Mortalin, Apoptosis, and Neurodegeneration

Abstract
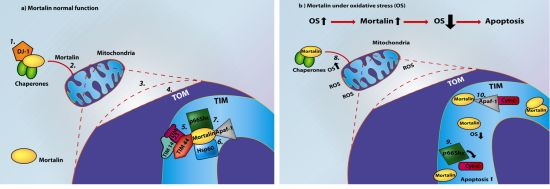
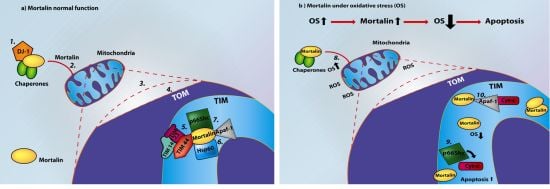
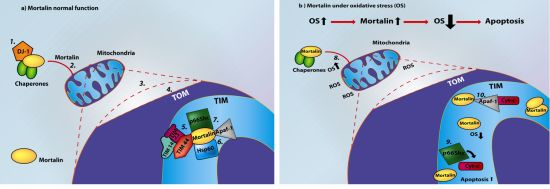
:
1. Introduction
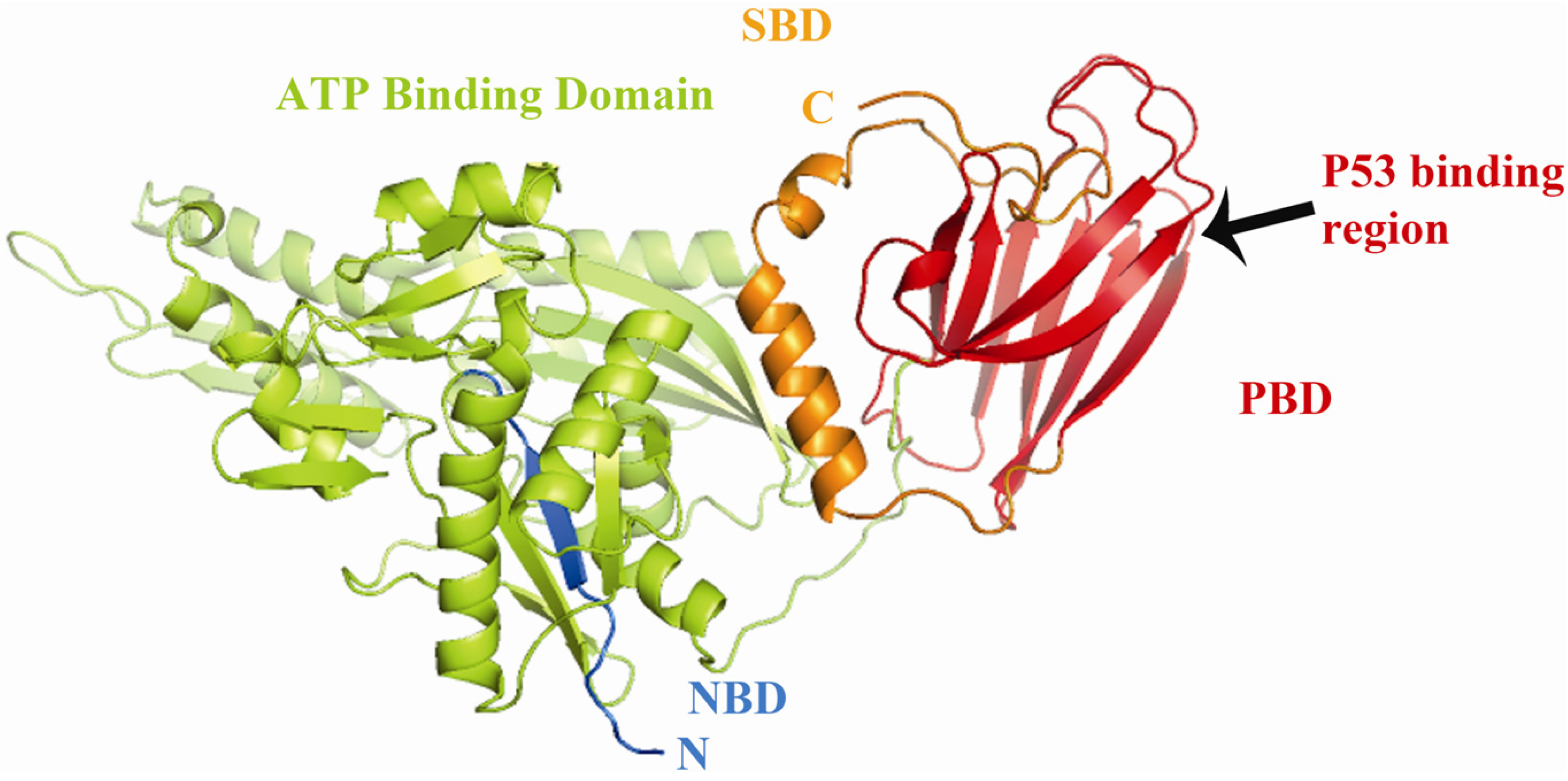
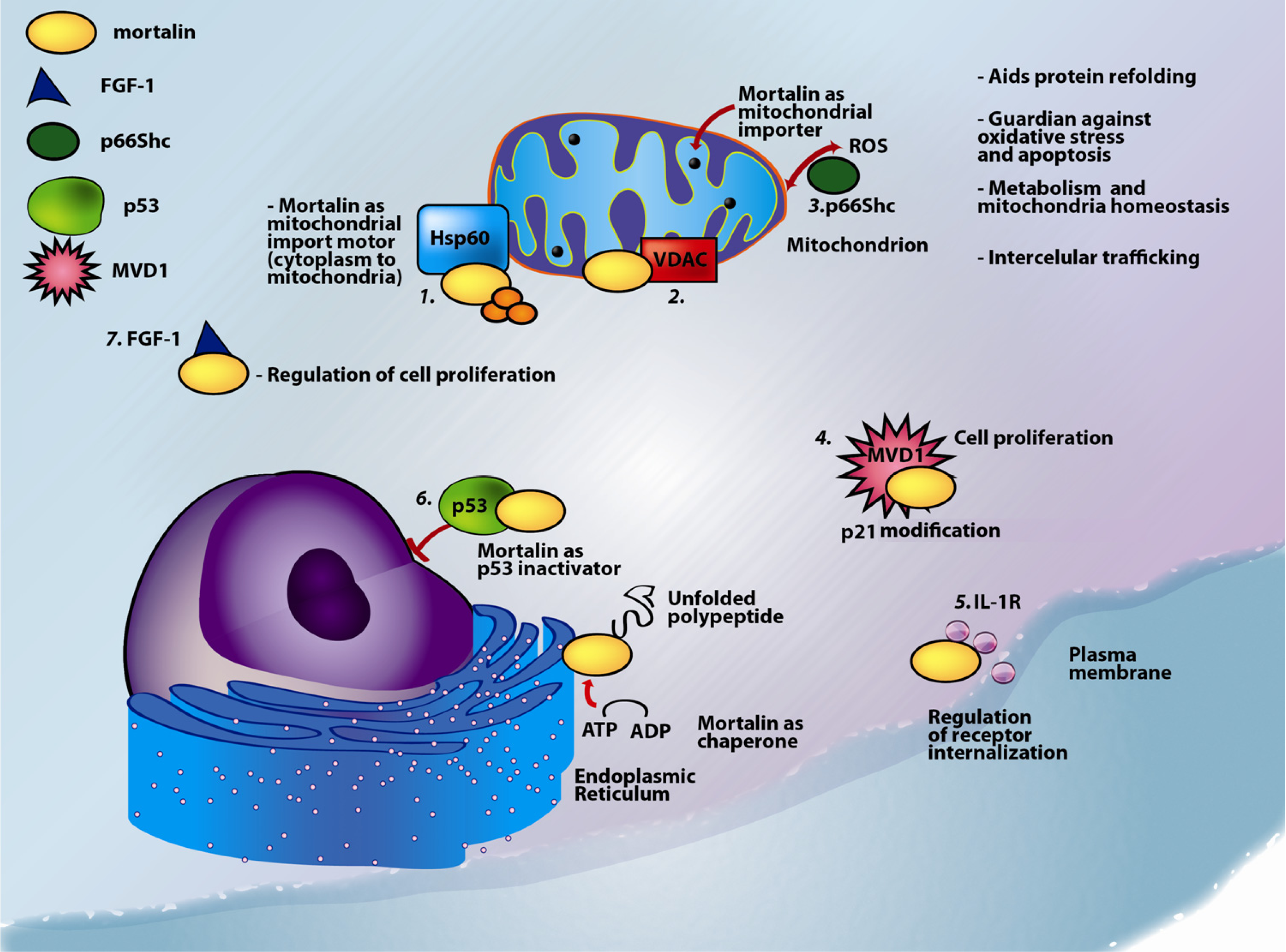
1.1. Mortalin: Structure and Known Functions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Subcellular location | Function | Reference |
|---|---|---|---|
| Amyloid precursor protein (APP) | Membrane. APP is an integral membrane protein expressed in many tissues and concentrated in the synapses of neurons. | Induces specific subsets of neuroprotective and anti-oxidative genes, mitochondrial regulatory genes and developmental genes.Activates mortalin expression | [43] |
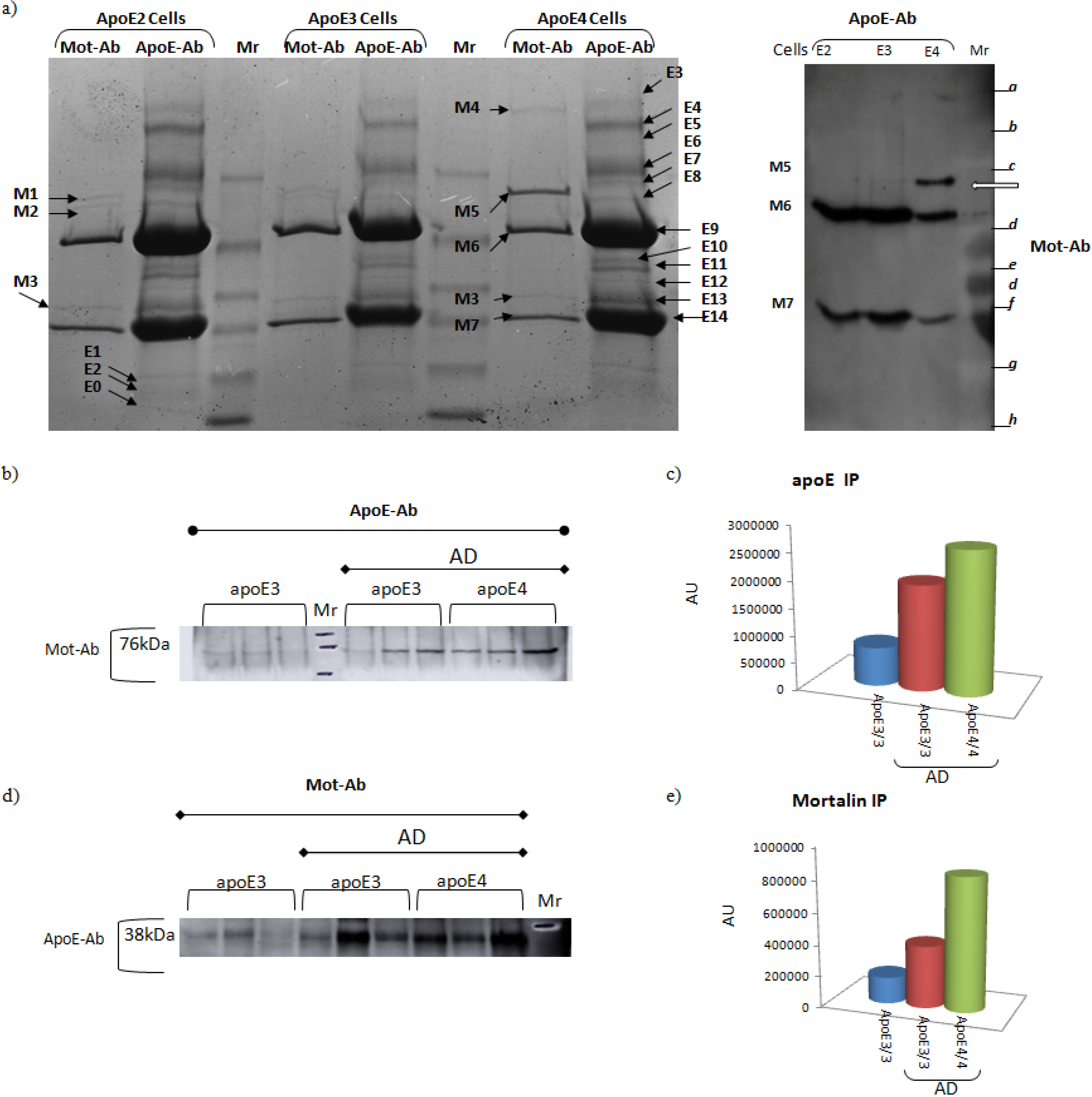
| Apolipoprotein E (ApoE) - Evidence suggesting that ApoE binds mortalin is shown in Figure 4 | Secreted | ApoE mediates the binding, internalization and catabolism of lipoprotein particles. It can serve as a ligand for the low density lipoprotein (ApoB/E) receptor and for the specific ApoE receptor (chylomicron remnant) of hepatic tissues. | See Figure 4 |
| CDK11p60 | CDK11p60 is the N-terminal portion of the cytosolic protein CDK11p110, that translocates into the mitochondria | Contributes to apoptosis directly at the mitochondria where it binds mortalin in vivo in cells undergoing Fas-induced apoptosis | [44] |
| Protein Dj-1 | Predominantly cytoplasmic, nucleus, and mitochondria | Dj-1 protects cells against oxidative stress and cell death.Associated with Parkinson’s Disease. | [45,46,47] |
