
Reflections on the Origin of Coded Protein Biosynthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Background
The First Functional Coded Polypeptides
3. The First Biocatalysts
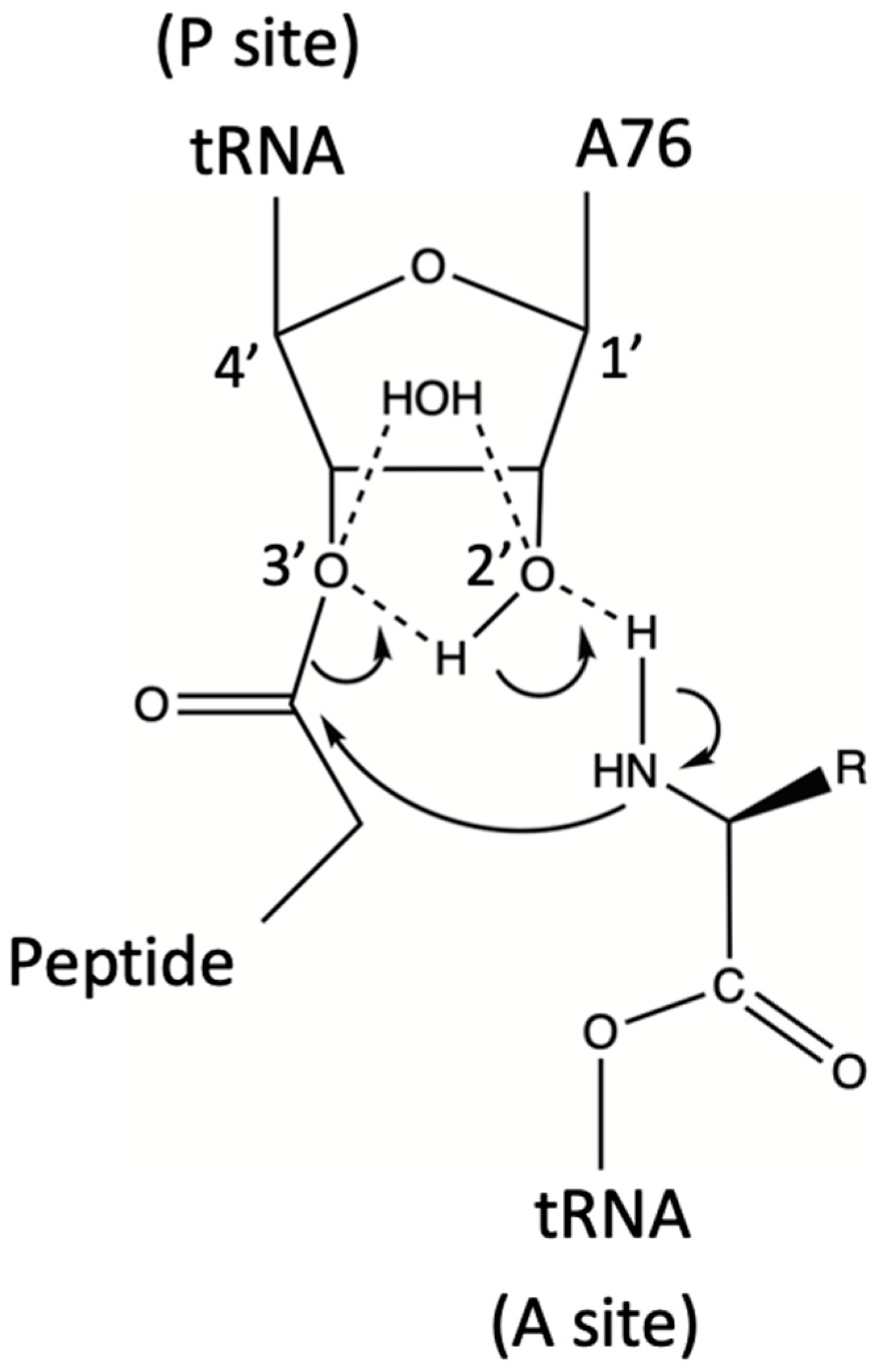
3.1. The Ribosome

3.2. Aminoacyl-tRNA Synthetases
- (a)
- aa + ATP ↔ aa-AMP + PPi
- (b)
- aa-AMP + tRNAaa ↔ aa-tRNAaa + AMP
3.3. DNA-Dependent RNA Polymerases
4. Conclusions

Funding
Acknowledgments
Conflicts of Interest
References
- Gilbert, W. Origin of Life: The RNA World. Nature 1986, 319, 618. [Google Scholar] [CrossRef]
- Tjhung, K.F.; Shokhirev, M.N.; Horning, D.P.; Joyce, G.F. An RNA Polymerase Ribozyme That Synthesizes Its Own Ancestor. Proc. Natl. Acad. Sci. USA 2020, 117, 2906–2913. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.W. What RNA World? Why a Peptide/RNA Partnership Merits Renewed Experimental Attention. Life 2015, 5, 294–320. [Google Scholar] [CrossRef] [PubMed]
- Wills, P.R.; Carter, C.W. Insuperable Problems of the Genetic Code Initially Emerging in an RNA World. Biosystems 2018, 164, 155–166. [Google Scholar] [CrossRef]
- Dale, T. Protein and Nucleic Acid Together: A Mechanism for the Emergence of Biological Selection. J. Theor. Biol. 2006, 240, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Chotera, A.; Sadihov, H.; Cohen-Luria, R.; Monnard, P.-A.; Ashkenasy, G. Functional Assemblies Emerging in Complex Mixtures of Peptides and Nucleic Acid-Peptide Chimeras. Chemistry 2018, 24, 10128–10135. [Google Scholar] [CrossRef] [PubMed]
- Taran, O.; Chen, C.; Omosun, T.O.; Hsieh, M.-C.; Rha, A.; Goodwin, J.T.; Mehta, A.K.; Grover, M.A.; Lynn, D.G. Expanding the Informational Chemistries of Life: Peptide/RNA Networks. Philos. Trans. A Math. Phys. Eng. Sci. 2017, 375, 20160356. [Google Scholar] [CrossRef]
- Dujardin, A.; Himbert, S.; Pudritz, R.; Rheinstädter, M.C. The Formation of RNA Pre-Polymers in the Presence of Different Prebiotic Mineral Surfaces Studied by Molecular Dynamics Simulations. Life 2023, 13, 112. [Google Scholar] [CrossRef] [PubMed]
- Bedoin, L.; Alves, S.; Lambert, J.-F. Origins of Life and Molecular Information: Selectivity in Mineral Surface-Induced Prebiotic Amino Acid Polymerization. ACS Earth Space Chem. 2020, 4, 1802–1812. [Google Scholar] [CrossRef]
- Namani, T.; Snyder, S.; Eagan, J.M.; Bevilacqua, P.C.; Wesdemiotis, C.; Sahai, N. Amino Acid Specific Nonenzymatic Montmorillonite-Promoted RNA Polymerization. ChemSystemsChem 2021, 3, e2000060. [Google Scholar] [CrossRef]
- El Samrout, O.; Fabbiani, M.; Berlier, G.; Lambert, J.-F.; Martra, G. Emergence of Order in Origin-of-Life Scenarios on Mineral Surfaces: Polyglycine Chains on Silica. Langmuir 2022, 38, 15516–15525. [Google Scholar] [CrossRef] [PubMed]
