
Repair of DNA Double-Strand Breaks in Heterochromatin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
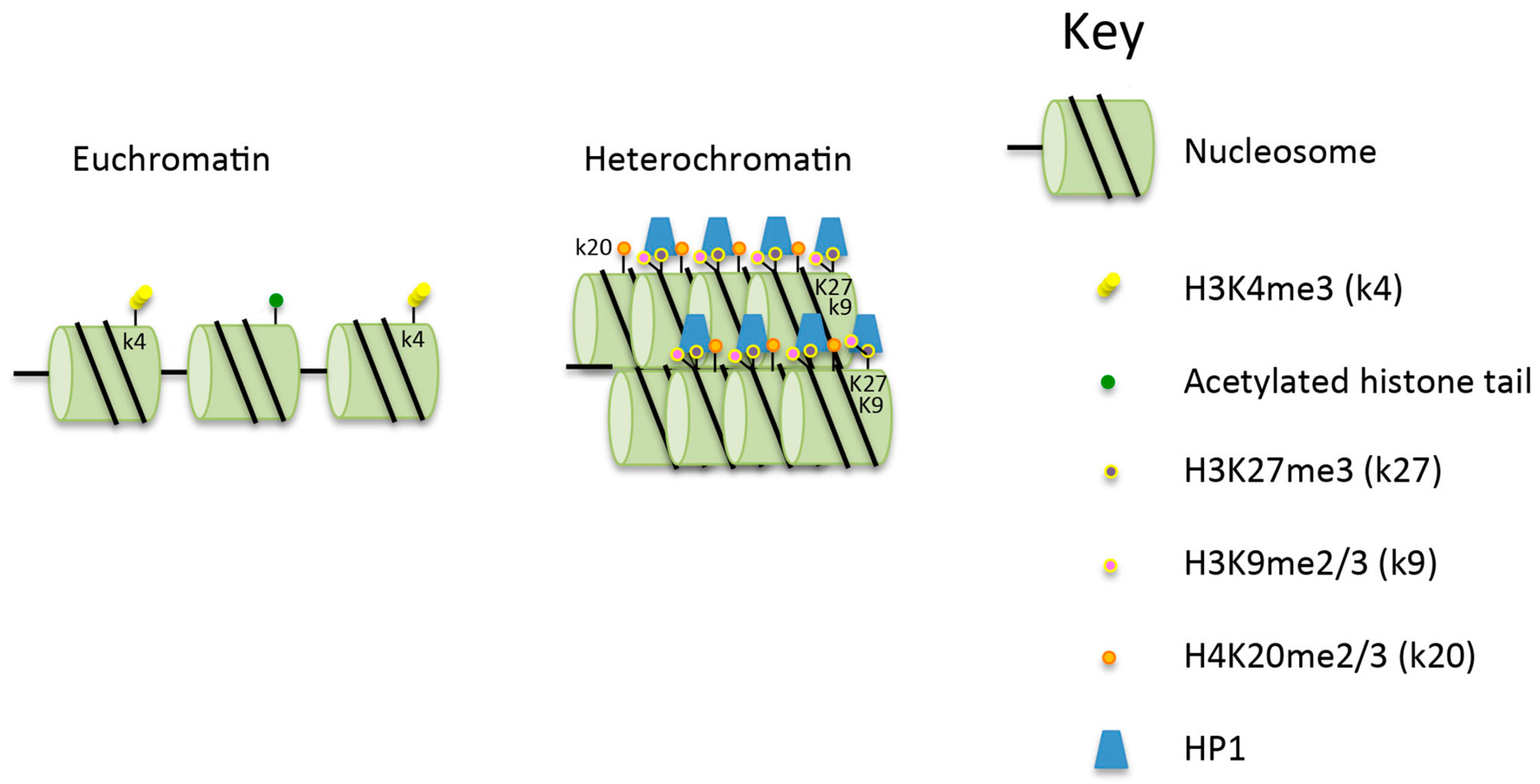
2. The Structure, Importance and Maintenance of Heterochromatin
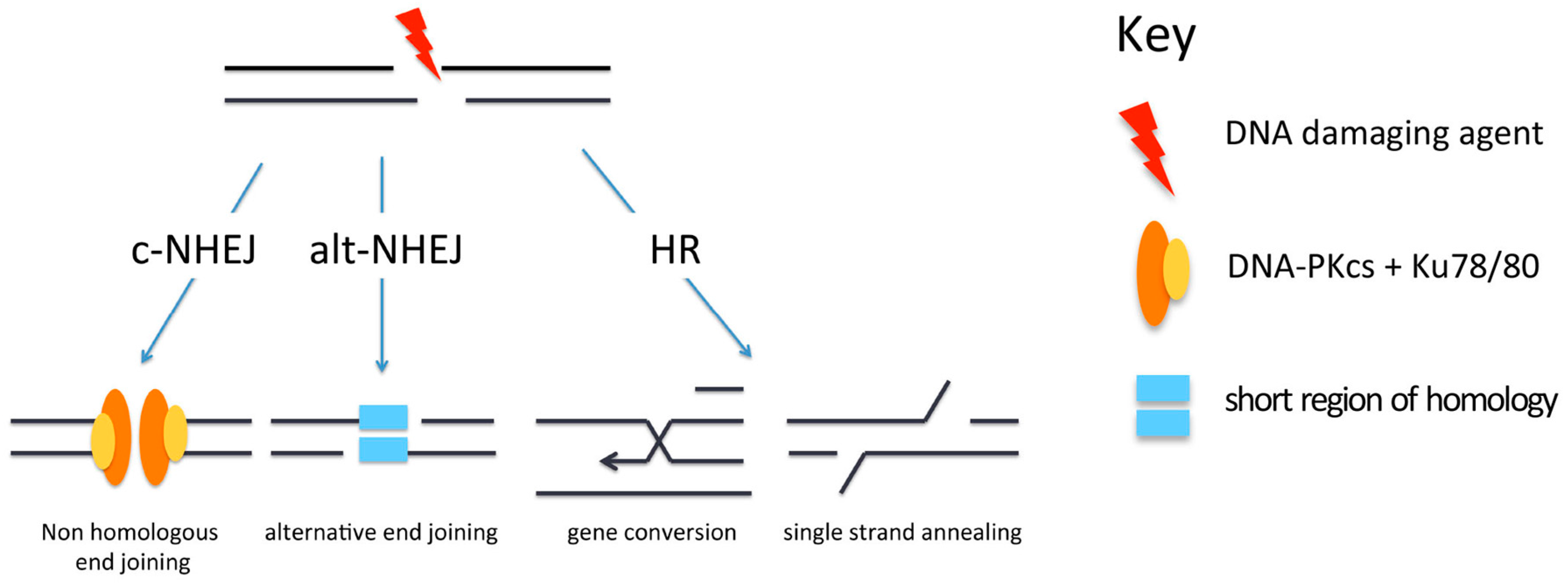
3. Repair of DSBs
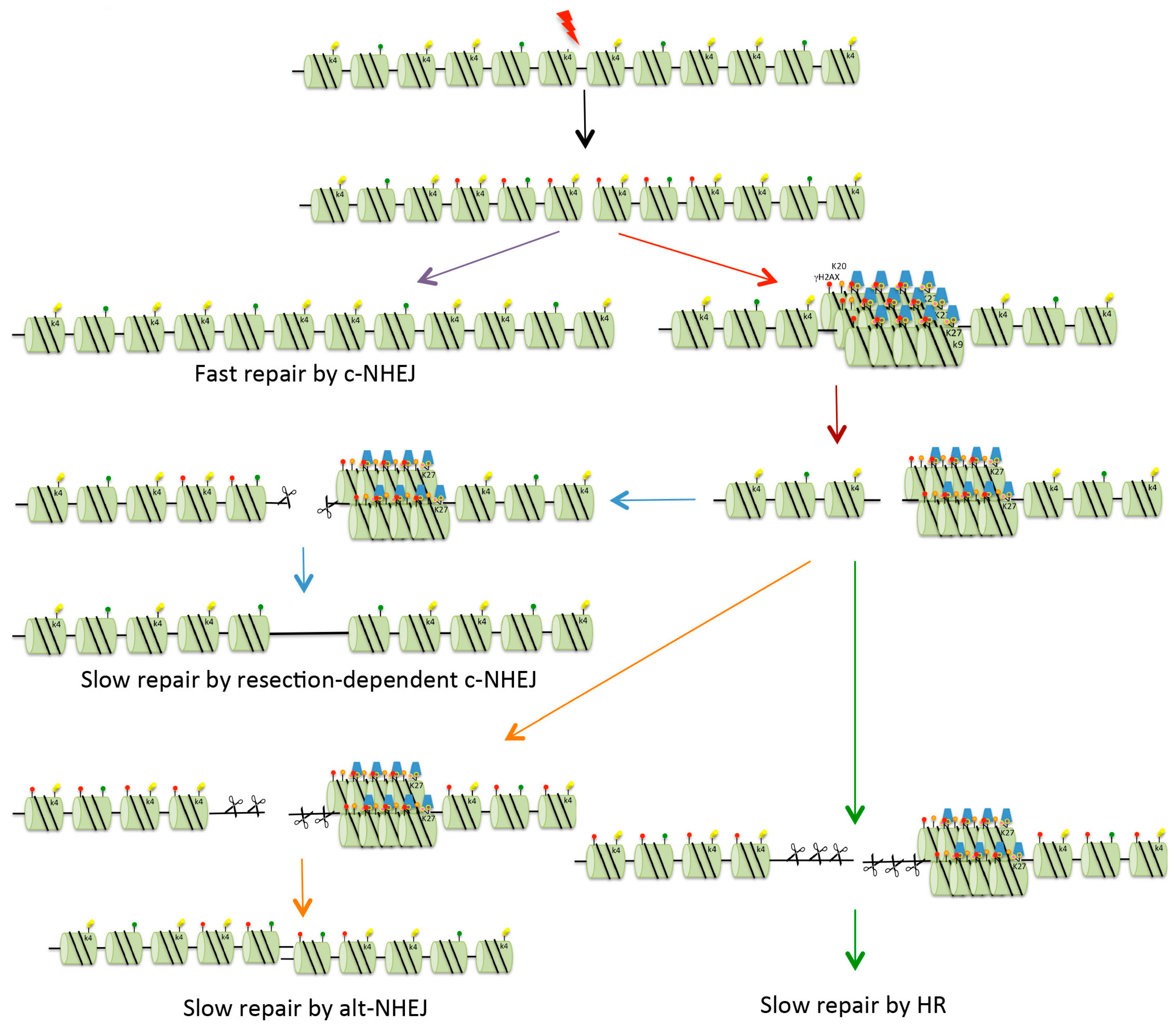
4. DSBs and Heterochromatin
4.1. Do DSBs Occur with Equal Frequency in EC and HC?
4.2. Once Created Are DSBs Protected in Heterochromatin?
4.3. Repair of DSBs in Heterochromatin
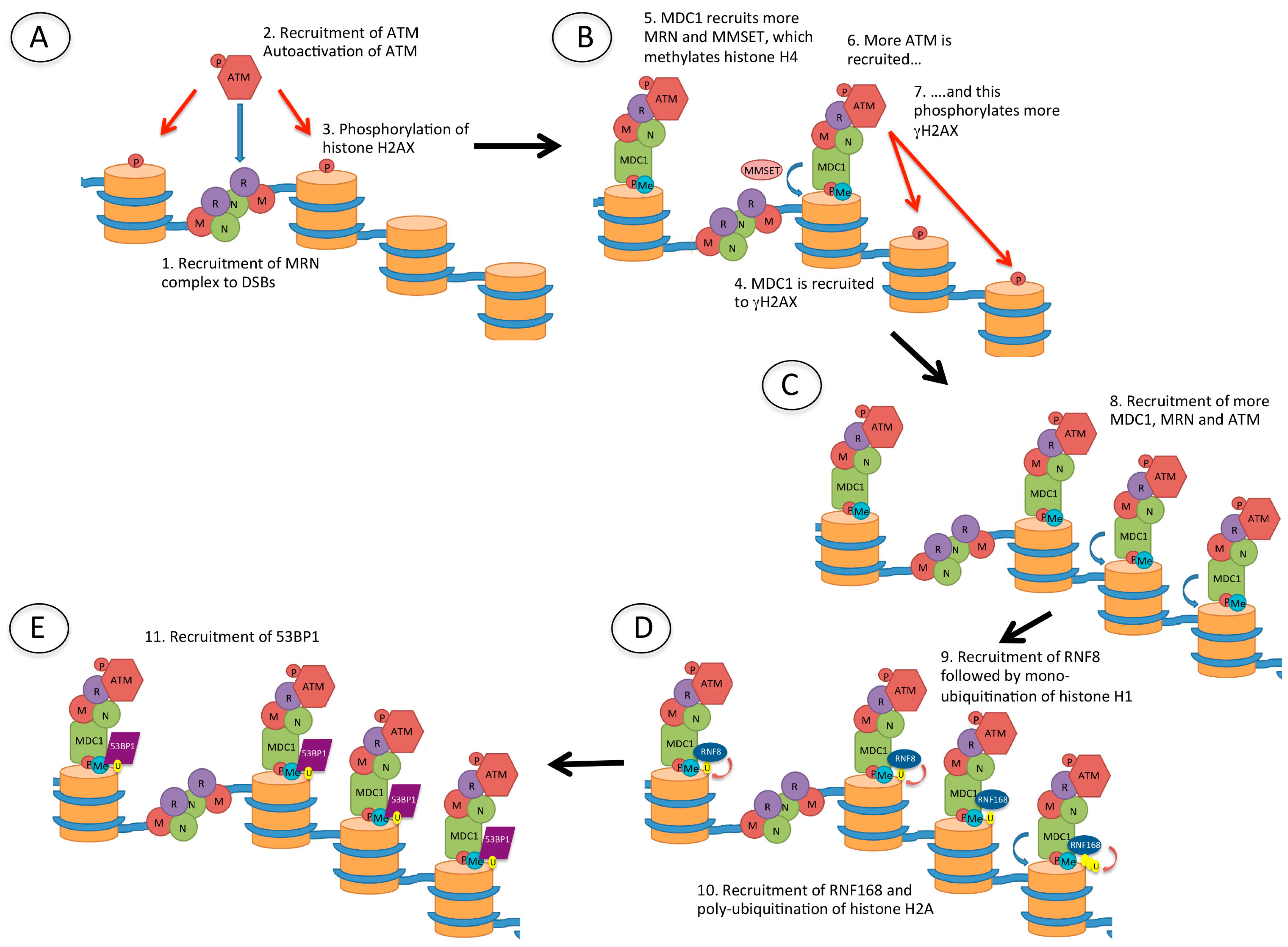
4.4. Role of 53BP1 in Repair of DSBs in Heterochromatin
4.5. Role of BRCA1
5. Summary
Acknowledgments
Conflicts of Interest
References
- Solovei, I.; Thanisch, K.; Feodorova, Y. How to rule the nucleus: Divide et impera. Curr. Opin. Cell Biol. 2016, 40, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.I.; Jia, S. Heterochromatin revisited. Nat. Rev. Genet. 2007, 8, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jia, S.T.; Jia, S. New insights into the regulation of heterochromatin. Trends Genet. 2016, 32, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Trojer, P.; Reinberg, D. Facultative heterochromatin: Is there a distinctive molecular signature? Mol. Cell 2007, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Binda, O. On your histone mark, SET, methylate! Epigenetics 2013, 8, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Towbin, B.D.; Gonzalez-Sandoval, A.; Gasser, S.M. Mechanisms of heterochromatin subnuclear localization. Trends Biochem. Sci. 2013, 38, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.C.; Friedman, J.R.; Rauscher, F.J. Targeting histone deacetylase complexes via KRAB-zinc finger proteins: The PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2α subunit of NuRD. Genes Dev. 2001, 15, 428–443. [Google Scholar] [CrossRef] [PubMed]
- Lyengar, S.; Ivanov, A.V.; Jin, V.X.; Rauscher, F.J.; Farnham, P.J. Functional analysis of KAP1 genomic recruitment. Mol. Cell. Biol. 2011, 31, 1833–1847. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, S.; Farnham, P.J. KAP1 Protein: An enigmatic master regulator of the genome. J. Biol. Chem. 2011, 286, 26267–26276. [Google Scholar] [CrossRef] [PubMed]
- Mermoud, J.E.; Rowbotham, S.P.; Varga-Weisz, P.D. Keeping chromatin quiet: How nucleosome remodeling restores heterochromatin after replication. Cell Cycle 2011, 10, 4017–4025. [Google Scholar] [CrossRef] [PubMed]
- Tyler, J.K. Nucleosomes find their place in life. Trends Genet. 2016, 12, 689–690. [Google Scholar] [CrossRef] [PubMed]
- Rowbotham, S.; Barki, L.; Neves-Costa, A.; Santos, F.; Dean, W.; Hawkes, N.; Choudhary, P.; Will, W.R.; Webster, J.; Oxley, D.; et al. Maintenance of silent chromatin through replication requires SWI/SNF-like chromatin remodeler SMARCAD1. Mol. Cell 2011, 42, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of single-strand annealing and its role in genome maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.C.; Kowalczykowski, S.C. Mechanics and single-molecule interrogation of DNA recombination. Ann. Rev. Biochem. 2016, 85, 193–226. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Guo, R.; Xu, D. Non-homologous end joining: Advances and frontiers. Acta Biochim. Biophys. Sin. 2016, 48, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Löbrich, M.; et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011, 30, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Löbrich, M. Artemis links ATM to double strand break rejoining. Cell Cycle 2005, 4, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Riballo, E.; Kühne, M.; Rief, N.; Doherty, A.; Smith, G.C.; Recio, M.J.; Reis, C.; Dahm, K.; Fricke, A.; Krempler, A.; et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to γ-H2AX foci. Mol. Cell 2004, 16, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Ghezraoui, H.; Piganeau, M.; Renouf, B.; Renaud, J.; Sallmyr, A.; Ruis, B.; Oh, S.; Tomkinson, A.E.; Hendrickson, E.; Giovannangeli, C.; et al. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol. Cell 2014, 55, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Rosidi, B.; Perrault, R.; Wang, M.; Zhang, L.; Windhofer, F.; Iliakis, G. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res. 2005, 65, 4020–4030. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Thorslund, T.; Ripplinger, A.; Hoffmann, S.; Wild, T.; Uckelmann, M.; Villumsen, B.; Narita, T.; Sixma, T.K.; Choudhary, C.; Bekker-Jensen, S.; et al. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 2015, 527, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Mattiroli, F.; Vissers, J.A.; van Dijk, W.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.; Sixma, T. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [PubMed]