| Fibroblast growth factor 1 (FGF-1) | Nucleus, cytoplasm, cytosol, and cytoplasmic vesicles | FGF-1 is involved in the regulation of cell proliferation, differentiation, and migration. | [35,48] |
| 94 kDa glucose-regulated protein (GRP94), tumor rejection antigen 1 | Endoplasmic reticulum (ER) | GRP94 is a molecular chaperone that functions in the processing and transport of secreted proteins. Functions in ER-associated protein degradation. | [49] |
| Heat shock protein 60 kDa (Hsp60) | Mitochondrial matrix | Hsp60 is implicated in mitochondrial protein import and macromolecular assembly, including facilitating proper folding of mitochondrial imported proteins. May also prevent protein misfolding and promote the refolding and proper assembly of unfolded polypeptides generated under stress conditions in the mitochondrial matrix. | [9] |
| Hyaluronan-mediated motility receptor (RHAMM) | Centrosomes and microtubules, cytoplasmic | Involved in cell motility. When hyaluronan binds to HMMR, the phosphorylation of a number of proteins occurs. May also be involved in cellular transformation and metastasis formation, and in regulating extracellular-regulated kinase (ERK) activity. | [50] |
| Interleukin-1 (IL-1)-α receptor | Secreted | Major proinflammatory cytokine mediating local and systemic responses of the immune system.An important protein during neuroinflammation and neurodegeneration. | [36] |
| Diphosphomevalonate decarboxylase (MVD1); previously known as MPD | Cytosol | MVD1 is involved in cholesterol biosynthesis, providing prenyl groups required for protein prenylation. | [51] |
| p53 | Cytosol, mitocondria | p53 is a tumor suppressor protein; it participates in apoptosis and genomic stability. | [23,52] |
