
Turning on the Radio: Epigenetic Inhibitors as Potential Radiopriming Agents

 ,
,
Abstract
:1. Introduction
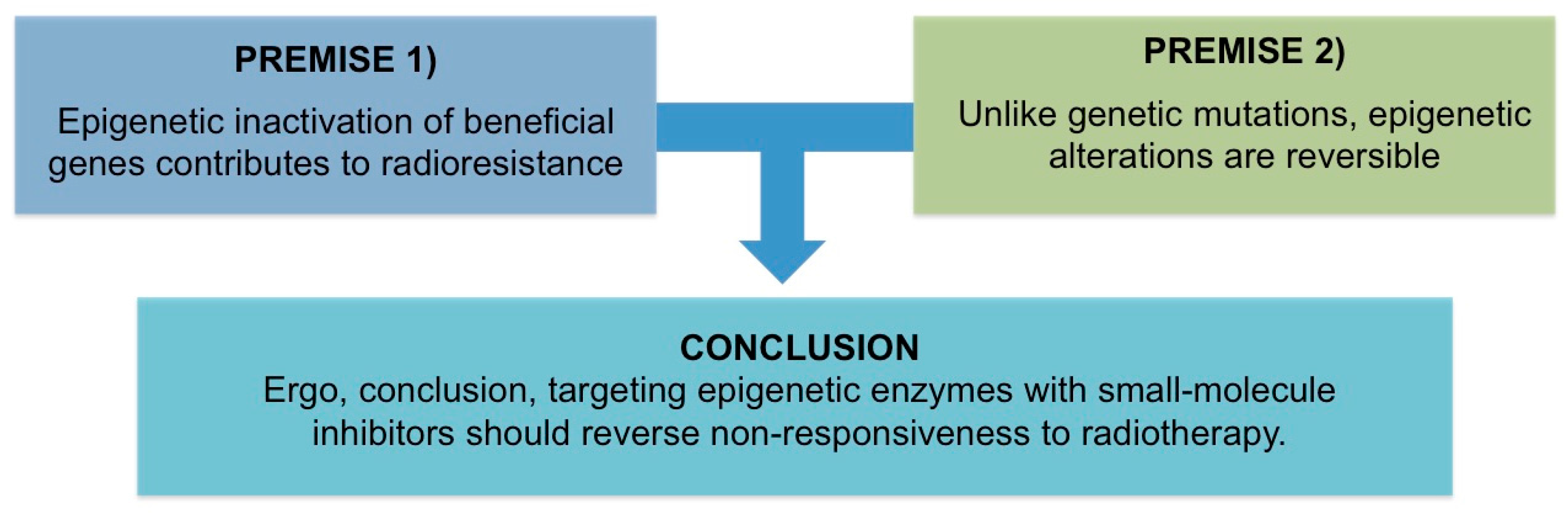
All Men Are Mortal. Socrates Is a Man. Therefore, All Men Are Socrates—Woody Allen
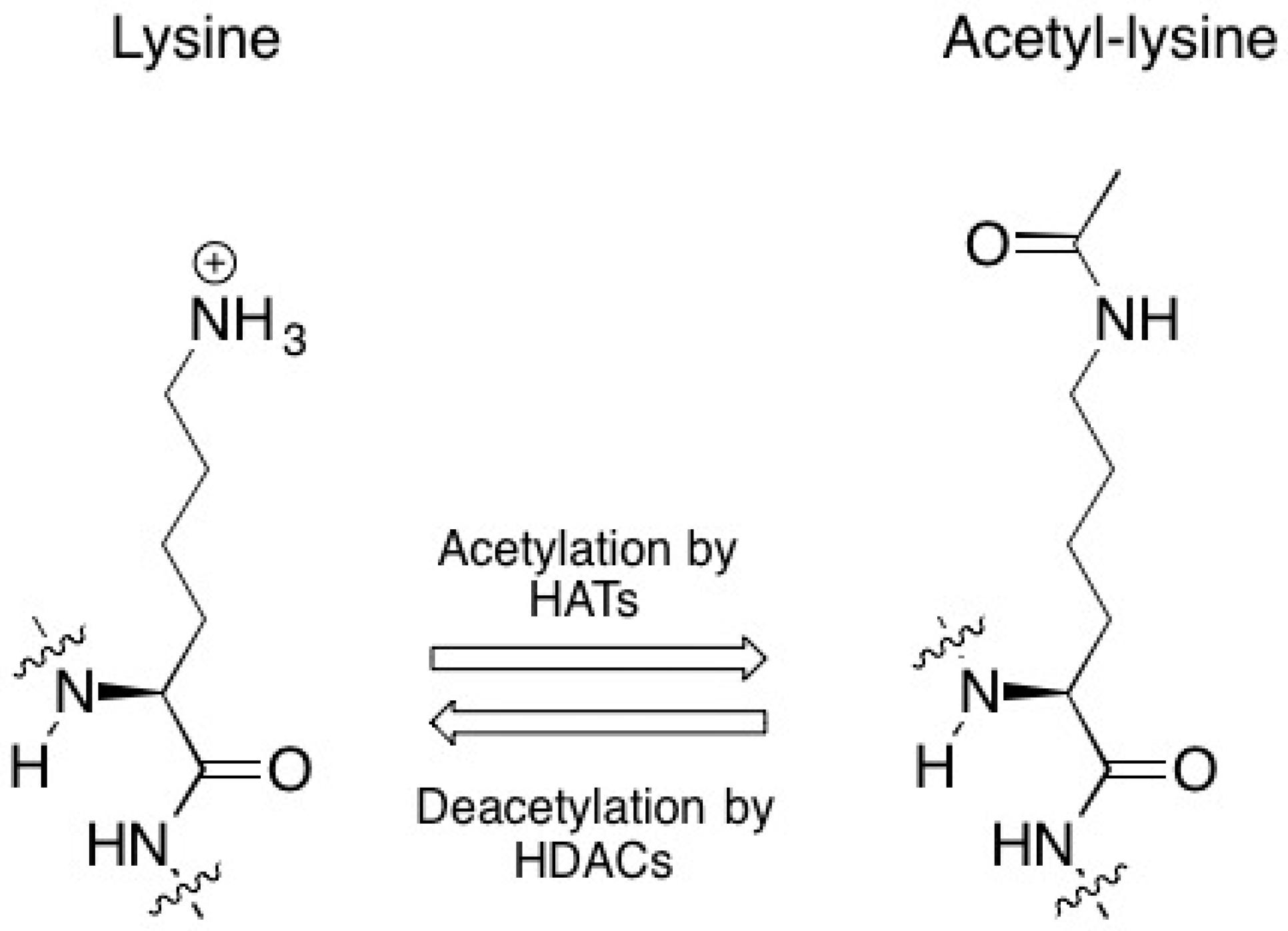
2. Premise #1: Epigenetic Inactivation of Beneficial Genes Contributes to Radioresistance
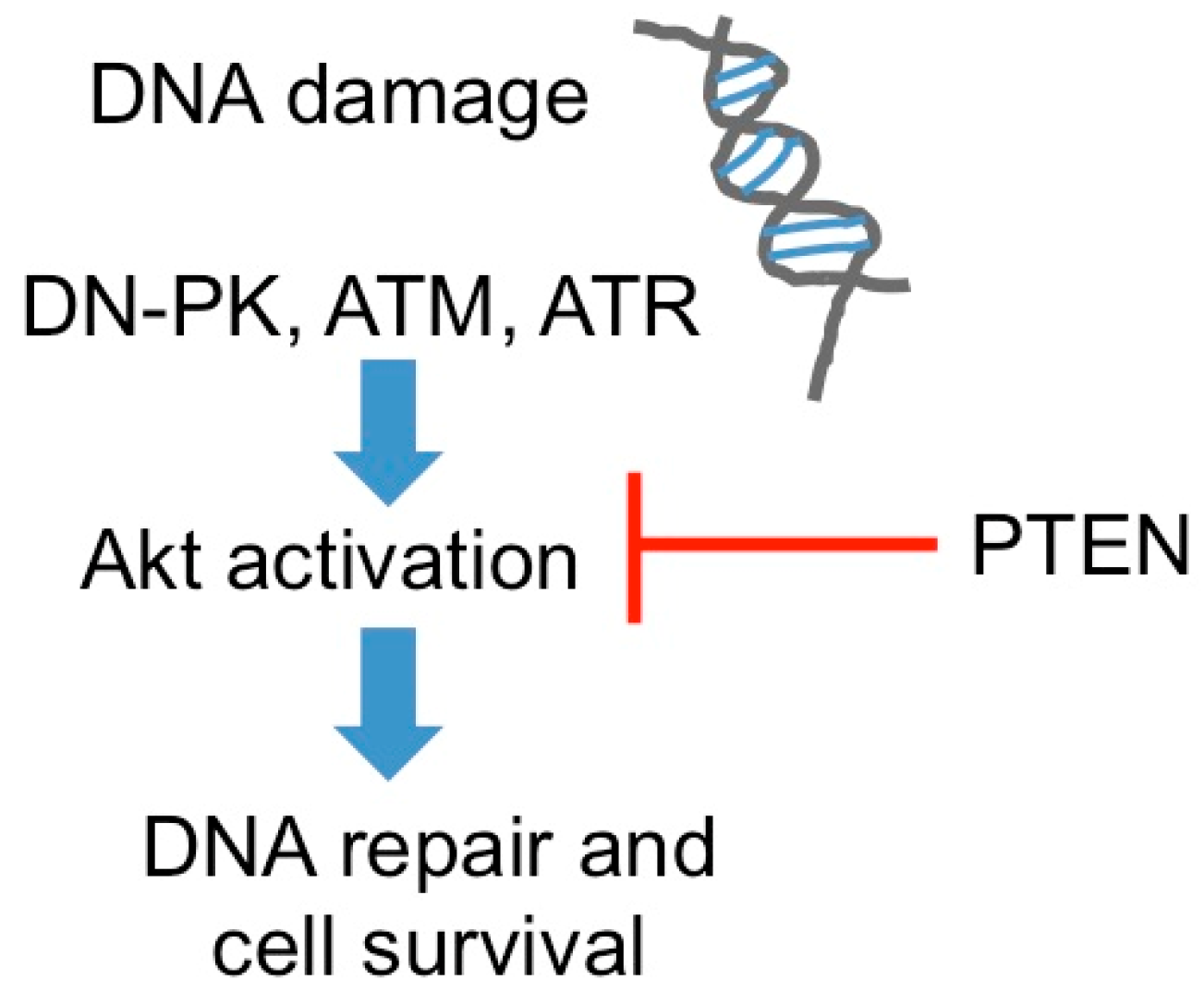
2.1. DNA Repair
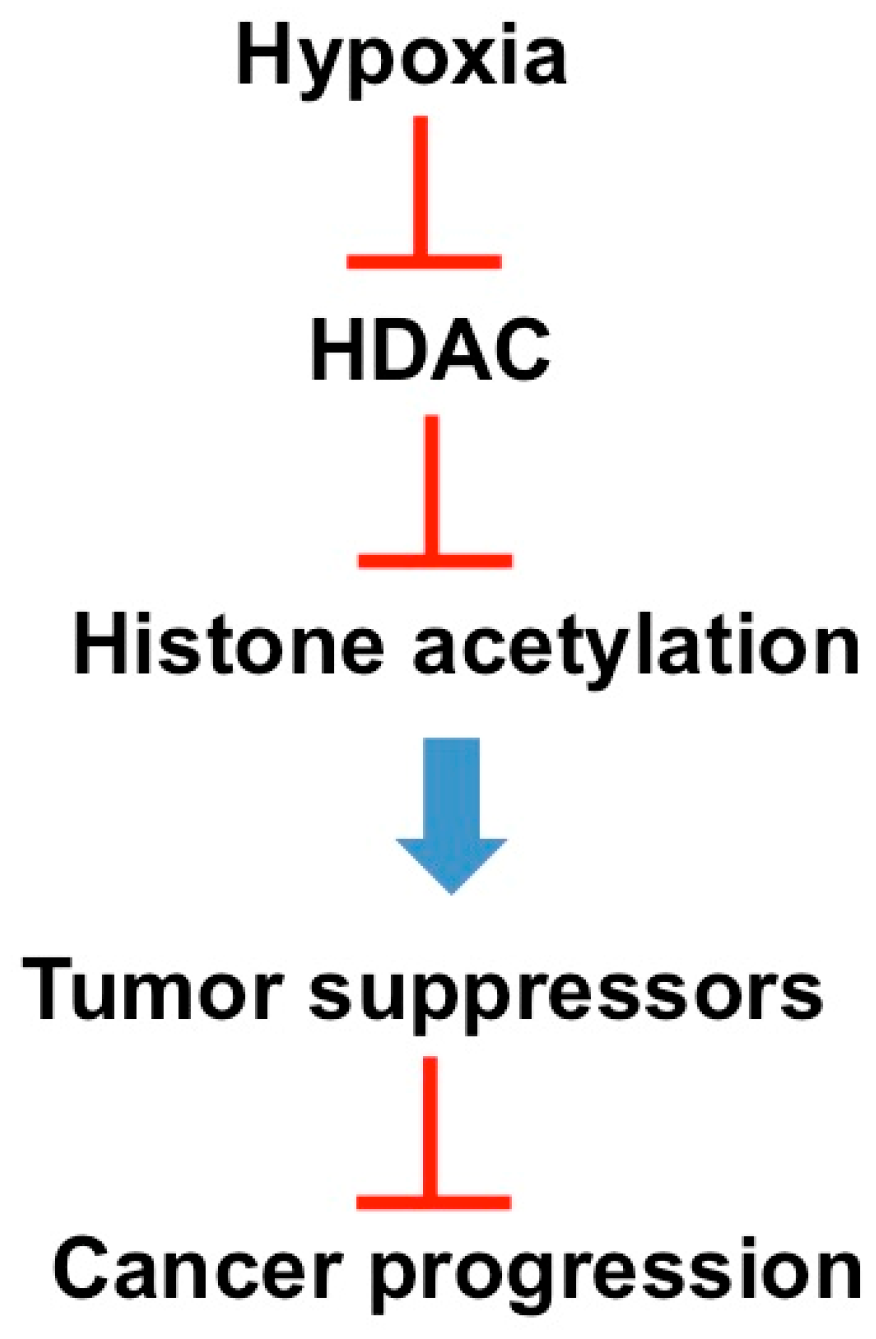
2.2. Hypoxia
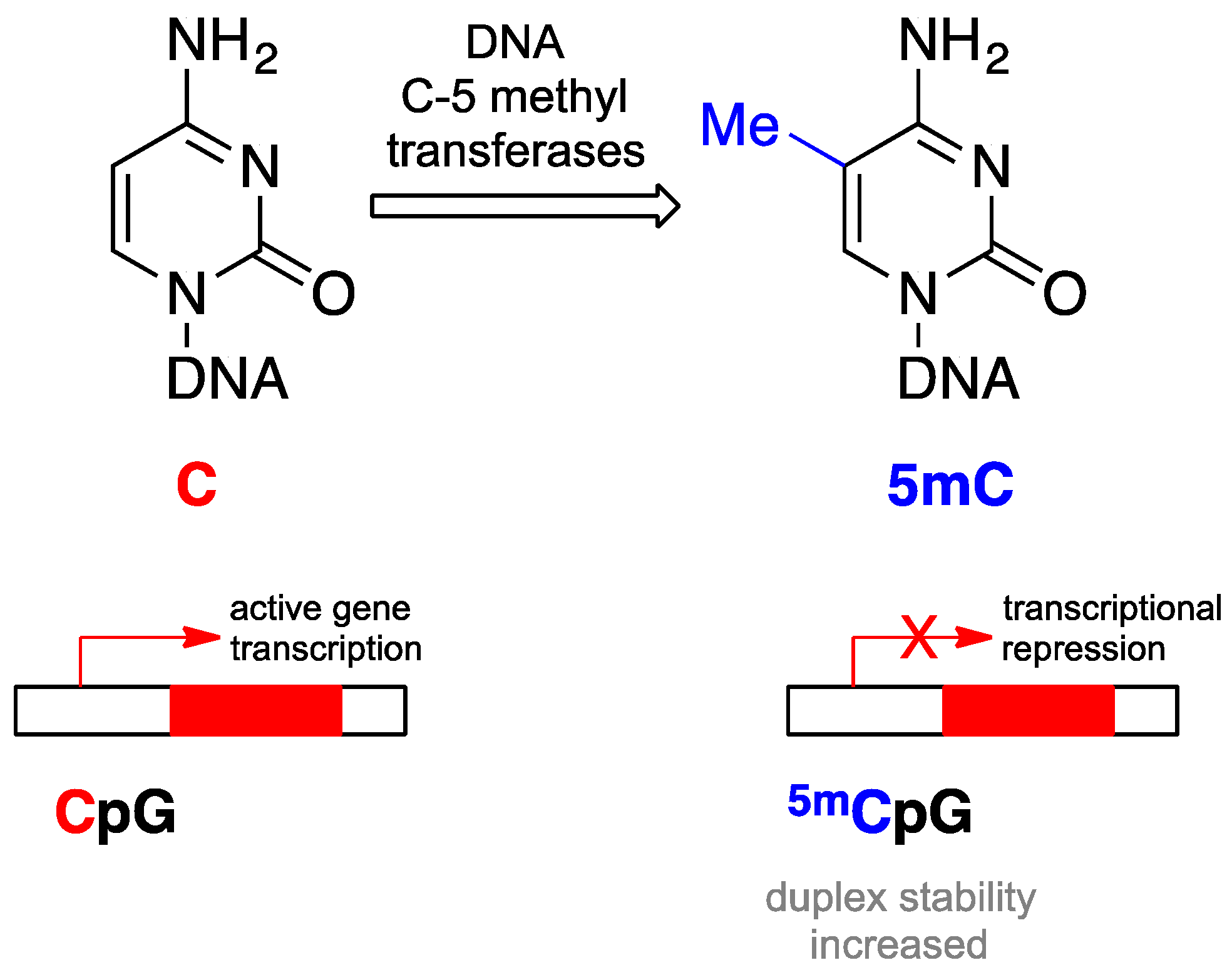
3. Premise #2: Epigenetic Alterations Are Reversible
4. Discussion of Analysis: Targeting Epigenetic Enzymes with Small-Molecule Inhibitors Should Reverse Non-Responsiveness to Radiotherapy
4.1. Epigenetic Inhibitors as Chemotherapy Primers
4.2. Epigenetic Inhibitors as Immunotherapy Primers
4.3. Epigenetic Inhibitors as Radiotherapy Primers
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brady, L.W.; Kramer, S.; Levitt, S.H.; Parker, R.G.; Powers, W.E. Radiation oncology: Contributions of the united states in the last years of the 20th century. Radiology 2001, 219, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Grubbé, E.H. Priority in the therapeutic use of X-rays. Radiology 1933, 21, 156–162. [Google Scholar] [CrossRef]
- Holsti, L.R. Development of clinical radiotherapy since 1896. Acta Oncol. 1995, 34, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Oronsky, B.T.; Oronsky, A.L.; Lybeck, M.; Oronsky, N.C.; Scicinski, J.J.; Carter, C.; Day, R.M.; Rodriguez Orengo, J.F.; Rodriguez-Torres, M.; Fanger, G.F.; et al. Episensitization: Defying time’s arrow. Front. Oncol. 2015. [Google Scholar] [CrossRef]
- Oronsky, B.; Oronsky, N.; Scicinski, J.; Fanger, G.; Lybeck, M.; Reid, T. Rewriting the epigenetic code for tumor resensitization: A review. Transl. Oncol. 2014, 7, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Pichierri, P.; Franchitto, A.; Palitti, F. Predisposition to cancer and radiosensitivity. Genet. Mol. Biol. 2000, 23, 1101–1105. [Google Scholar] [CrossRef]