- Gatti, M.; Pinato, S.; Maspero, E.; Soffientini, P.; Polo, S.; Penengo, L. A novel ubiquitin mark at the N-terminal tail of histone H2As targeted by RNF168 ubiquitin ligase. Cell Cycle 2012, 11, 2538–2544. [Google Scholar] [CrossRef] [PubMed]
- Moyal, L.; Lerenthal, Y.; Gana-Weisz, M.; Mass, G.; So, S.; Wang, S.; Eppink, B.; Chung, Y.; Shalev, G.; Shema, E.; et al. Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol. Cell 2011, 41, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Shema, E.; Moyal, L.; Oren, M. RNF20–RNF40: A ubiquitin-driven link between gene expression and the DNA damage response. FEBS Lett. 2011, 585, 2795–2802. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.D.; Benlekbir, S.; Fradet-Turcotte, A.; Sherker, A.; Julien, J.P.; McEwan, A.; Noordermeer, S.M.; Sicheri, F.; Rubinstein, J.L.; Durocher, D. The structural basis of modified nucleosome recognition by 53BP1. Nature 2016, 536, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Diaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.; Tkac, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; de Lange, T. 53BP1: Pro choice in DNA repair. Trends Cell Biol. 2013, 24, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Lottersberger, F.; Buonomo, S.B.; Sfeir, A.; de Lange, T. 53BP1 regulates DSB repair using Rif1 to control 5' end resection. Science 2013, 339, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Diaz, C.; Durocher, D. DNA repair pathway choice—A PTIP of the hat to 53BP1. EMBO Rep. 2013, 14, 665–666. [Google Scholar] [CrossRef] [PubMed]