| SHC-transforming protein 1 - p66 isoform, p66Shc | mitochondrion | The 66 kDa isoform of the SHC-transforming protein regulates lifespan in mammals, and is a critical component of the apoptotic response to oxidative stress. | [53,54] |
| NADH dehydrogenase | Mitochondrial inner membrane. | Core subunit of the mitochondrial membrane respiratory chain. NADH dehydrogenase - complex I, functions in the transfer of electrons from NADH to the respiratory chain. | [2] |
| E3 ubiquitin-protein ligase, Parkin | Mainly cytosolic, nucleus, ER, and mitochondria. | Parkin is involved in the regulation of mitochondrial morphology, antagonizing oxidative damage to mtDNA and activating mitochondrial self-repair mechanisms. | [15,55] |
| Tid1 (DnaJ (Hsp40) homolog, subfamily A, member 3) | Mitochondrial matrix | Nucleotide exchange factor.Heat shock protein co-chaperone. | [14,56] |
| TNF receptor-associated protein (TRAP-1) | Mitochondrial matrix | Chaperone, preserves mitochondrial membrane potential, maintains ATP levels and cell viability during stress. | [57] |
| Voltage-dependent anion-selective channel (VDAC) | Mitochondrial outer membrane, cell membrane | Participates in energy metabolism, mitochondrial homeostasis, and apoptosis. It also may participate in the formation of the permeability transition pore complex (PTPC) responsible for the release of mitochondrial products that triggers apoptosis. | [58] |
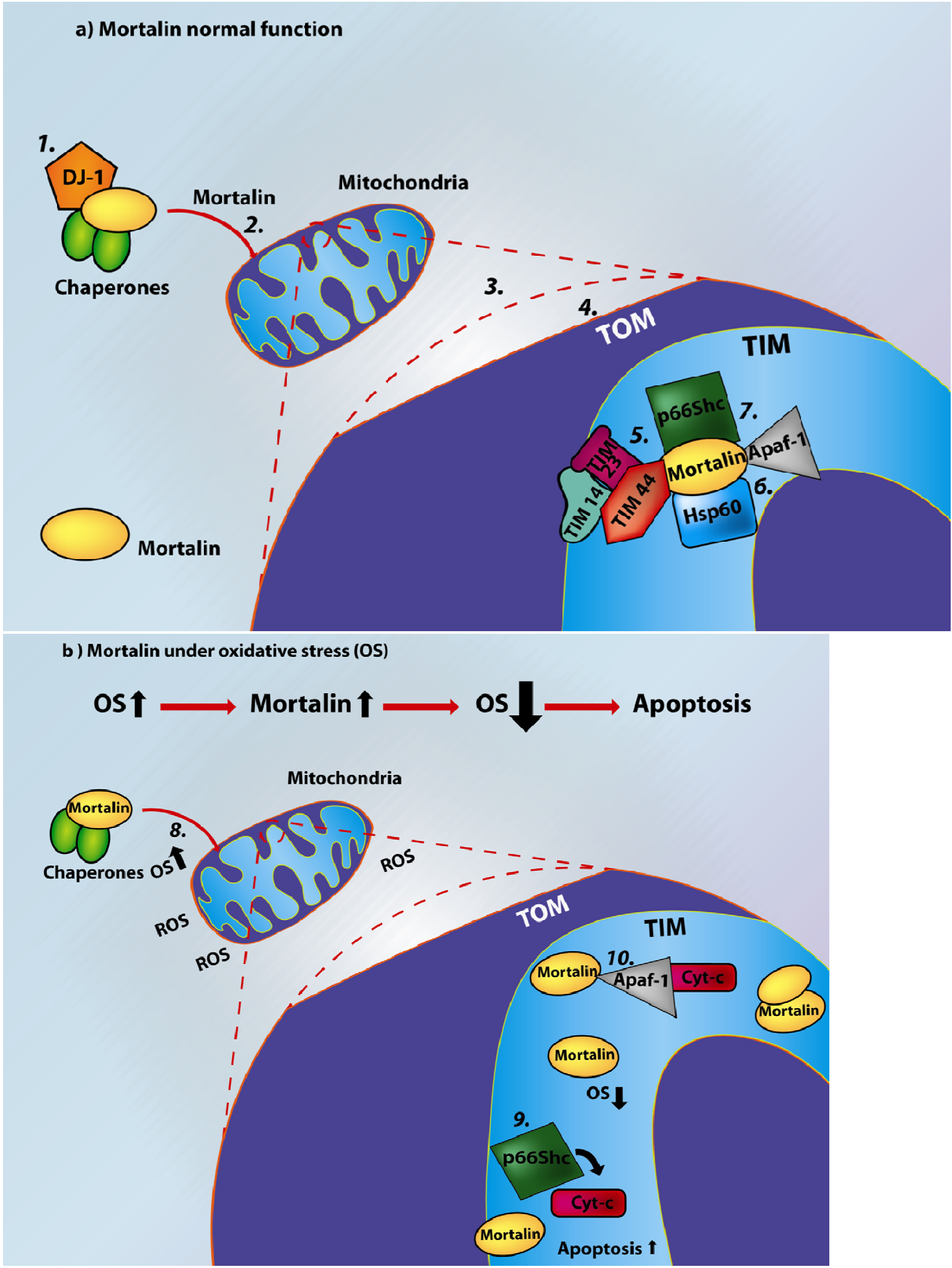
1.2. Mortalin and Mitochondrial Function
- Tim14 (Pam18 or DNAJC19), a J-domain protein, stimulates mortalin’s ATPase activity [74].
