
Transcription Blockage Leads to New Beginnings

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Transcription Elongation—What is Blocking the Path?

2.1. DNA Damage on the Track
2.2. Transcription Meets Replication
2.3. Why are Some Genes so Long?
3. Recovery of RNA Synthesis
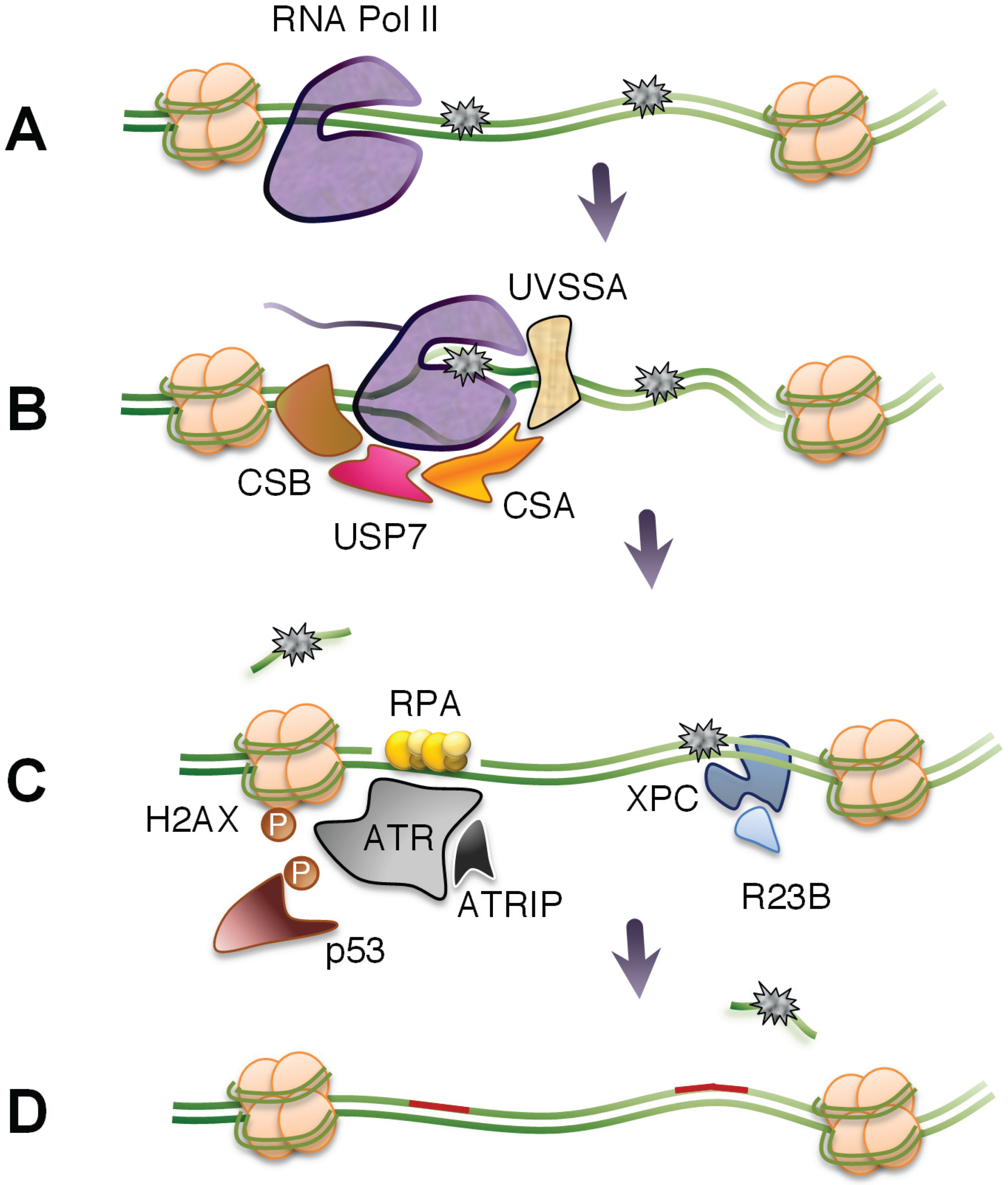
3.1. TC-NER
3.2. Assessment of DNA Repair Genome-Wide
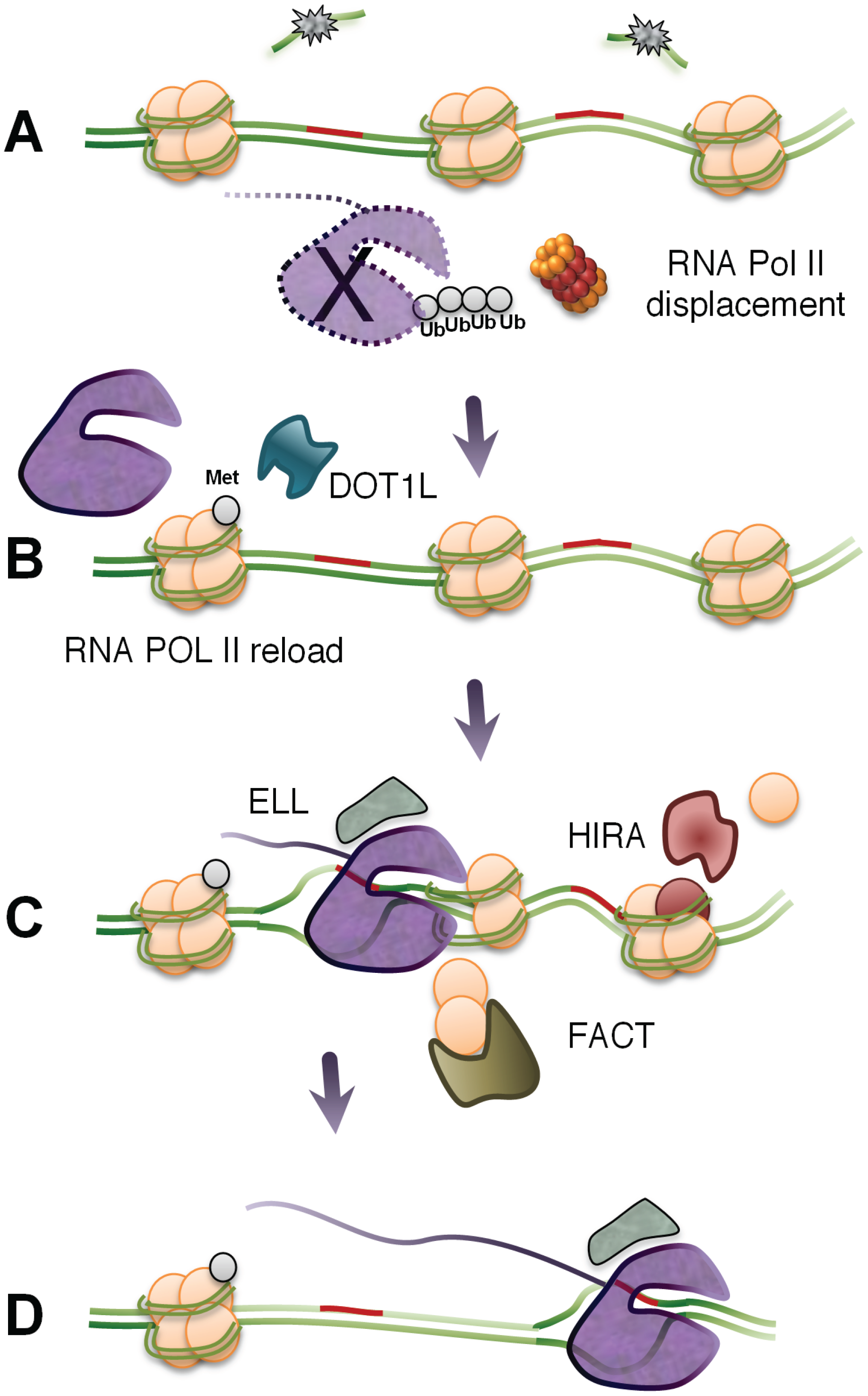
3.3. The Fate of Stalled RNA Polymerases

3.4. Factors Promoting the Recovery of RNA Synthesis

3.5. Resumption or Restart of Transcription?
4. Conclusions
Acknowledgements
Conflict of Interest
References
- Voss, T.C.; Hager, G.L. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat. Rev. Genet. 2014, 15, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Muse, G.W.; Gilchrist, D.A.; Nechaev, S.; Shah, R.; Parker, J.S.; Grissom, S.F.; Zeitlinger, J.; Adelman, K. RNA polymerase is poised for activation across the genome. Nat. Genet. 2007, 39, 1507–1511. [Google Scholar] [CrossRef] [PubMed]
- Adelman, K.; Lis, J.T. Promoter-proximal pausing of RNA polymerase II: Emerging roles in metazoans. Nat. Rev. Genet. 2012, 13, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Veloso, A.; Kirkconnell, K.S.; Magnuson, B.; Biewen, B.; Paulsen, M.T.; Wilson, T.E.; Ljungman, M. Rate of elongation by RNA polymerase II is associated with specific gene features and epigenetic modifications. Genome Res. 2014, 24, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Kuehner, J.N.; Pearson, E.L.; Moore, C. Unravelling the means to an end: RNA polymerase II transcription termination. Nat. Rev. Mol. Cell Biol. 2011, 12, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Maniatis, T.; Reed, R. An extensive network of coupling among gene expression machines. Nature 2002, 416, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Schoenberg, D.R.; Maquat, L.E. Regulation of cytoplasmic mrna decay. Nat. Rev. Genet. 2012, 13, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Gudipati, R.K.; Xu, Z.; Lebreton, A.; Seraphin, B.; Steinmetz, L.M.; Jacquier, A.; Libri, D. Extensive degradation of RNA precursors by the exosome in wild-type cells. Mol. Cell 2012, 48, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Tornaletti, S. Transcription arrest at DNA damage sites. Mutat. Res. 2005, 577, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Khobta, A.; Epe, B. Interactions between DNA damage, repair, and transcription. Mutat. Res. 2012, 736, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Lima, L.C.; Veloso, A.; Paulsen, M.T.; Menck, C.F.; Ljungman, M. DNA repair and recovery of RNA synthesis following exposure to ultraviolet light are delayed in long genes. Nucleic Acids Res. 2015, 43, 2744–2756. [Google Scholar] [CrossRef] [PubMed]
- Veloso, A.; Biewen, B.; Paulsen, M.T.; Berg, N.; Carmo de Andrade Lima, L.; Prasad, J.; Bedi, K.; Magnuson, B.; Wilson, T.E.; Ljungman, M. Genome-wide transcriptional effects of the anti-cancer agent camptothecin. PLoS ONE 2013, 8, e78190. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Ryan, M.C.; Martin, S.E.; Varma, S.; Kohn, K.W.; Liu, H.; Zeeberg, B.R.; Pommier, Y. Transcription poisoning by topoisomerase I is controlled by gene length, splice sites, and miR-142-3p. Cancer Res. 2013, 73, 4830–4839. [Google Scholar] [CrossRef] [PubMed]
- King, I.F.; Yandava, C.N.; Mabb, A.M.; Hsiao, J.S.; Huang, H.S.; Pearson, B.L.; Calabrese, J.M.; Starmer, J.; Parker, J.S.; Magnuson, T.; et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature 2013, 501, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, M.; Zhang, F. Blockage of RNA polymerase as a possible trigger for UV Light-induced apoptosis. Oncogene 1996, 13, 823–831. [Google Scholar] [PubMed]
