro, and Luisa Politano. 2024. "Phenotypic Variability of Andersen–Tawil Syndrome Due to Allelic Mutation c.652C>T in the KCNJ2 Gene—A New Family Case Report" Biomolecules 14, no. 4: 507. https://doi.org/10.3390/biom14040507
APA StyleOnore, M. E., Picillo, E., D’Ambrosio, P., Morra, S., Nigro, V., & Politano, L. (2024). Phenotypic Variability of Andersen–Tawil Syndrome Due to Allelic Mutation c.652C>T in the KCNJ2 Gene—A New Family Case Report. Biomolecules, 14(4), 507. https://doi.org/10.3390/biom14040507