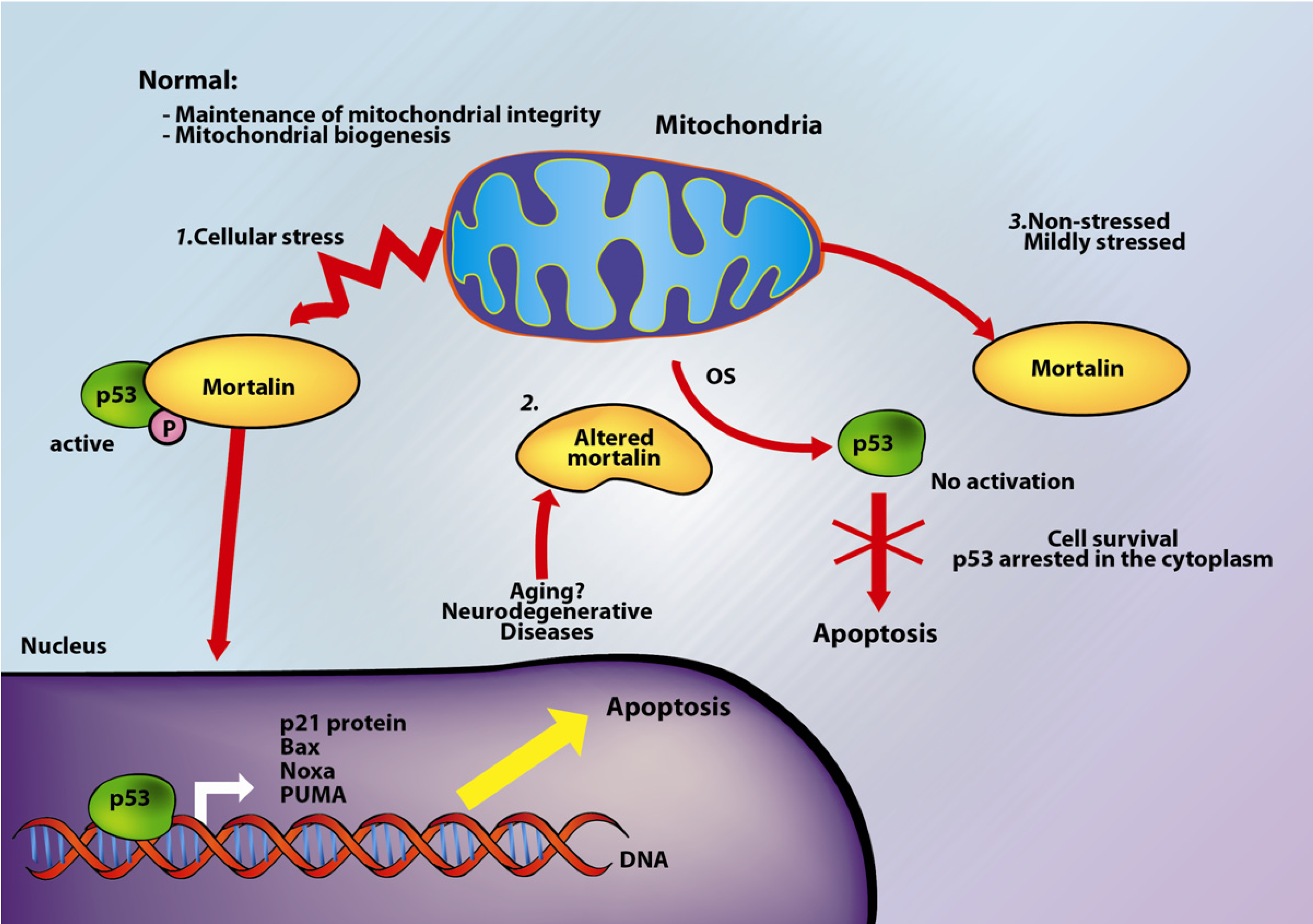
2. Mortalin and Apoptosis

3. Mortalin and Neurodegeneration

4. Mortalin, Apoptosis and Neurodegeneration

5. Concluding Remarks
Acknowledgement
References
- Wadhwa, R.; Kaul, S.C.; Ikawa, Y.; Sugimoto, Y. Identification of a novel member of mouse hsp70 family. Its association with cellular mortal phenotype. J. Biol. Chem. 1993, 268, 6615–6621. [Google Scholar]
- Bhattacharyya, T.; Karnezis, A.N.; Murphy, S.P.; Hoang, T.; Freeman, B.C.; Phillips, B.; Morimoto, R.I. Cloning and subcellular localization of human mitochondrial hsp70. J. Biol. Chem. 1995, 270, 1705–1710. [Google Scholar]
- Domanico, S.Z.; DeNagel, D.C.; Dahlseid, J.N.; Green, J.M.; Pierce, S.K. Cloning of the gene encoding peptide-binding protein 74 shows that it is a new member of the heat shock protein 70 family. Mol. Cell. Biol. 1993, 13, 3598–3610. [Google Scholar]
- Webster, T.J.; Naylor, D.J.; Hartman, D.J.; Hoj, P.B.; Hoogenraad, N.J. cDNA cloning and efficient mitochondrial import of pre-mtHSP70 from rat liver. DNA Cell Biol. 1994, 13, 1213–1220. [Google Scholar]
- Deocaris, C.C.; Kaul, S.C.; Wadhwa, R. On the brotherhood of the mitochondrial chaperones mortalin and heat shock protein 60. Cell Stress Chaperones 2006, 11, 116–128. [Google Scholar]
- Czarnecka, A.M.; Campanella, C.; Zummo, G.; Cappello, F. Mitochondrial chaperones in cancer: From molecular biology to clinical diagnostics. Cancer Biol. Ther. 2006, 5, 714–720. [Google Scholar]
- Dundas, S.R.; Lawrie, L.C.; Rooney, P.H.; Murray, G.I. Mortalin is over-expressed by colorectal adenocarcinomas and correlates with poor survival. J. Pathol. 2005, 205, 74–81. [Google Scholar]
- Ohashi, M.; Oyanagi, M.; Hatakeyama, K.; Inoue, M.; Kominami, R. The gene encoding PBP74/CSA/motalin-1, a novel mouse hsp70, maps to mouse chromosome 18. Genomics 1995, 30, 406–407. [Google Scholar]
- Wadhwa, R.; Takano, S.; Kaur, K.; Aida, S.; Yaguchi, T.; Kaul, Z.; Hirano, T.; Taira, K.; Kaul, S.C. Identification and characterization of molecular interactions between mortalin/mthsp70 and hsp60. Biochem. J. 2005, 391, 185–190. [Google Scholar]
- Xie, H.; Hu, Z.; Chyna, B.; Horrigan, S.K.; Westbrook, C.A. Human mortalin (HSPA9): A candidate for the myeloid leukemia tumor suppressor gene on 5q31. Leukemia 2000, 14, 2128–2134. [Google Scholar]
- Deocaris, C.C.; Kaul, S.C.; Wadhwa, R. The versatile stress protein mortalin as a chaperone therapeutic agent. Protein Pept. Lett. 2009, 16, 517–529. [Google Scholar]
- Wadhwa, R.; Taira, K.; Kaul, S.C. An Hsp70 family chaperone, mortalin/mthsp70/PBP74/Grp75: What, when, and where? Cell Stress Chaperones 2002, 581, 3702–3710. [Google Scholar]
- Daugaard, M.; Rohde, M.; Jaattela, M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007, 581, 3702–3710. [Google Scholar]
- Iosefson, O.; Sharon, S.; Goloubinoff, P.; Azem, A. Reactivation of protein aggregates by mortalin and Tid1-the human mitochondrial Hsp70 chaperone system. Cell Stress Chaperones 2012, 17, 57–66. [Google Scholar]
- Yang, H.; Zhou, X.; Liu, X.; Yang, L.; Chen, Q.; Zhao, D.; Zuo, J.; Liu, W. Mitochondrial dysfunction induced by knockdown of mortalin is rescued by Parkin. Biochem. Biophys. Res. Commun. 2011, 410, 114–120. [Google Scholar]
- Yokoyama, K.; Fukumoto, K.; Murakami, T.; Harada, S.; Hosono, R.; Wadhwa, R.; Mitsui, Y.; Ohkuma, S. Extended longevity of Caenorhabditis elegans by knocking in extra copies of hsp70F, a homolog of mot-2 (mortalin)/mthsp70/Grp75. FEBS Lett. 2002, 516, 53–57. [Google Scholar]
- Ran, Q.; Wadhwa, R.; Kawai, R.; Kaul, S.C.; Sifers, R.N.; Bick, R.J.; Smith, J.R.; Pereira-Smith, O.M. Extramitochondrial localization of mortalin/mthsp70/PBP74/GRP75. Biochem. Biophys. Res. Commun. 2000, 275, 174–179. [Google Scholar]
- Wadhwa, R.; Ando, H.; Kawasaki, H.; Taira, K.; Kaul, S.C. Targeting mortalin using conventional and RNA-helicase-coupled hammerhead ribozymes. EMBO Rep. 2003, 4, 595–601. [Google Scholar]
- Deocaris, C.C.; Kaul, S.C.; Wadhwa, R. From proliferative to neurological role of an hsp70 stress chaperone, mortalin. Biogerontology 2008, 9, 391–403. [Google Scholar]
- Osorio, C.; Sullivan, P.M.; He, D.N.; Mace, B.E.; Ervin, J.F.; Strittmatter, W.J.; Alzate, O. Mortalin is regulated by APOE in hippocampus of AD patients and by human APOE in TR mice. Neurobiol. Aging 2007, 28, 1853–1862. [Google Scholar]
- DeKroon, R.M.; Osorio, C.; Robinette, J.B.; Mocanu, M.; Winnik, W.M.; Alzate, O. Simultaneous detection of changes in protein expression and oxidative modification as a function of age and APOE genotype. J. Proteome Res. 2011, 10, 1632–1644. [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC. Available online: http://www.pymol.org/ (accessed on 30 October 2011).
- Iosefson, O.; Azem, A. Reconstitution of the mitochondrial Hsp70 (mortalin)-p53 interaction using purified proteins—Identification of additional interacting regions. FEBS Lett. 2010, 584, 1080–1084. [Google Scholar]