- Lempiainen, H.; Halazonetis, T.D. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009, 28, 3067–3073. [Google Scholar] [CrossRef] [PubMed]
- Durocher, D.; Jackson, S.P. DNA-PK, ATM and ATR as sensors of DNA damage: Variations on a theme? Curr. Opin. Cell Biol. 2001, 13, 225–231. [Google Scholar] [CrossRef]
- Jackson, S.P. Sensing and repairing DNA double-strand breaks. Carcinogenesis 2002, 23, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, R.; Ahmed, S.U.; Strathdee, K.; Gomez-Roman, N.; Amoah-Buahin, E.; Watts, C.; Chalmers, A.J. Abrogation of radioresistance in glioblastoma stem-like cells by inhibition of atm kinase. Mol. Oncol. 2015, 9, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Ming, M.; He, Y.Y. PTEN in DNA damage repair. Cancer Lett. 2012, 319, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Mirmohammadsadegh, A.; Marini, A.; Nambiar, S.; Hassan, M.; Tannapfel, A.; Ruzicka, T.; Hengge, U.R. Epigenetic silencing of the pten gene in melanoma. Cancer Res. 2006, 66, 6546–6552. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.H.; Lee, H.S.; Kim, W.H. Promoter methylation and silencing of pten in gastric carcinoma. Lab. Investig. 2002, 82, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Byler, S.; Sarkar, S. Do epigenetic drug treatments hold the key to killing cancer progenitor cells? Epigenomics 2014, 6, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Thomlinson, R.H.; Gray, L.H. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br. J. Cancer 1955, 9, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Kalns, J.; Krock, L.; Piepmeier, E., Jr. The effect of hyperbaric oxygen on growth and chemosensitivity of metastatic prostate cancer. Anticancer Res. 1998, 18, 363–367. [Google Scholar] [PubMed]