- Ljungman, M.; Zhang, F.F.; Chen, F.; Rainbow, A.J.; McKay, B.C. Inhibition of RNA polymerase II as a trigger for the p53 response. Oncogene 1999, 18, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, M.; Lane, D.P. Transcription-guarding the genome by sensing DNA damage. Nat. Rev. Cancer 2004, 4, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Derheimer, F.A.; O'Hagan H, M.; Krueger, H.M.; Hanasoge, S.; Paulsen, M.T.; Ljungman, M. RPA and ATR link transcriptional stress to p53. Proc. Natl. Acad. Sci. USA 2007, 104, 12778–12783. [Google Scholar] [CrossRef] [PubMed]
- Sordet, O.; Redon, C.E.; Guirouilh-Barbat, J.; Smith, S.; Solier, S.; Douarre, C.; Conti, C.; Nakamura, A.J.; Das, B.B.; Nicolas, E.; et al. Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. EMBO Rep. 2009, 10, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Tresini, M.; Warmerdam, D.O.; Kolovos, P.; Snijder, L.; Vrouwe, M.G.; Demmers, J.A.; van IJcken, I.W.F.J.; Grosveld, F.G.; Medema, R.H.; Hoeijmakers, J.H.; et al. The core spliceosome as target and effector of non-canonical atm signalling. Nature 2015, 523, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Mellon, I.; Spivak, G.; Hanawalt, P.C. Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell 1987, 51, 241–249. [Google Scholar] [CrossRef]
- Derheimer, F.A.; Chang, C.W.; Ljungman, M. Transcription inhibition: A potential strategy for cancer therapeutics. Eur. J. Cancer 2005, 41, 2569–2576. [Google Scholar] [CrossRef] [PubMed]
- Jonkers, I.; Lis, J.T. Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Padgett, R.A. Rates of in situ transcription and splicing in large human genes. Nat. Struct. Mol. Biol. 2009, 16, 1128–1133. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, G.; Voichek, Y.; Benjamin, S.; Gilad, S.; Amit, I.; Oren, M. 4sudrb-seq: Measuring genomewide transcriptional elongation rates and initiation frequencies within cells. Genome Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Jonkers, I.; Kwak, H.; Lis, J.T. Genome-wide dynamics of pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. eLife 2014. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.; Fuda, N.J.; Core, L.J.; Lis, J.T. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 2013, 339, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Nojima, T.; Gomes, T.; Grosso, A.R.; Kimura, H.; Dye, M.J.; Dhir, S.; Carmo-Fonseca, M.; Proudfoot, N.J. Mammalian net-seq reveals genome-wide nascent transcription coupled to RNA processing. Cell 2015, 161, 526–540. [Google Scholar] [CrossRef] [PubMed]
- Ditlevson, J.V.; Tornaletti, S.; BelotserkovskII, B.P.; Teijeiro, V.; Wang, G.; Vasquez, K.M.; Hanawalt, P.C. Inhibitory effect of a short z-DNA forming sequence on transcription elongation by T7 RNA polymerase. Nucleic Acids Res. 2008, 36, 3163–3170. [Google Scholar] [CrossRef] [PubMed]
- Salinas-Rios, V.; BelotserkovskII, B.P.; Hanawalt, P.C. DNA slip-outs cause RNA polymerase II arrest in vitro: Potential implications for genetic instability. Nucleic Acids Res. 2011, 39, 7444–7454. [Google Scholar] [CrossRef] [PubMed]
- Tornaletti, S.; Park-Snyder, S.; Hanawalt, P.C. G4-forming sequences in the non-transcribed DNA strand pose blocks to T7 RNA polymerase and mammalian RNA polymerase II. J. Biol. Chem. 2008, 283, 12756–12762. [Google Scholar] [CrossRef] [PubMed]