- Liu, Y.; Liu, W.; Song, X.D.; Zuo, J. Effect of GRP75/mthsp70/PBP74/mortalin overexpression on intracellular ATP level, mitochondrial membrane potential and ROS accumulation following glucose deprivation in PC12 cells. Mol. Cell. Biochem. 2005, 268, 45–51. [Google Scholar]
- Merrick, B.A.; Walker, V.R.; He, C.; Patterson, R.M.; Selkirk, J.K. Induction of novel Grp75 isoforms by 2-deoxyglucose in human and murine fibroblasts. Cancer Lett. 1997, 119, 185–190. [Google Scholar]
- Resendez, E., Jr.; Attenello, J.W.; Grafsky, A.; Chang, C.S.; Lee, A.S. Calcium ionophore A23187 induces expression of glucose-regulated genes and their heterologous fusion genes. Mol. Cell. Biol. 1985, 5, 1212–1219. [Google Scholar]
- Craig, E.E.; Chesley, A.; Hood, D.A. Thyroid hormone modifies mitochondrial phenotype by increasing protein import without altering degradation. Am. J. Physiol. 1998, 275, C1508–C1515. [Google Scholar]
- Sadekova, S.; Lehnert, S.; Chow, T.Y. Induction of PBP74/mortalin/Grp75, a member of the hsp70 family, by low doses of ionizing radiation: A possible role in induced radioresistance. Int. J. Radiat. Biol. 1997, 72, 653–660. [Google Scholar] [CrossRef]
- Kaul, S.C.; Reddel, R.R.; Sugihara, T.; Mitsui, Y.; Wadhwa, R. Inactivation of p53 and life span extension of human diploid fibroblasts by mot-2. FEBS Lett. 2000, 474, 159–164. [Google Scholar]
- Qu, M.; Zhou, Z.; Xu, S.; Chen, C.; Yu, Z.; Wang, D. Mortalin overexpression attenuates beta-amyloid-induced neurotoxicity in SH-SY5Y cells. Brain Res. 2011, 1368, 336–345. [Google Scholar] [CrossRef]
- Xu, L.; Voloboueva, L.A.; Ouyang, Y.; Emery, J.F.; Giffard, R.G. Overexpression of mitochondrial Hsp70/Hsp75 in rat brain protects mitochondria, reduces oxidative stress, and protects from focal ischemia. J. Cereb. Blood Flow Metab. 2009, 29, 365–374. [Google Scholar]
- Taurin, S.; Seyrantepe, V.; Orlov, S.N.; Tremblay, T.L.; Thibault, P.; Bennett, M.R.; Hamet, P.; Pshezhetsky, A.V. Proteome analysis and functional expression identify mortalin as an antiapoptotic gene induced by elevation of [Na+]i/[K+]i ratio in cultured vascular smooth muscle cells. Circ. Res. 2002, 91, 915–922. [Google Scholar]
- Kaul, S.C.; Yaguchi, T.; Taira, K.; Reddel, R.R.; Wadhwa, R. Overexpressed mortalin (mot-2)/mthsp70/GRP75 and hTERT cooperate to extend the in vitro lifespan of human fibroblasts. Exp. Cell Res. 2003, 286, 96–101. [Google Scholar] [CrossRef]
- Kaul, S.C.; Deocaris, C.C.; Wadhwa, R. Three faces of mortalin: A housekeeper, guardian and killer. Exp. Gerontol. 2007, 42, 263–274. [Google Scholar]
- Mizukoshi, E.; Suzuki, M.; Loupatov, A.; Uruno, T.; Hayashi, H.; Misono, T.; Kaul, S.C.; Wadhwa, R.; Imamura, T. Fibroblast growth factor-1 interacts with the glucose-regulated protein GRP75/mortalin. Biochem. J. 1999, 343, 461–466. [Google Scholar]
- Sacht, G.; Brigelius-Flohe, R.; Kiess, M.; Sztajer, H.; Flohe, L. ATP-sensitive association of mortalin with the IL-1 receptor type I. Biofactors 1999, 9, 49–60. [Google Scholar]
- Pilzer, D.; Fishelson, Z. Mortalin/GRP75 promotes release of membrane vesicles from immune attacked cells and protection from complement-mediated lysis. Int. Immunol. 2005, 17, 1239–1248. [Google Scholar]
- Carette, J.; Lehnert, S.; Chow, T.Y. Implication of PBP74/mortalin/GRP75 in the radio-adaptive response. Int. J. Radiat. Biol. 2002, 78, 183–190. [Google Scholar]
- Ibi, T.; Sahashi, K.; Ling, J.; Marui, K.; Mitsuma, T. Immunostaining of mitochondrial heat shock proteins (mtHSPs) in skeletal muscle fibers of mitochondrial cytopathy. Rinsho Shinkeigaku 1996, 36, 61–64. [Google Scholar]
- Rivolta, M.N.; Holley, M.C. Asymmetric segregation of mitochondria and mortalin correlates with the multi-lineage potential of inner ear sensory cell progenitors in vitro. Brain Res. Dev. Brain Res. 2002, 133, 49–56. [Google Scholar] [CrossRef]
- Kaul, S.C.; Taira, K.; Pereira-Smith, O.M.; Wadhwa, R. Mortalin: Present and prospective. Exp. Gerontol. 2002, 37, 1157–1164. [Google Scholar]
- Wadhwa, R.; Yaguchi, T.; Hasan, M.K.; Mitsui, Y.; Reddel, R.R.; Kaul, S.C. Hsp70 family member, mot-2/mthsp70/GRP75, binds to the cytoplasmic sequestration domain of the p53 protein. Exp. Cell Res. 2002, 274, 246–253. [Google Scholar] [CrossRef]
- Kogel, D.; Schomburg, R.; Copanaki, E.; Prehn, J.H. Regulation of gene expression by the amyloid precursor protein: Inhibition of the JNK/c-Jun pathway. Cell Death Differ. 2005, 12, 1–9. [Google Scholar]
- Feng, Y.; Ariza, M.E.; Goulet, A.C.; Shi, J.; Nelson, M.A. Death-signal-induced relocalization of cyclin-dependent kinase 11 to mitochondria. Biochem. J. 2005, 392, 65–73. [Google Scholar]
- Burbulla, L.F.; Schelling, C.; Kato, H.; Rapaport, D.; Woitalla, D.; Schiesling, C.; Schulte, C.; Sharma, M.; Illig, T.; Bauer, P.; et al. Dissecting the role of the mitochondrial chaperone mortalin in Parkinson’s disease: Functional impact of disease-related variants on mitochondrial homeostasis. Hum. Mol. Genet. 2010, 19, 4437–4452. [Google Scholar]