- Vaupel, P.; Schlenger, K.; Knoop, C.; Hockel, M. Oxygenation of human tumors: Evaluation of tissue oxygen distribution in breast cancers by computerized O2 tension measurements. Cancer Res. 1991, 51, 3316–3322. [Google Scholar] [PubMed]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Oronsky, B.T.; Knox, S.J.; Scicinski, J. Six degrees of separation: The oxygen effect in the development of radiosensitizers. Transl. Oncol. 2011, 4, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Palcic, B.; Skarsgard, L.D. Reduced oxygen enhancement ratio at low doses of ionizing radiation. Radiat. Res. 1984, 100, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ledaki, I.; Turley, H.; Gatter, K.C.; Montero, J.C.; Li, J.L.; Harris, A.L. Role of hypoxia-inducible factors in epigenetic regulation via histone demethylases. Ann. N. Y. Acad. Sci. 2009, 1177, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J.; Collier, P.; Tea, I.; Neary, R.; Watson, J.A.; Robinson, C.; Phelan, D.; Ledwidge, M.T.; McDonald, K.M.; McCann, A.; et al. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum. Mol. Genet. 2014, 23, 2176–2188. [Google Scholar] [CrossRef] [PubMed]
- Shmakova, A.; Batie, M.; Druker, J.; Rocha, S. Chromatin and oxygen sensing in the context of JmjC histone demethylases. Biochem J. 2014, 462, 385–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohse, B.; Kristensen, J.L.; Kristensen, L.H.; Agger, K.; Helin, K.; Gajhede, M.; Clausen, R.P. Inhibitors of histone demethylases. Bioorg. Med. Chem. 2011, 19, 3625–3636. [Google Scholar] [CrossRef] [PubMed]
- Pollard, P.J.; Loenarz, C.; Mole, D.R.; McDonough, M.A.; Gleadle, J.M.; Schofield, C.J.; Ratcliffe, P.J. Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1alpha. Biochem. J. 2008, 416, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.H.; Zhang, W.; Chen, X.; George, J.; Ng, H.H. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 2007, 21, 2545–2557. [Google Scholar] [CrossRef] [PubMed]