- BelotserkovskII, B.P.; Liu, R.; Tornaletti, S.; Krasilnikova, M.M.; Mirkin, S.M.; Hanawalt, P.C. Mechanisms and implications of transcription blockage by guanine-rich DNA sequences. Proc. Natl. Acad. Sci. USA 2010, 107, 12816–12821. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.T.; Vallur, A.C.; Eddy, J.; Maizels, N. G quadruplexes are genomewide targets of transcriptional helicases XPB and XPD. Nat. Chem. Biol. 2014, 10, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.N.; Fox, D., 3rd; Guo, R.; Enomoto, T.; Wang, W. RecQL5 promotes genome stabilization through two parallel mechanisms—Interacting with RNA polymerase II and acting as a helicase. Mol. Cell. Biol. 2010, 30, 2460–2472. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, M.; Kantidakis, T.; Mitter, R.; Kelly, G.P.; Heron, M.; Williams, H.; Soding, J.; Stewart, A.; Svejstrup, J.Q. RecQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell 2014, 157, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A. The connection between transcription and genomic instability. EMBO J. 2002, 21, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Jinks-Robertson, S. Transcription as a source of genome instability. Nat. Rev. Genet. 2012, 13, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; Herrera-Moyano, E.; Aguilera, A. Transcription-associated genome instability. Chem. Rev. 2013, 113, 8638–8661. [Google Scholar] [CrossRef] [PubMed]
- Gottipati, P.; Helleday, T. Transcription-associated recombination in eukaryotes: Link between transcription, replication and recombination. Mutagenesis 2009. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C. A brief history of the DNA repair field. Cell Res. 2008, 18, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Masters, M.; Pardee, A.B. Failure of ultraviolet-irradiated Escherichia coli to produce a cross-reacting protein. Biochim. Biophys. Acta 1962, 56, 609–611. [Google Scholar] [CrossRef]
- Brueckner, F.; Hennecke, U.; Carell, T.; Cramer, P. CPD damage recognition by transcribing RNA polymerase II. Science 2007, 315, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Damsma, G.E.; Alt, A.; Brueckner, F.; Carell, T.; Cramer, P. Mechanism of transcriptional stalling at cisplatin-damaged DNA. Nat. Struct. Mol. Biol. 2007, 14, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhu, G.; Huang, X.; Lippard, S.J. X-ray structure and mechanism of RNA polymerase II stalled at an antineoplastic monofunctional platinum-DNA adduct. Proc. Natl. Acad. Sci. USA 2010, 17, 9584–9589. [Google Scholar] [CrossRef] [PubMed]
- Kellinger, M.W.; Park, G.Y.; Chong, J.; Lippard, S.J.; Wang, D. Effect of a monofunctional phenanthriplatin-DNA adduct on RNA polymerase II transcriptional fidelity and translesion synthesis. J. Am. Chem. Soc. 2013, 135, 13054–13061. [Google Scholar] [CrossRef] [PubMed]
- Schinecker, T.M.; Perlow, R.A.; Broyde, S.; Geacintov, N.E.; Scicchitano, D.A. Human RNA polymerase II is partially blocked by DNA adducts derived from tumorigenic benzo[c]phenanthrene diol epoxides: Relating biological consequences to conformational preferences. Nucleic Acids Res. 2003, 31, 6004–6015. [Google Scholar] [CrossRef] [PubMed]