- Canet-Aviles, R.M.; Wilson, M.A.; Miller, D.W.; Ahmad, R.; McLendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar]
- Jin, J.; Hulette, C.; Wang, Y.; Zhang, T.; Pan, C.; Wadhwa, R.; Zhang, J. Proteomic identification of a stress protein, mortalin/mthsp70/GRP75: Relevance to Parkinson disease. Mol. Cell. Proteomics 2006, 5, 1193–1204. [Google Scholar]
- Mizukoshi, E.; Suzuki, M.; Misono, T.; Loupatov, A.; Munekata, E.; Kaul, S.C.; Wadhwa, R.; Imamura, T. Cell-cycle dependent tyrosine phosphorylation on mortalin regulates its interaction with fibroblast growth factor-1. Biochem. Biophys. Res. Commun. 2001, 280, 1203–1209. [Google Scholar]
- Takano, S.; Wadhwa, R.; Mitsui, Y.; Kaul, S.C. Identification and characterization of molecular interactions between glucose-regulated proteins (GRPs) mortalin/GRP75/peptide-binding protein 74 (PBP74) and GRP94. Biochem. J. 2001, 357, 393–398. [Google Scholar]
- Kuwabara, H.; Yoneda, M.; Hayasaki, H.; Nakamura, T.; Mori, H. Glucose regulated proteins 78 and 75 bind to the receptor for hyaluronan mediated motility in interphase microtubules. Biochem. Biophys. Res. Commun. 2006, 339, 971–976. [Google Scholar]
- Wadhwa, R.; Yaguchi, T.; Hasan, M.K.; Taira, K.; Kaul, S.C. Mortalin-MPD (mevalonate pyrophosphate decarboxylase) interactions and their role in control of cellular proliferation. Biochem. Biophys. Res. Commun. 2003, 302, 735–742. [Google Scholar]
- Wadhwa, R.; Takano, S.; Robert, M.; Yoshida, A.; Nomura, H.; Reddel, R.R.; Mitsui, Y.; Kaul, S.C. Inactivation of tumor suppressor p53 by mot-2, a hsp70 family member. J. Biol. Chem. 1998, 273, 29586–29591. [Google Scholar]
- Orsini, F.; Migliaccio, E.; Moroni, M.; Contursi, C.; Raker, V.A.; Piccini, D.; Martin-Padura, I.; Pelliccia, G.; Trinei, M.; Bono, M.; et al. The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. J. Biol. Chem. 2004, 279, 25689–25695. [Google Scholar]
- Pellegrini, M.; Pacini, S.; Baldari, C.T. p66SHC: The apoptotic side of Shc proteins. Apoptosis 2005, 10, 13–18. [Google Scholar]
- Grunewald, A.; Voges, L.; Rakovic, A.; Kasten, M.; Vandebona, H.; Hemmelmann, C.; Lohmann, K.; Orolicki, S.; Ramirez, A.; Schapira, A.H.; et al. Mutant Parkin impairs mitochondrial function and morphology in human fibroblasts. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Goswami, A.V.; Chittoor, B.; D’Silva, P. Understanding the functional interplay between mammalian mitochondrial Hsp70 chaperone machine components. J. Biol. Chem. 2010, 285, 19472–19482. [Google Scholar]
- Voloboueva, L.A.; Duan, M.; Ouyang, Y.; Emery, J.F.; Stoy, C.; Giffard, R.G. Overexpression of mitochondrial Hsp70/Hsp75 protects astrocytes against ischemic injury in vitro. J. Cereb. Blood Flow Metab. 2008, 28, 1009–1016. [Google Scholar] [CrossRef]
- Schwarzer, C.; Barnikol-Watanabe, S.; Thinnes, F.P.; Hilschmann, N. Voltage-dependent anion-selective channel (VDAC) interacts with the dynein light chain Tctex1 and the heat-shock protein PBP74. Int. J. Biochem. Cell Biol. 2002, 34, 1059–1070. [Google Scholar]
- Qu, M.; Zhou, Z.; Chen, C.; Li, M.; Pei, L.; Yang, J.; Wang, Y.; Li, L.; Liu, C.; Zhang, G.; et al. Inhibition of mitochondrial permeability transition pore opening is involved in the protective effects of mortalin overexpression against beta-amyloid-induced apoptosis in SH-SY5Y cells. Neurosci. Res. 2012, 72, 94–102. [Google Scholar] [CrossRef]
- Ornatsky, O.I.; Connor, M.K.; Hood, D.A. Expression of stress proteins and mitochondrial chaperonins in chronically stimulated skeletal muscle. Biochem. J. 1995, 311, 119–123. [Google Scholar]
- Takahashi, M.; Chesley, A.; Freyssenet, D.; Hood, D.A. Contractile activity-induced adaptations in the mitochondrial protein import system. Am. J. Physiol. 1998, 274, C1380–C1387. [Google Scholar]
- Brunner, M.; Schneider, H.C.; Lill, R.; Neupert, W. Dissection of protein translocation across the mitochondrial outer and inner membranes. Cold Spring Harb. Symp. Quant. Biol. 1995, 60, 619–627. [Google Scholar]
- Schneider, H.C.; Berthold, J.; Bauer, M.F.; Dietmeier, K.; Guiard, B.; Brunner, M.; Neupert, W. Mitochondrial Hsp70/MIM44 complex facilitates protein import. Nature 1994, 371, 768–774. [Google Scholar]
- Voos, W. Mitochondrial protein homeostasis: The cooperative roles of chaperones and proteases. Res. Microbiol. 2009, 160, 718–725. [Google Scholar]
- Voos, W.; Martin, H.; Krimmer, T.; Pfanner, N. Mechanisms of protein translocation into mitochondria. Biochim. Biophys. Acta 1999, 1422, 235–254. [Google Scholar]
- Harner, M.; Neupert, W.; Deponte, M. Lateral release of proteins from the TOM complex into the outer membrane of mitochondria. EMBO J. 2011, 30, 3232–3241. [Google Scholar]
- Voos, W.; Rottgers, K. Molecular chaperones as essential mediators of mitochondrial biogenesis. Biochim. Biophys. Acta 2002, 1592, 51–62. [Google Scholar] [CrossRef]