- Shahrzad, S.; Bertrand, K.; Minhas, K.; Coomber, B.L. Induction of DNA hypomethylation by tumor hypoxia. Epigenetics 2007, 2, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Srivastava, T.; Sharma, M.K.; Mehndiratta, M.; Das, P.; Sinha, S.; Chattopadhyay, P. Aberrant methylation and associated transcriptional mobilization of alu elements contributes to genomic instability in hypoxia. J. Cell. Mol. Med. 2010, 14, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Thirlwell, C.; Schulz, L.; Dibra, H.; Beck, S. Suffocating cancer: Hypoxia-associated epimutations as targets for cancer therapy. Clin. Epigenet. 2011. [Google Scholar] [CrossRef] [PubMed]
- Rockwell, S. Oxygen delivery: Implications for the biology and therapy of solid tumors. Oncol. Res. 1997, 9, 383–390. [Google Scholar] [PubMed]
- Semenza, G.L. Hypoxia-inducible factor 1: Master regulator of O2 homeostasis. Curr. Opin. Genet. Dev. 1998, 8, 588–594. [Google Scholar] [CrossRef]
- Qian, D.Z.; Kato, Y.; Shabbeer, S.; Wei, Y.; Verheul, H.M.; Salumbides, B.; Sanni, T.; Atadja, P.; Pili, R. Targeting tumor angiogenesis with histone deacetylase inhibitors: The hydroxamic acid derivative LBH589. Clin. Cancer Res. 2006, 12, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Hammers, H.; Pili, R. Targeting tumor angiogenesis with histone deacetylase inhibitors. Cancer Lett. 2009, 280, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.B.; Denko, N.; Barton, M.C. Hypoxia induces a novel signature of chromatin modifications and global repression of transcription. Mutat. Res. 2008, 640, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Alleman, W.G.; Tabios, R.L.; Chandramouli, G.V.; Aprelikova, O.N.; Torres-Cabala, C.; Mendoza, A.; Rogers, C.; Sopko, N.A.; Linehan, W.M.; Vasselli, J.R. The in vitro and in vivo effects of re-expressing methylated von hippel-lindau tumor suppressor gene in clear cell renal carcinoma with 5-aza-2′-deoxycytidine. Clin. Cancer Res. 2004, 10, 7011–7021. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sang, N. Histone deacetylase inhibitors: The epigenetic therapeutics that repress hypoxia-inducible factors. J. Biomed. Biotechnol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Goldgar, S.; Byler, S.; Rosenthal, S.; Heerboth, S. Demethylation and re-expression of epigenetically silenced tumor suppressor genes: Sensitization of cancer cells by combination therapy. Epigenomics 2013, 5, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Fath, D.M.; Kong, X.; Liang, D.; Lin, Z.; Chou, A.; Jiang, Y.; Fang, J.; Caro, J.; Sang, N. Histone deacetylase inhibitors repress the transactivation potential of hypoxia-inducible factors independently of direct acetylation of HIF-alpha. J. Biol. Chem. 2006, 281, 13612–13619. [Google Scholar] [CrossRef] [PubMed]
- Mie Lee, Y.; Kim, S.H.; Kim, H.S.; Jin Son, M.; Nakajima, H.; Jeong Kwon, H.; Kim, K.W. Inhibition of hypoxia-induced angiogenesis by FK228, a specific histone deacetylase inhibitor, via suppression of HIF-1alpha activity. Biochem. Biophys. Res. Commun. 2003, 300, 241–246. [Google Scholar] [CrossRef]
- Fischer, C.; Leithner, K.; Wohlkoenig, C.; Quehenberger, F.; Bertsch, A.; Olschewski, A.; Olschewski, H.; Hrzenjak, A. Panobinostat reduces hypoxia-induced cisplatin resistance of non-small cell lung carcinoma cells via HIF-1alpha destabilization. Mol. Cancer 2015. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Lin, B.; Chang, H.; Chang, C. Radio resistance mechanisms of cancers: An overview and future perspectives. Biol. Med. 2015. [Google Scholar] [CrossRef]