- You, C.; Wang, J.; Dai, X.; Wang, Y. Transcriptional inhibition and mutagenesis induced by N-nitroso compound-derived carboxymethylated thymidine adducts in DNA. Nucleic Acids Res. 2015, 43, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Cline, S.D.; Riggins, J.N.; Tornaletti, S.; Marnett, L.J.; Hanawalt, P.C. Malondialdehyde adducts in DNA arrest transcription by T7 RNA polymerase and mammalian RNA polymerase II. Proc. Natl. Acad. Sci. USA 2004, 101, 7275–7280. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.J. The 8,5'-cyclopurine-2'-deoxynucleosides: Candidate neurodegenerative DNA lesions in xeroderma pigmentosum, and unique probes of transcription and nucleotide excision repair. DNA repair 2008, 7, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Kitsera, N.; Stathis, D.; Luhnsdorf, B.; Muller, H.; Carell, T.; Epe, B.; Khobta, A. 8-oxo-7,8-dihydroguanine in DNA does not constitute a barrier to transcription, but is converted into transcription-blocking damage by Ogg1. Nucleic Acids Res. 2011, 39, 5926–5934. [Google Scholar] [CrossRef] [PubMed]
- Yanamadala, S.; Ljungman, M. Potential role of MLH1 in the induction of p53 and apoptosis by blocking transcription on damaged DNA templates. Mol. Cancer Res. 2003, 1, 747–754. [Google Scholar] [PubMed]
- Tornaletti, S.; Maeda, L.S.; Hanawalt, P.C. Transcription arrest at an abasic site in the transcribed strand of template DNA. Chem. Res. Toxicol. 2006, 19, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Hanawalt, P.C.; Spivak, G. Comet-fish with strand-specific probes reveals transcription-coupled repair of 8-oxoguanine in human cells. Nucleic Acids Res. 2013, 41, 7700–7712. [Google Scholar] [CrossRef] [PubMed]
- Hanawalt, P.C.; Spivak, G. Transcription-coupled DNA repair: Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008, 9, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Svejstrup, J.Q. Contending with transcriptional arrest during RNAPII transcript elongation. Trends Biochem. Sci. 2007. [Google Scholar] [CrossRef] [PubMed]
- Tornaletti, S. DNA repair in mammalian cells: Transcription-coupled DNA repair: Directing your effort where it’s most needed. Cell. Mol. Life Sci. 2009, 66, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Pope, B.D.; Ryba, T.; Dileep, V.; Yue, F.; Wu, W.; Denas, O.; Vera, D.L.; Wang, Y.; Hansen, R.S.; Canfield, T.K.; et al. Topologically associating domains are stable units of replication-timing regulation. Nature 2014, 515, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Meryet-Figuiere, M.; Alaei-Mahabadi, B.; Ali, M.M.; Mitra, S.; Subhash, S.; Pandey, G.K.; Larsson, E.; Kanduri, C. Temporal separation of replication and transcription during S-phase progression. Cell Cycle 2014, 13, 3241–3248. [Google Scholar] [CrossRef] [PubMed]
- Wansink, D.G.; Manders, E.E.; van der Kraan, I.; Aten, J.A.; van Driel, R.; de Jong, L. RNA polymerase II transcription is concentrated outside replication domains throughout S-phase. J. Cell Sci. 1994, 107, 1449–1456. [Google Scholar] [PubMed]
- McKay, B.; Becerril, C.; Spronck, J.; Ljungman, M. Ultraviolet light-induced apoptosis is associated with S-phase in primary human fibroblasts. DNA Repair 2002, 1, 811–820. [Google Scholar] [CrossRef]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Miron, K.; Golan-Lev, T.; Dvir, R.; Ben-David, E.; Kerem, B. Oncogenes create a unique landscape of fragile sites. Nat. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.E.; Arlt, M.F.; Park, S.H.; Rajendran, S.; Paulsen, M.; Ljungman, M.; Glover, T.W. Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 2015, 25, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Sauerbier, W.; Hercules, K. Gene and transcription unit mapping by radiation effects. Annu. Rev. Genet. 1978, 12, 329–363. [Google Scholar] [CrossRef] [PubMed]