- Lim, J.H.; Martin, F.; Guiard, B.; Pfanner, N.; Voos, W. The mitochondrial Hsp70-dependent import system actively unfolds preproteins and shortens the lag phase of translocation. EMBO J. 2001, 20, 941–950. [Google Scholar]
- Marom, M.; Azem, A.; Mokranjac, D. Understanding the molecular mechanism of protein translocation across the mitochondrial inner membrane: Still a long way to go. Biochim. Biophys. Acta 2011, 1808, 990–1001. [Google Scholar] [CrossRef]
- Neupert, W.; Brunner, M. The protein import motor of mitochondria. Nat. Rev. Mol. Cell. Biol. 2002, 3, 555–565. [Google Scholar]
- Scherer, P.E.; Manning-Krieg, U.C.; Jeno, P.; Schatz, G.; Horst, M. Identification of a 45-kDa protein at the protein import site of the yeast mitochondrial inner membrane. Proc. Natl. Acad. Sci. USA 1992, 89, 11930–11934. [Google Scholar]
- D’Silva, P.; Liu, Q.; Walter, W.; Craig, E.A. Regulated interactions of mtHsp70 with Tim44 at the translocon in the mitochondrial inner membrane. Nat. Struct. Mol. Biol. 2004, 11, 1084–1091. [Google Scholar]
- Elsner, S.; Simian, D.; Iosefson, O.; Marom, M.; Azem, A. The mitochondrial protein translocation motor: Structural conservation between the human and yeast Tim14/Pam18-Tim16/Pam16 co-chaperones. Int. J. Mol. Sci. 2009, 10, 2041–2053. [Google Scholar]
- Mokranjac, D.; Sichting, M.; Neupert, W.; Hell, K. Tim14, a novel key component of the import motor of the TIM23 protein translocase of mitochondria. EMBO J. 2003, 22, 4945–4956. [Google Scholar]
- Sastry, P.S.; Rao, K.S. Apoptosis and the nervous system. J. Neurochem. 2000, 74, 1–20. [Google Scholar]
- Reed, T.T.; Sultana, R.; Butterfield, D.A. Redox Proteomics of Oxidatively Modified Brain Proteins in Mild Cognitive Impairmen. In Neuroproteomics; Alzate, O., Ed.; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Kimura, K.; Tanaka, N.; Nakamura, N.; Takano, S.; Ohkuma, S. Knockdown of mitochondrial heat shock protein 70 promotes progeria-like phenotypes in caenorhabditis elegans. J. Biol. Chem. 2007, 282, 5910–5918. [Google Scholar]
- Chipuk, J.E.; Green, D.R. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008, 18, 157–164. [Google Scholar] [CrossRef]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar]
- Strasser, A.; Cory, S.; Adams, J.M. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011, 30, 3667–3683. [Google Scholar]
- Deshmukh, M.; Johnson, E.M., Jr. Staurosporine-induced neuronal death: Multiple mechanisms and methodological implications. Cell Death Differ. 2000, 7, 250–261. [Google Scholar]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar]
- Woo, M.; Hakem, R.; Mak, T.W. Executionary pathway for apoptosis: Lessons from mutant mice. Cell Res. 2000, 10, 267–278. [Google Scholar]
- Bratton, S.B.; Salvesen, G.S. Regulation of the Apaf-1-caspase-9 apoptosome. J. Cell Sci. 2010, 123, 3209–3214. [Google Scholar]
- Chandra, J.; Samali, A.; Orrenius, S. Triggering and modulation of apoptosis by oxidative stress. Free Radic. Biol. Med. 2000, 29, 323–333. [Google Scholar]
- Llambi, F.; Green, D.R. Apoptosis and oncogenesis: Give and take in the BCL-2 family. Curr. Opin. Genet. Dev. 2011, 21, 12–20. [Google Scholar]
- Llambi, F.; Moldoveanu, T.; Tait, S.W.; Bouchier-Hayes, L.; Temirov, J.; McCormick, L.L.; Dillon, C.P.; Green, D.R. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol. Cell 2011, 44, 517–531. [Google Scholar]
- Riedl, S.J.; Li, W.; Chao, Y.; Schwarzenbacher, R.; Shi, Y. Structure of the apoptotic protease-activating factor 1 bound to ADP. Nature 2005, 434, 926–933. [Google Scholar]
- Zou, H.; Henzel, W.J.; Liu, X.; Lutschg, A.; Wang, X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 1997, 90, 405–413. [Google Scholar] [CrossRef]
- Beere, H.M.; Wolf, B.B.; Cain, K.; Mosser, D.D.; Mahboubi, A.; Kuwana, T.; Tailor, P.; Morimoto, R.I.; Cohen, G.M.; Green, D.R. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat. Cell Biol. 2000, 2, 469–475. [Google Scholar]
- Ott, M.; Robertson, J.D.; Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA 2002, 99, 1259–1263. [Google Scholar]
- Lu, W.J.; Lee, N.P.; Kaul, S.C.; Lan, F.; Poon, R.T.; Wadhwa, R.; Luk, J.M. Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ. 2011, 18, 1046–1056. [Google Scholar]
- Zhang, H.; Reed, J.C. Studies of apoptosis proteins in yeast. Methods Cell Biol. 2001, 66, 453–468. [Google Scholar]
- Liu, S.; Li, J.; Tao, Y.; Xiao, X. Small heat shock protein alphaB-crystallin binds to p53 to sequester its translocation to mitochondria during hydrogen peroxide-induced apoptosis. Biochem. Biophys. Res. Commun. 2007, 354, 109–114. [Google Scholar]
- Nikolaev, A.Y.; Li, M.; Puskas, N.; Qin, J.; Gu, W. Parc: A cytoplasmic anchor for p53. Cell 2003, 112, 29–40. [Google Scholar]
- Zhao, L.Y.; Liao, D. Sequestration of p53 in the cytoplasm by adenovirus type 12 E1B 55-kilodalton oncoprotein is required for inhibition of p53-mediated apoptosis. J. Virol. 2003, 77, 13171–13181. [Google Scholar]
- Zilfou, J.T.; Lowe, S.W. Tumor suppressive functions of p53. Cold Spring Harb. Perspect. Biol. 2009. [Google Scholar] [CrossRef]