- Akouche, M.; Jaber, M.; Maurel, M.-C.; Lambert, J.-F.; Georgelin, T. Phosphoribosyl Pyrophosphate: A Molecular Vestige of the Origin of Life on Minerals. Angew. Chem. Int. Ed. Engl. 2017, 56, 7920–7923. [Google Scholar] [CrossRef] [PubMed]
- Kunnev, D.; Gospodinov, A. Possible Emergence of Sequence Specific RNA Aminoacylation via Peptide Intermediary to Initiate Darwinian Evolution and Code Through Origin of Life. Life 2018, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Nissen, P.; Hansen, J.; Ban, N.; Moore, P.B.; Steitz, T.A. The Structural Basis of Ribosome Activity in Peptide Bond Synthesis. Science 2000, 289, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Krupkin, M.; Matzov, D.; Tang, H.; Metz, M.; Kalaora, R.; Belousoff, M.J.; Zimmerman, E.; Bashan, A.; Yonath, A. A Vestige of a Prebiotic Bonding Machine Is Functioning within the Contemporary Ribosome. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 2972–2978. [Google Scholar] [CrossRef]
- Crick, F.H. The Origin of the Genetic Code. J. Mol. Biol. 1968, 38, 367–379. [Google Scholar] [CrossRef]
- Yarus, M.; Widmann, J.J.; Knight, R. RNA-Amino Acid Binding: A Stereochemical Era for the Genetic Code. J. Mol. Evol. 2009, 69, 406–429. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.F.; Wang, L. Imprints of the Genetic Code in the Ribosome. Proc. Natl. Acad. Sci. USA 2010, 107, 8298–8303. [Google Scholar] [CrossRef]
- Krüger, D.M.; Neubacher, S.; Grossmann, T.N. Protein–RNA Interactions: Structural Characteristics and Hotspot Amino Acids. RNA 2018, 24, 1457–1465. [Google Scholar] [CrossRef]
- Fontecilla-Camps, J.C. Reflections on the Origin and Early Evolution of the Genetic Code. ChemBioChem 2023, 24, e202300048. [Google Scholar] [CrossRef]
- Carter, C.W. Coding of Class I and II Aminoacyl-tRNA Synthetases. Adv. Exp. Med. Biol. 2017, 966, 103–148. [Google Scholar] [CrossRef] [PubMed]
- Frenkel-Pinter, M.; Haynes, J.W.; Mohyeldin, A.M.; Martin, C.; Sargon, A.B.; Petrov, A.S.; Krishnamurthy, R.; Hud, N.V.; Williams, L.D.; Leman, L.J. Mutually Stabilizing Interactions between Proto-Peptides and RNA. Nat. Commun. 2020, 11, 3137. [Google Scholar] [CrossRef] [PubMed]
- Lake, J.A.; Larsen, J.; Tran, D.T.; Sinsheimer, J.S. Uncovering the Genomic Origins of Life. Genome Biol. Evol. 2018, 10, 1705–1714. [Google Scholar] [CrossRef]
- Bruice, T.C. Proximity Effects and Enzyme Catalysis. In The Enzymes; Boyer, P.D., Ed.; Academic Press: Cambridge, MA, USA, 1970; Volume 2, pp. 217–279. [Google Scholar]