- Kim, S.H.; Joshi, K.; Ezhilarasan, R.; Myers, T.R.; Siu, J.; Gu, C.; Nakano-Okuno, M.; Taylor, D.; Minata, M.; Sulman, E.P.; et al. EZH2 protects glioma stem cells from radiation-induced cell death in a MELK/FOXM1-dependent manner. Stem Cell Rep. 2015, 4, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Azad, N.; Zahnow, C.A.; Rudin, C.M.; Baylin, S.B. The future of epigenetic therapy in solid tumours—Lessons from the past. Nat. Rev. Clin. Oncol. 2013, 10, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; Buckles, E.; Estrada, J.; Koochekpour, S. Aberrant DNA methylation and prostate cancer. Curr. Genom. 2011, 12, 486–505. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.C.; Baylin, S.B. Cancer epigenetics: Linking basic biology to clinical medicine. Cell Res. 2011, 21, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.K.; O’Donnell, A.H.; Bestor, T.H. Mammalian cytosine methylation at a glance. J. Cell Sci. 2009, 122, 2787–2791. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P. CPG island methylator phenotype in cancer. Nat. Rev. Cancer 2004, 4, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, Y.A.; Khamis, A.M.; Kulakovskiy, I.V.; Ba-Alawi, W.; Bhuyan, M.S.; Kawaji, H.; Lassmann, T.; Harbers, M.; Forrest, A.R.; Bajic, V.B.; et al. Effects of cytosine methylation on transcription factor binding sites. BMC Genom. 2014. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Gowher, H.; Jeltsch, A. Biochemistry and biology of mammalian DNA methyltransferases. Cell. Mol. Life Sci. 2004, 61, 2571–2587. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J. Chromosomal DNA and Its Packaging in the Chromatin Fiber. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002; Available online: http://www.ncbi.nlm.nih.gov/books/NBK26834/ (accessed on 29 June 2016).