- McKay, B.C.; Stubbert, L.J.; Fowler, C.C.; Smith, J.M.; Cardamore, R.A.; Spronck, J.C. Regulation of ultraviolet light-induced gene expression by gene size. Proc. Natl. Acad. Sci. USA 2004, 101, 6582–6586. [Google Scholar] [CrossRef] [PubMed]
- Fousteri, M.; Vermeulen, W.; van Zeeland, A.A.; Mullenders, L.H. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol. cell 2006, 23, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Smerdon, M.J. RPB4 and RPB9 mediate subpathways of transcription-coupled DNA repair in saccharomyces cerevisiae. EMBO J. 2002, 21, 5921–5929. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; Wellinger, R.E.; Aguilera, A. A new connection of MRNP biogenesis and export with transcription-coupled repair. Nucleic Acids Res. 2007, 35, 3893–3906. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; Tous, C.; Botet, J.; Gonzalez-Aguilera, C.; Quintero, M.J.; Viladevall, L.; Garcia-Rubio, M.L.; Rodriguez-Gil, A.; Marin, A.; Arino, J.; et al. Genome-wide analysis of factors affecting transcription elongation and DNA repair: A new role for PAF and Ccr4-not in transcription-coupled repair. PLoS Genet. 2009, 5, e1000364. [Google Scholar] [CrossRef] [PubMed]
- Kruk, J.A.; Dutta, A.; Fu, J.; Gilmour, D.S.; Reese, J.C. The multifunctional Ccr4-not complex directly promotes transcription elongation. Genes Dev. 2011, 25, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, Y.; Asahina, H.; Citterio, E.; Rademakers, S.; Vermeulen, W.; Kamiuchi, S.; Yeo, J.P.; Khaw, M.C.; Saijo, M.; Kodo, N.; et al. XAB2, a novel tetratricopeptide repeat protein involved in transcription-coupled DNA repair and transcription. J. Biol. Chem. 2000, 275, 34931–34937. [Google Scholar] [CrossRef] [PubMed]
- Kamileri, I.; Karakasilioti, I.; Garinis, G.A. Nucleotide excision repair: New tricks with old bricks. Trends Genet. 2012, 28, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Hanasoge, S.; Ljungman, M. H2AX phosphorylation after UV irradiation is triggered by DNA repair intermediates and is mediated by the ATR kinase. Carcinogenesis 2007, 28, 2298–2304. [Google Scholar] [CrossRef] [PubMed]
- Marti, T.M.; Hefner, E.; Feeney, L.; Natale, V.; Cleaver, J.E. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc. Natl. Acad. Sci. USA 2006, 103, 9891–9896. [Google Scholar] [CrossRef] [PubMed]
- Sertic, S.; Pizzi, S.; Cloney, R.; Lehmann, A.R.; Marini, F.; Plevani, P.; Muzi-Falconi, M. Human exonuclease 1 connects nucleotide excision repair (NER) processing with checkpoint activation in response to UV irradiation. Proc. Natl. Acad. Sci. USA 2011, 108, 13647–13652. [Google Scholar] [CrossRef] [PubMed]
- Zavala, A.G.; Morris, R.T.; Wyrick, J.J.; Smerdon, M.J. High-resolution characterization of CPD hotspot formation in human fibroblasts. Nucleic Acids Res. 2014, 42, 893–905. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Bennett, M.; Evans, K.E.; Zhuang-Jackson, H.; Higgs, A.; Reed, S.H.; Waters, R. A novel method for the genome-wide high resolution analysis of DNA damage. Nucleic Acids Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.R.; Bennett, M.R.; Evans, K.E.; Yu, S.; Webster, R.M.; Waters, R.; Skinner, N.; Reed, S.H. 3D-DIP-Chip: A microarray-based method to measure genomic DNA damage. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Bryan, D.S.; Ransom, M.; Adane, B.; York, K.; Hesselberth, J.R. High resolution mapping of modified DNA nucleobases using excision repair enzymes. Genome Res. 2014, 24, 1534–1542. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Adar, S.; Selby, C.P.; Lieb, J.D.; Sancar, A. Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev. 2015, 29, 948–960. [Google Scholar] [CrossRef] [PubMed]