- Noor, E.; Flamholz, A.I.; Jayaraman, V.; Ross, B.L.; Cohen, Y.; Patrick, W.M.; Gruic-Sovulj, I.; Tawfik, D.S. Uniform Binding and Negative Catalysis at the Origin of Enzymes. Protein Sci. 2022, 31, e4381. [Google Scholar] [CrossRef] [PubMed]
- Wächtershäuser, G. Before Enzymes and Templates: Theory of Surface Metabolism. Microbiol. Rev. 1988, 52, 452–484. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.K.Y.; Suslov, N.; Tuttle, N.; Sengupta, R.; Piccirilli, J.A. The Mechanism of Peptidyl Transfer Catalysis by the Ribosome. Annu. Rev. Biochem. 2011, 80, 527–555. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.B.; Steitz, T.A. After the Ribosome Structures: How Does Peptidyl Transferase Work? RNA 2003, 9, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Selmer, M.; Dunham, C.M.; Murphy, F.V.; Weixlbaumer, A.; Petry, S.; Kelley, A.C.; Weir, J.R.; Ramakrishnan, V. Structure of the 70S Ribosome Complexed with mRNA and tRNA. Science 2006, 313, 1935–1942. [Google Scholar] [CrossRef] [PubMed]
- Sievers, A.; Beringer, M.; Rodnina, M.V.; Wolfenden, R. The Ribosome as an Entropy Trap. Proc. Natl. Acad. Sci. USA 2004, 101, 7897–7901. [Google Scholar] [CrossRef]
- Changalov, M.M.; Ivanova, G.D.; Rangelov, M.A.; Acharya, P.; Acharya, S.; Minakawa, N.; Földesi, A.; Stoineva, I.B.; Yomtova, V.M.; Roussev, C.D.; et al. 2′/3′-O-Peptidyl Adenosine as a General Base Catalyst of Its Own External Peptidyl Transfer: Implications for the Ribosome Catalytic Mechanism. ChemBioChem 2005, 6, 992–996. [Google Scholar] [CrossRef]
- Hansen, J.L.; Schmeing, T.M.; Moore, P.B.; Steitz, T.A. Structural Insights into Peptide Bond Formation. Proc. Natl. Acad. Sci. USA 2002, 99, 11670–11675. [Google Scholar] [CrossRef]
- Weinger, J.S.; Parnell, K.M.; Dorner, S.; Green, R.; Strobel, S.A. Substrate-Assisted Catalysis of Peptide Bond Formation by the Ribosome. Nat. Struct. Mol. Biol. 2004, 11, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Youngman, E.M.; Brunelle, J.L.; Kochaniak, A.B.; Green, R. The Active Site of the Ribosome Is Composed of Two Layers of Conserved Nucleotides with Distinct Roles in Peptide Bond Formation and Peptide Release. Cell 2004, 117, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Polacek, N.; Gaynor, M.; Yassin, A.; Mankin, A.S. Ribosomal Peptidyl Transferase Can Withstand Mutations at the Putative Catalytic Nucleotide. Nature 2001, 411, 498–501. [Google Scholar] [CrossRef]
- Beringer, M.; Adio, S.; Wintermeyer, W.; Rodnina, M. The G2447A Mutation Does Not Affect Ionization of a Ribosomal Group Taking Part in Peptide Bond Formation. RNA 2003, 9, 919–922. [Google Scholar] [CrossRef]