- Miremadi, A.; Oestergaard, M.Z.; Pharoah, P.D.; Caldas, C. Cancer genetics of epigenetic genes. Hum. Mol. Genet. 2007, 16, R28–R49. [Google Scholar] [CrossRef] [PubMed]
- Sterner, D.E.; Berger, S.L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.; Reid, T.; Fisher, G.; Cho-Phan, C.; Kunz, P.; Kaiser, H.; Oronsky, B.; Fanger, G.; Caroen, S.; Parker, C.; et al. O3.8early results: “Rocket” a phase II study of RRX-001, a novel triple epigenetic inhibitor, resensitization to irinotecan in colorectal cancer. Ann. Oncol. 2015. [Google Scholar] [CrossRef]
- Frew, A.J.; Lindemann, R.K.; Martin, B.P.; Clarke, C.J.; Sharkey, J.; Anthony, D.A.; Banks, K.M.; Haynes, N.M.; Gangatirkar, P.; Stanley, K.; et al. Combination therapy of established cancer using a histone deacetylase inhibitor and a trail receptor agonist. Proc. Natl. Acad. Sci. USA 2008, 105, 11317–11322. [Google Scholar] [CrossRef] [PubMed]
- Cacan, E.; Ali, M.W.; Boyd, N.H.; Hooks, S.B.; Greer, S.F. Inhibition of HDAC1 and DNMT1 modulate RGS10 expression and decrease ovarian cancer chemoresistance. PLoS ONE 2014, 9, e87455. [Google Scholar] [CrossRef] [PubMed]
- Wrangle, J.; Wang, W.; Koch, A.; Easwaran, H.; Mohammad, H.P.; Vendetti, F.; Vancriekinge, W.; Demeyer, T.; Du, Z.; Parsana, P.; et al. Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget 2013, 4, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Sigalotti, L.; Fratta, E.; Coral, S.; Maio, M. Epigenetic drugs as immunomodulators for combination therapies in solid tumors. Pharmacol. Ther. 2014, 142, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Juergens, R.A.; Vendetti, F.; Coleman, B.; Sebree, R.S.; Rudek, M.A.; Belinsky, S.A. Phase I trial of 5-azacitidine (5AC) and SNDX-275 in advanced lung cancer (NSCLC). J. Clin. Oncol. 2008, 26. abstract No. 19036. [Google Scholar]
- Juergens, R.A.; Wrangle, J.; Vendetti, F.P.; Murphy, S.C.; Zhao, M.; Coleman, B.; Sebree, R.; Rodgers, K.; Hooker, C.M.; Franco, N.; et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011, 1, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.; Rozwarski, J.; Murtha Cancer Center, Walter Reed National Military Medical Center, Bethesda, MD, USA. Unpublished data. 2016.
- Candelaria, M.; Cetina, L.; Garcia, A.; Wegman-Ostrosky, T.; Robles, E.; González-Fierro, A.; López-Graniel, C.; González, A.; Cantú, D.; Ribera, L.; et al. Epigenetic therapy with hydralazine and valproate associated to cisplatin chemoradiation in FIGO stage IIIB. A phase II study. BMC Cancer 2007. [Google Scholar] [CrossRef]
- Herceg, Z.; Hainaut, P. Genetic and epigenetic alterations as biomarkers for cancer detection, diagnosis and prognosis. Mol. Oncol. 2007, 1, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Keating, P.; Cambrosio, A.; Nelson, N.C.; Mogoutov, A.; Cointet, J.P. Therapy’s shadow: A short history of the study of resistance to cancer chemotherapy. Front. Pharmacol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.; Timmerman, R.; Foster, R.S.; Roth, B.J.; Einhorn, L.H.; Nichols, C.R. Long-term toxicity of radiation therapy. In Holland-Frei Cancer Medicine, 6th ed.; Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Bast, R.C., Jr., Gansler, T.S., Holland, J.F., Frei, E., III, Eds.; Hamilton (ON): BC Decker, 2003; Available online: http://www.ncbi.nlm.nih.gov/books/NBK13384/ (accessed on 29 June 2016).