- Deocaris, C.C.; Widodo, N.; Ishii, T.; Kaul, S.C.; Wadhwa, R. Functional significance of minor structural and expression changes in stress chaperone mortalin. Ann. N. Y. Acad. Sci. 2007, 1119, 165–175. [Google Scholar]
- Kaul, S.C.; Aida, S.; Yaguchi, T.; Kaur, K.; Wadhwa, R. Activation of wild type p53 function by its mortalin-binding, cytoplasmically localizing carboxyl terminus peptides. J. Biol. Chem. 2005, 280, 39373–39379. [Google Scholar]
- Liu, B.; Chen, Y.; St Clair, D.K. ROS and p53: A versatile partnership. Free Radic. Biol. Med. 2008, 44, 1529–1535. [Google Scholar]
- Soti, C.; Csermely, P. Chaperones and aging: Role in neurodegeneration and in other civilizational diseases. Neurochem. Int. 2002, 41, 383–389. [Google Scholar]
- Berlett, B.S.; Stadtman, E.R. Protein oxidation in aging, disease, and oxidative stres. J. Biol. Chem. 1997, 272, 20313–20316. [Google Scholar]
- Choi, J.; Forster, M.J.; McDonald, S.R.; Weintraub, S.T.; Carroll, C.A.; Gracy, R.W. Proteomic identification of specific oxidized proteins in ApoE-knockout mice: Relevance to Alzheimer’s disease. Free Radic. Biol. Med. 2004, 36, 1155–1162. [Google Scholar]
- Yaguchi, T.; Aida, S.; Kaul, S.C.; Wadhwa, R. Involvement of mortalin in cellular senescence from the perspective of its mitochondrial import, chaperone, and oxidative stress management function. Ann. N. Y. Acad. Sci. 2007, 1100, 306–311. [Google Scholar] [CrossRef]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5644–5651. [Google Scholar]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J.; et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993, 43, 1467–1472. [Google Scholar]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar]
- Strittmatter, W.J.; Weisgraber, K.H.; Huang, D.Y.; Dong, L.M.; Salvesen, G.S.; Pericak-Vance, M.; Schmechel, D.; Saunders, A.M.; Goldgaber, D.; Roses, A.D. Binding of human apolipoprotein E to synthetic amyloid beta peptide: Isoform-specific effects and implications for late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 8098–8102. [Google Scholar]
- Beal, M.F. Aging, energy, and oxidative stress in neurodegenerative disease. Ann. Neurol. 1995, 38, 357–366. [Google Scholar]
- McNaught, K.S.; Olanow, C.W.; Halliwell, B.; Isacson, O.; Jenner, P. Failure of the ubiquitin-proteasome system in Parkinson’s disease. Nat. Rev. Neurosci. 2001, 2, 589–594. [Google Scholar]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell. Biol. 2011, 12, 9–14. [Google Scholar]
- Samii, A.; Nutt, J.G.; Ransom, B.R. Parkinson’s disease. Lancet 2004, 363, 1783–1793. [Google Scholar]
- Van Laar, V.S.; Dukes, A.A.; Cascio, M.; Hastings, T.G. Proteomic analysis of rat brain mitochondria following exposure to dopamine quinone: Implications for Parkinson disease. Neurobiol. Dis. 2008, 29, 477–489. [Google Scholar]
- Tieu, K.; Ischiropoulos, H.; Przedborski, S. Nitric oxide and reactive oxygen species in Parkinson’s disease. IUBMB Life 2003, 55, 329–335. [Google Scholar]
- Geissler, A.; Rassow, J.; Pfanner, N.; Voos, W. Mitochondrial import driving forces: Enhanced trapping by matrix Hsp70 stimulates translocation and reduces the membrane potential dependence of loosely folded preproteins. Mol. Cell. Biol. 2001, 21, 7097–7104. [Google Scholar]
- Narendra, D.; Kane, L.A.; Hauser, D.N.; Fearnley, I.M.; Youle, R.J. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010, 6, 1090–1106. [Google Scholar] [CrossRef]
- Malgieri, G.; Eliezer, D. Structural effects of Parkinson’s disease linked DJ-1 mutations. Protein Sci. 2008, 17, 855–868. [Google Scholar]
- Li, H.M.; Niki, T.; Taira, T.; Iguchi-Ariga, S.M.; Ariga, H. Association of DJ-1 with chaperones and enhanced association and colocalization with mitochondrial Hsp70 by oxidative stress. Free Radic. Res. 2005, 39, 1091–1099. [Google Scholar]
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell. Biol. 2000, 1, 120–129. [Google Scholar]
- Oppenheim, R.W. Cell death during development of the nervous system. Annu. Rev. Neurosci. 1991, 14, 453–501. [Google Scholar]
- Mattson, M.P. Metal-catalyzed disruption of membrane protein and lipid signaling in the pathogenesis of neurodegenerative disorders. Ann. N. Y. Acad. Sci. 2004, 1012, 37–50. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Londono, C.; Osorio, C.; Gama, V.; Alzate, O. Mortalin, Apoptosis, and Neurodegeneration. Biomolecules 2012, 2, 143-164. https://doi.org/10.3390/biom2010143
Londono C, Osorio C, Gama V, Alzate O. Mortalin, Apoptosis, and Neurodegeneration. Biomolecules. 2012; 2(1):143-164. https://doi.org/10.3390/biom2010143
Chicago/Turabian StyleLondono, Carolina, Cristina Osorio, Vivian Gama, and Oscar Alzate. 2012. "Mortalin, Apoptosis, and Neurodegeneration" Biomolecules 2, no. 1: 143-164. https://doi.org/10.3390/biom2010143
APA StyleLondono, C., Osorio, C., Gama, V., & Alzate, O. (2012). Mortalin, Apoptosis, and Neurodegeneration. Biomolecules, 2(1), 143-164. https://doi.org/10.3390/biom2010143