- Trobro, S.; Åqvist, J. Mechanism of Peptide Bond Synthesis on the Ribosome. Proc. Natl. Acad. Sci. USA 2005, 102, 12395–12400. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.K.; Xiang, Y.; Kato, M.; Warshel, A. What Are the Roles of Substrate-Assisted Catalysis and Proximity Effects in Peptide Bond Formation by the Ribosome? Biochemistry 2005, 44, 11307–11314. [Google Scholar] [CrossRef]
- Damer, B.; Deamer, D. Coupled Phases and Combinatorial Selection in Fluctuating Hydrothermal Pools: A Scenario to Guide Experimental Approaches to the Origin of Cellular Life. Life 2015, 5, 872–887. [Google Scholar] [CrossRef] [PubMed]
- Fares, H.M.; Marras, A.E.; Ting, J.M.; Tirrell, M.V.; Keating, C.D. Impact of Wet-Dry Cycling on the Phase Behavior and Compartmentalization Properties of Complex Coacervates. Nat. Commun. 2020, 11, 5423. [Google Scholar] [CrossRef]
- Rimola, A.; Sodupe, M.; Ugliengo, P. Amide and Peptide Bond Formation: Interplay between Strained Ring Defects and Silanol Groups at Amorphous Silica Surfaces. J. Phys. Chem. 2016, 120, 24817–24826. [Google Scholar] [CrossRef]
- Deiana, C.; Sakhno, Y.; Fabbiani, M.; Pazzi, M.; Vincenti, M.; Martra, G. Direct Synthesis of Amides from Carboxylic Acids and Amines by Using Heterogeneous Catalysts: Evidence of Surface Carboxylates as Activated Electrophilic Species. ChemCatChem 2013, 5, 2832–2834. [Google Scholar] [CrossRef]
- Nazarea, A.D.; Bloch, D.P.; Semrau, A.C. Detection of a Fundamental Modular Format Common to Transfer and Ribosomal RNAs: Second-Order Spectral Analysis. Proc. Natl. Acad. Sci. USA 1985, 82, 5337–5341. [Google Scholar] [CrossRef]
- Bloch, D.P.; McArthur, B.; Widdowson, R.; Spector, D.; Guimaraes, R.C.; Smith, J. tRNA-rRNA Sequence Homologies: Evidence for a Common Evolutionary Origin? J. Mol. Evol. 1983, 19, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M.; Winkler-Oswatitsch, R. Transfer-RNA, an Early Gene? Naturwissenschaften 1981, 68, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.W.; Hendrickson, T.L. Putting Amino Acids onto tRNAs: The Aminoacyl-tRNA Synthetases as Catalysts. In The Enzymes; Ribas de Pouplana, L., Kaguni, L.S., Eds.; Biology of Aminoacyl-tRNA Synthetases; Academic Press: Cambridge, MA, USA, 2020; Volume 48, pp. 39–68. [Google Scholar]
- Eriani, G.; Delarue, M.; Poch, O.; Gangloff, J.; Moras, D. Partition of tRNA Synthetases into Two Classes Based on Mutually Exclusive Sets of Sequence Motifs. Nature 1990, 347, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Weinger, J.S.; Strobel, S.A. Participation of the tRNA A76 Hydroxyl Groups throughout Translation. Biochemistry 2006, 45, 5939–5948. [Google Scholar] [CrossRef] [PubMed]