- Jagsi, R. Postmastectomy Radiation Therapy: An Overview for the Practicing Surgeon. ISRN Surg. 2013. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, T.A.; Strom, E.A.; Perkins, G.H.; McNeese, M.D. Controversies regarding the use of radiation after mastectomy in breast cancer. Oncologist 2002, 7, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, C.L.; Recht, A. Side effects of adjuvant treatment of breast cancer. N. Engl. J. Med. 2001, 344, 1997–2008. [Google Scholar] [PubMed]
- Haffty, B.G.; Hunt, K.K.; Harris, J.R.; Buchholz, T.A. Positive sentinel nodes without axillary dissection: Implications for the radiation oncologist. J. Clin. Oncol. 2011, 29, 4479–4481. [Google Scholar] [CrossRef] [PubMed]
- Oronsky, B.; Oronsky, N.; Knox, S.; Fanger, G.; Scicinski, J. Episensitization: Therapeutic tumor resensitization by epigenetic agents: A review and reassessment. Anticancer Agents Med. Chem. 2014, 14, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.; Figg, W.D. Epigenetic approaches to overcoming chemotherapy resistance. Lancet Oncol. 2015, 16, 1013–1015. [Google Scholar] [CrossRef]
- Rosato, R.R.; Almenara, J.A.; Maggio, S.C.; Coe, S.; Atadja, P.; Dent, P.; Grant, S. Role of histone deacetylase inhibitor-induced reactive oxygen species and DNA damage in LAQ-824/fludarabine antileukemic interactions. Mol. Cancer Ther. 2008, 7, 3285–3297. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.Y.; Park, Y.S.; Yang, K.; Kim, G.Y.; Kim, W.J.; Han, M.H.; Kang, H.S.; Choi, Y.H. Decitabine, a DNA methyltransferase inhibitor, induces apoptosis in human leukemia cells through intracellular reactive oxygen species generation. Int. J. Oncol. 2012, 41, 910–918. [Google Scholar] [PubMed]
- Ning, S.; Bednarski, M.; Oronsky, B.; Scicinski, J.; Saul, G.; Knox, S.J. Dinitroazetidines are a novel class of anticancer agents and hypoxia-activated radiation sensitizers developed from highly energetic materials. Cancer Res. 2012, 72, 2600–2608. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.A.; Gius, D.; Wink, D.A.; Krishna, M.C.; Russo, A.; Mitchell, J.B. Oxidative stress, redox, and the tumor microenvironment. Semin. Radiat. Oncol. 2004, 14, 259–266. [Google Scholar] [CrossRef] [PubMed]
- De Schutter, H.; Nuyts, S. Radiosensitizing potential of epigenetic anticancer drugs. Anticancer Agents Med. Chem. 2009, 9, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Rada-Iglesias, A. Pioneering barren land: Mitotic bookmarking by transcription factors. Dev. Cell 2013, 24, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Kadauke, S.; Blobel, G.A. Mitotic bookmarking by transcription factors. Epigenet. Chromatin 2013. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012, 21, 430–446. [Google Scholar] [CrossRef] [PubMed]
- Oronsky, B.T.; Oronsky, N.C.; Fanger, G.R.; Lybeck, M.; Caroen, S.Z.; Parker, C.W.; Scicinski, J. A review of two promising radiosensitizers in brain metastases: RRx-001 and 2-deoxyglucose. J. Cancer Sci. Ther. 2015, 7, 137–141. [Google Scholar] [CrossRef]
- Muller, B.A.; Dhalla, N.S. Mechanisms of the beneficial actions of ischemic preconditioning on subcellular remodeling in ischemic-reperfused heart. Curr. Cardiol. Rev. 2010, 6, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Doi, H.; Saar, M.; Santos, J.; Li, X.; Peehl, D.M.; Knox, S.J. Radioprotection and cell cycle arrest of intestinal epithelial cells by darinaparsin, a tumor radiosensitizer. Int. J. Radiat. Oncol. Biol. Phys. 2013, 87, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Scicinski, J.; Oronsky, B.; Ning, S.; Knox, S.; Peehl, D.; Kim, M.M.; Langecker, P.; Fanger, G. No to cancer: The complex and multifaceted role of nitric oxide and the epigenetic nitric oxide donor, RRx-001. Redox Biol. 2015, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Influence Gene Expression | |||||
|---|---|---|---|---|---|
| Change Type | Susceptible to Induced Change | Reproducible | Reversible | Persistent after Stressor Is Removed | Heritable |
| Genetic | Limited | Partially | No | Yes | Yes |
| Epigenetic | Yes | Yes | Yes | Yes | Yes |
| Metabolic | Yes | Yes | Yes | No | No |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oronsky, B.; Scicinski, J.; Kim, M.M.; Cabrales, P.; Salacz, M.E.; Carter, C.A.; Oronsky, N.; Lybeck, H.; Lybeck, M.; Larson, C.; et al. Turning on the Radio: Epigenetic Inhibitors as Potential Radiopriming Agents. Biomolecules 2016, 6, 32. https://doi.org/10.3390/biom6030032
Oronsky B, Scicinski J, Kim MM, Cabrales P, Salacz ME, Carter CA, Oronsky N, Lybeck H, Lybeck M, Larson C, et al. Turning on the Radio: Epigenetic Inhibitors as Potential Radiopriming Agents. Biomolecules. 2016; 6(3):32. https://doi.org/10.3390/biom6030032
Chicago/Turabian StyleOronsky, Bryan, Jan Scicinski, Michelle M. Kim, Pedro Cabrales, Michael E. Salacz, Corey A. Carter, Neil Oronsky, Harry Lybeck, Michelle Lybeck, Christopher Larson, and et al. 2016. "Turning on the Radio: Epigenetic Inhibitors as Potential Radiopriming Agents" Biomolecules 6, no. 3: 32. https://doi.org/10.3390/biom6030032
APA StyleOronsky, B., Scicinski, J., Kim, M. M., Cabrales, P., Salacz, M. E., Carter, C. A., Oronsky, N., Lybeck, H., Lybeck, M., Larson, C., Reid, T. R., & Oronsky, A. (2016). Turning on the Radio: Epigenetic Inhibitors as Potential Radiopriming Agents. Biomolecules, 6(3), 32. https://doi.org/10.3390/biom6030032