- Tornaletti, S.; Reines, D.; Hanawalt, P.C. Structural characterization of RNA polymerase II complexes arrested by a cyclobutane pyrimidine dimer in the transcribed strand of template DNA. J. Biol. Chem. 1999, 274, 24124–24130. [Google Scholar] [CrossRef] [PubMed]
- Selby, C.; Sancar, A. Molecular mecahanism of transcription-repair coupling. Science 1993, 260, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Howan, K.; Smith, A.J.; Westblade, L.F.; Joly, N.; Grange, W.; Zorman, S.; Darst, S.A.; Savery, N.J.; Strick, T.R. Initiation of transcription-coupled repair characterized at single-molecule resolution. Nature 2012, 490, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Epshtein, V.; Kamarthapu, V.; McGary, K.; Svetlov, V.; Ueberheide, B.; Proshkin, S.; Mironov, A.; Nudler, E. UvrD facilitates DNA repair by pulling RNA polymerase backwards. Nature 2014, 505, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.E.; Lewis, C.A.; Mooney, R.A.; Kohanski, M.A.; Collins, J.J.; Landick, R.; Walker, G.C. Roles for the transcription elongation factor NusA in both DNA repair and damage tolerance pathways in Escherichia coli. Proc. Natl. Acad. Sci. USA 2010, 107, 15517–15522. [Google Scholar] [CrossRef] [PubMed]
- Kamarthapu, V.; Nudler, E. Rethinking transcription coupled DNA repair. Curr. Opin. Microbiol. 2015, 24, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Beerens, N.; Hoeijmakers, J.H.; Kanaar, R.; Vermeulen, W.; Wyman, C. The csb protein actively wraps DNA. J. Biol. Chem. 2005, 280, 4722–4729. [Google Scholar] [CrossRef] [PubMed]
- Selby, C.P.; Sancar, A. Human transcription-repair coupling factor CSB/ERCC6 is a DNA-stimulated atpase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J. Biol. Chem. 1997, 272, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Selby, C.P.; Sancar, A. Cockayne syndrome group b protein enhances elongation by RNA polymerase II. Proc. Natl. Acad. Sci. USA 1997, 94, 11205–11209. [Google Scholar] [CrossRef] [PubMed]
- Bregman, D.B.; Halaban, R.; Vangool, A.J.; Henning, K.A.; Friedberg, E.C.; Warren, S.L. UV-induced ubiquitination of RNA polymerase II: A novel modification deficient in Cockayne syndrome cells. Proc. Natl. Acad. Sci. USA 1996, 93, 11586–11590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratner, J.N.; Balasubramanian, B.; Corden, J.; Warren, S.L.; Bregman, D.B. Ultraviolet radiation-induced ubiquitination and proteasomal degradation of the large subunit of RNA polymerase II Implications for transcription-coupled DNA repair. J. Biol. Chem. 1998, 273, 5184–5189. [Google Scholar] [CrossRef] [PubMed]
- McKay, B.C.; Chen, F.; Clarke, S.T.; Wiggin, H.E.; Harley, L.M.; Ljungman, M. UV light-induced degradation of RNA polymerase II is dependent on the Cockayne's syndrome A and B proteins but not p53 or MLH1. Mutat. Res. 2001, 485, 93–105. [Google Scholar] [CrossRef]
- Anindya, R.; Aygun, O.; Svejstrup, J.Q. Damage-induced ubiquitylation of human RNA polymerase II by the ubiquitin ligase Nedd4, but not Cockayne syndrome proteins or BRCA1. Mol. cell 2007, 28, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Harreman, M.; Taschner, M.; Sigurdsson, S.; Anindya, R.; Reid, J.; Somesh, B.; Kong, S.E.; Banks, C.A.; Conaway, R.C.; Conaway, J.W.; et al. Distinct ubiquitin ligases act sequentially for RNA polymerase II polyubiquitylation. Proc. Natl. Acad. Sci. USA 2009, 106, 20705–20710. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.D.; Harreman, M.; Svejstrup, J.Q. Ubiquitylation and degradation of elongating RNA polymerase II: The last resort. Biochim. Biophys. Acta 2013, 1829, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Sarker, A.H.; Tsutakawa, S.E.; Kostek, S.; Ng, C.; Shin, D.S.; Peris, M.; Campeau, E.; Tainer, J.A.; Nogales, E.; Cooper, P.K. Recognition of RNA polymerase II and transcription bubbles by XPG, CSB, and TFIIH: Insights for transcription-coupled repair and Cockayne Syndrome. Mol. Cell 2005, 20, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, N.; Fonseca, D.; Mohammed, S.; Cevher, M.A.; Manley, J.L.; Kleiman, F.E. The 3' processing factor CstF functions in the DNA repair response. Nucleic Acids Res. 2008, 36, 1792–1804. [Google Scholar] [CrossRef] [PubMed]
- Schwertman, P.; Lagarou, A.; Dekkers, D.H.; Raams, A.; van der Hoek, A.C.; Laffeber, C.; Hoeijmakers, J.H.; Demmers, J.A.; Fousteri, M.; Vermeulen, W.; et al. UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat. Genet. 2012, 44, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Horibata, K.; Saijo, M.; Ishigami, C.; Ukai, A.; Kanno, S.; Tahara, H.; Neilan, E.G.; Honma, M.; Nohmi, T.; et al. Mutations in UVSSA cause UV-sensitive syndrome and destabilize ERCC6 in transcription-coupled DNA repair. Nat. Genet. 2012, 44, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, Y.; Sasaki, K.; Mitsutake, N.; Matsuse, M.; Shimada, M.; Nardo, T.; Takahashi, Y.; Ohyama, K.; Ito, K.; Mishima, H.; et al. Mutations in uvssa cause UV-sensitive syndrome and impair RNA polymerase IIo processing in transcription-coupled nucleotide-excision repair. Nat. Genet. 2012, 44, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Donahue, B.A.; Yin, S.; Taylor, J.S.; Reines, D.; Hanawalt, P.C. Transcript cleavage by RNA polymerase II arrested by a cyclobutane pyrimidine dimer in the DNA template. Proc. Natl. Acad. Sci. USA 1994, 91, 8502–8506. [Google Scholar] [CrossRef] [PubMed]
- Mourgues, S.; Gautier, V.; Lagarou, A.; Bordier, C.; Mourcet, A.; Slingerland, J.; Kaddoum, L.; Coin, F.; Vermeulen, W.; Gonzales de Peredo, A.; et al. ELL, a novel TFIIH partner, is involved in transcription restart after DNA repair. Proc. Natl. Acad. Sci. USA 2013, 110, 17927–17932. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.; Mullenders, L.H. Transcription factor IIS impacts UV-inhibited transcription. DNA Repair 2010, 9, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Babbarwal, V.; Fu, J.; Brunke-Reese, D.; Libert, D.M.; Willis, J.; Reese, J.C. Ccr4-not and TFIIS function cooperatively to rescue arrested RNA polymerase II. Mol. Cell. Biol. 2015, 35, 1915–1925. [Google Scholar] [CrossRef] [PubMed]
- Oksenych, V.; Zhovmer, A.; Ziani, S.; Mari, P.O.; Eberova, J.; Nardo, T.; Stefanini, M.; Giglia-Mari, G.; Egly, J.M.; Coin, F. Histone methyltransferase DOT1L drives recovery of gene expression after a genotoxic attack. PLoS Genet. 2013, 9, e1003611. [Google Scholar] [CrossRef] [PubMed]
- Adam, S.; Polo, S.E.; Almouzni, G. Transcription recovery after DNA damage requires chromatin priming by the H3.3 histone chaperone HIRA. Cell 2013, 155, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Dinant, C.; Ampatziadis-Michailidis, G.; Lans, H.; Tresini, M.; Lagarou, A.; Grosbart, M.; Theil, A.F.; van Cappellen, W.A.; Kimura, H.; Bartek, J.; et al. Enhanced chromatin dynamics by fact promotes transcriptional restart after UV-induced DNA damage. Mol. Cell 2013, 51, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Mandemaker, I.; Vermeulen, W.; Marteijn, J. Gearing up chromatin: A role of chromatin remodeling during the transcriptional restart upon DNA damage. Nucleus 2014. [Google Scholar] [CrossRef] [PubMed]