- Ruff, M.; Krishnaswamy, S.; Boeglin, M.; Poterszman, A.; Mitschler, A.; Podjarny, A.; Rees, B.; Thierry, J.C.; Moras, D. Class II Aminoacyl Transfer RNA Synthetases: Crystal Structure of Yeast Aspartyl-tRNA Synthetase Complexed with tRNA(Asp). Science 1991, 252, 1682–1689. [Google Scholar] [CrossRef]
- Rodin, S.N.; Ohno, S. Two Types of Aminoacyl-tRNA Synthetases Could Be Originally Encoded by Complementary Strands of the Same Nucleic ACID. Orig. Life. Evol. Biosph. 1995, 25, 565–589. [Google Scholar] [CrossRef] [PubMed]
- Fontecilla-Camps, J.C. Primordial Bioenergy Sources: The Two Facets of Adenosine Triphosphate. J. Inorg. Biochem. 2020, 216, 111347. [Google Scholar] [CrossRef]
- Medvedev, K.E.; Kinch, L.N.; Schaeffer, R.D.; Grishin, N.V. Functional Analysis of Rossmann-like Domains Reveals Convergent Evolution of Topology and Reaction Pathways. PLoS Comput. Biol. 2019, 15, e1007569. [Google Scholar] [CrossRef]
- Raanan, H.; Poudel, S.; Pike, D.H.; Nanda, V.; Falkowski, P.G. Small Protein Folds at the Root of an Ancient Metabolic Network. Proc. Natl. Acad. Sci. USA 2020, 117, 7193–7199. [Google Scholar] [CrossRef] [PubMed]
- Westheimer, F.H. Why Nature Chose Phosphates. Science 1987, 235, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Guth, E.; Connolly, S.H.; Bovee, M.; Francklyn, C.S. A Substrate-Assisted Concerted Mechanism for Aminoacylation by a Class II Aminoacyl-tRNA Synthetase. Biochemistry 2005, 44, 3785–3794. [Google Scholar] [CrossRef] [PubMed]
- Belogurov, G.A.; Artsimovitch, I. The Mechanisms of Substrate Selection, Catalysis, and Translocation by the Elongating RNA Polymerase. J. Mol. Biol. 2019, 431, 3975–4006. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.K.; Kao, C.C. Analysis of RNA-Dependent RNA Polymerase Structure and Function as Guided by Known Polymerase Structures and Computer Predictions of Secondary Structure. Virology 1998, 252, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Dall’Acqua, W.; Carter, P. Substrate-Assisted Catalysis: Molecular Basis and Biological Significance. Protein Sci. 2000, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A.; Pleckaityte, M.; Selent, U.; Wolfes, H.; Siksnys, V.; Pingoud, A. Evidence for Substrate-Assisted Catalysis in the DNA Cleavage of Several Restriction Endonucleases. Gene 1995, 157, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Schweins, T.; Geyer, M.; Scheffzek, K.; Warshel, A.; Kalbitzer, H.R.; Wittinghofer, A. Substrate-Assisted Catalysis as a Mechanism for GTP Hydrolysis of P21ras and Other GTP-Binding Proteins. Nat. Struct. Biol. 1995, 2, 36–44. [Google Scholar] [CrossRef]