- McKay, B.; Ljungman, M. Role for p53 in the recovery of transcription and protection against apoptosis induced by ultraviolet light. Neoplasia 1999, 1, 276–284. [Google Scholar] [CrossRef] [PubMed]
- McKay, B.C.; Chen, F.; Perumalswami, C.R.; Zhang, F.F.; Ljungman, M. The tumor suppressor p53 can both stimulate and inhibit ultraviolet light-induced apoptosis. Mol. Biol. Cell 2000, 11, 2543–2551. [Google Scholar] [CrossRef] [PubMed]
- McKay, B.C.; Becerril, C.; Ljungman, M. P53 plays a protective role against UV-and cisplatin-induced apoptosis in transcription-coupled repair proficient fibroblasts. Oncogene 2001, 20, 6805–6808. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.M.; Hanawalt, P.C. Li-fraumeni syndrome fibroblasts homozygous for p53 mutations are deficient in global DNA repair but exhibit normal transcription-coupled repair and enhanced UV resistance. Proc. Natl. Acad. Sci. USA 1995, 92, 8876–8880. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.M.; Hanawalt, P.C. Expression of wild-type p53 is required for efficient global genomic nucleotide excision repair in UV-irradiated human fibroblasts. J. Biol. Chem. 1997, 272, 28073–28080. [Google Scholar] [CrossRef] [PubMed]
- Therrien, J.P.; Drouin, R.; Baril, C.; Drobetsky, E.A. Human cells compromised for p53 function exhibit defective global and transcription-coupled nucleotide excision repair, whereas cells compromised for PRB function are defective only in global repair. Proc. Natl. Acad. Sci. USA 1999, 96, 15038–15043. [Google Scholar] [CrossRef] [PubMed]
- Rubbi, C.P.; Milner, J. P53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J. 2003, 22, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, M.T.; Veloso, A.; Prasad, J.; Bedi, K.; Ljungman, E.A.; Magnuson, B.; Wilson, T.E.; Ljungman, M. Use of Bru-Seq and Bruchase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods 2014, 67, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, M.T.; Veloso, A.; Prasad, J.; Bedi, K.; Ljungman, E.A.; Tsan, Y.C.; Chang, C.W.; Tarrier, B.; Washburn, J.G.; Lyons, R.; et al. Coordinated regulation of synthesis and stability of RNA during the acute TNF-induced proinflammatory response. Proc. Natl. Acad. Sci. USA 2013, 110, 2240–2245. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, M.; Hanawalt, P.C. The anti-cancer drug camptothecin inhibits elongation but stimulates initiation of RNA polymerase II transcription. Carcinogenesis 1996, 17, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, M. The DNA damage response—Repair or despair? Environ. Mol. Mutagen. 2010, 51, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, M. The transcription stress response. Cell Cycle 2007, 6, 2252–2257. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrade-Lima, L.C.; Veloso, A.; Ljungman, M. Transcription Blockage Leads to New Beginnings. Biomolecules 2015, 5, 1600-1617. https://doi.org/10.3390/biom5031600
Andrade-Lima LC, Veloso A, Ljungman M. Transcription Blockage Leads to New Beginnings. Biomolecules. 2015; 5(3):1600-1617. https://doi.org/10.3390/biom5031600
Chicago/Turabian StyleAndrade-Lima, Leonardo C., Artur Veloso, and Mats Ljungman. 2015. "Transcription Blockage Leads to New Beginnings" Biomolecules 5, no. 3: 1600-1617. https://doi.org/10.3390/biom5031600
APA StyleAndrade-Lima, L. C., Veloso, A., & Ljungman, M. (2015). Transcription Blockage Leads to New Beginnings. Biomolecules, 5(3), 1600-1617. https://doi.org/10.3390/biom5031600