- Schweins, T.; Langen, R.; Warshel, A. Why Have Mutagenesis Studies Not Located the General Base in Ras P21. Nat. Struct. Biol. 1994, 1, 476–484. [Google Scholar] [CrossRef]
- Setlik, R.F.; Garduno-Juarez, R.; Manchester, J.I.; Shibata, M.; Ornstein, R.L.; Rein, R. Modeling Study on the Cleavage Step of the Self-Splicing Reaction in Group I Introns. J. Biomol. Struct. Dyn. 1993, 10, 945–972. [Google Scholar] [CrossRef]
- Fontecilla-Camps, J.C. The Complex Roles of Adenosine Triphosphate in Bioenergetics. ChemBioChem 2022, 23, e202200064. [Google Scholar] [CrossRef] [PubMed]
- Hanson, R.W. The Role of ATP in Metabolism. Biochem. Educ. 1989, 17, 86–92. [Google Scholar] [CrossRef]
- Banfalvi, G. Ribose Selected as Precursor to Life. DNA Cell Biol. 2020, 39, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Akouche, M.; Jaber, M.; Zins, E.-L.; Maurel, M.-C.; Lambert, J.-F.; Georgelin, T. Thermal Behavior of D-Ribose Adsorbed on Silica: Effect of Inorganic Salt Coadsorption and Significance for Prebiotic Chemistry. Chemistry 2016, 22, 15834–15846. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.A.; Turchyn, A.V.; Ralser, M. Non-enzymatic glycolysis and pentose phosphate pathway-like reactions in a plausible Archean ocean. Mol. Syst. Biol. 2014, 10, 725. [Google Scholar] [CrossRef] [PubMed]
- Greene, B.L.; Kang, G.; Cui, C.; Bennati, M.; Nocera, D.G.; Drennan, C.L.; Stubbe, J. Ribonucleotide Reductases (RNRs): Structure, Chemistry, and Metabolism Suggest New Therapeutic Targets. Annu. Rev. Biochem. 2020, 89, 45–75. [Google Scholar] [CrossRef] [PubMed]
- Torrents, E. Ribonucleotide Reductases: Essential Enzymes for Bacterial Life. Front. Cell. Infect. Microbiol. 2014, 4, 52. [Google Scholar] [CrossRef]
- Fontecilla-Camps, J.C.; Volbeda, A. Quinolinate Synthase: An Example of the Roles of the Second and Outer Coordination Spheres in Enzyme Catalysis. Chem. Rev. 2022, 122, 12110–12131. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontecilla-Camps, J.C. Reflections on the Origin of Coded Protein Biosynthesis. Biomolecules 2024, 14, 518. https://doi.org/10.3390/biom14050518
Fontecilla-Camps JC. Reflections on the Origin of Coded Protein Biosynthesis. Biomolecules. 2024; 14(5):518. https://doi.org/10.3390/biom14050518
Chicago/Turabian StyleFontecilla-Camps, Juan Carlos. 2024. "Reflections on the Origin of Coded Protein Biosynthesis" Biomolecules 14, no. 5: 518. https://doi.org/10.3390/biom14050518
APA StyleFontecilla-Camps, J. C. (2024). Reflections on the Origin of Coded Protein Biosynthesis. Biomolecules, 14(5), 518. https://doi.org/10.3390/biom14050518