- Ochs, F.; Somyajit, K.; Altmeyer, M.; Rask, M.; Lukas, J.; Lukas, C. 53BP1 fosters fidelity of homology-directed DNA repair. Nat. Struct. Mol. Biol. 2016, 23, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Kakarougkas, A.; Ismail, A.; Klement, K.; Goodarzi, A.A.; Conrad, S.; Freire, R.; Shibata, A.; Lobrich, M.; Jeggo, P.A. Opposing roles for 53BP1 during homologous recombination. Nucleic Acids Res. 2013, 41, 9719–9731. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Sossick, A.J.; Boulton, S.J.; Jackson, S.P. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J. Cell Sci. 2012, 125, 3529–3534. [Google Scholar] [CrossRef] [PubMed]
- Schuster-Böckler, B.; Lehner, B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 2012, 488, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Gordenin, D.A. Hypermutation in human cancer genomes: Footprints and mechanisms. Nat. Rev. Cancer 2014, 14, 786–800. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Capetillo, O.; Nussenzweig, A. ATM Breaks into Heterochromatin. Mol. Cell 2008, 31, 303–304. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, P.; Panyutin, I.V.; Remeeva, E.; Neumann, R.D.; Panyutin, I.G.; Panyutin, I.; Neumann, R.; Panyutin, I. Effect of Chromatin Structure on the Extent and Distribution of DNA Double Strand Breaks Produced by Ionizing Radiation; Comparative Study of hESC and Differentiated Cells Lines. Int. J. Mol. Sci. 2016, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Yakovlev, V.A. Cells redox environment modulates BRCA1 expression and DNA homologous recombination repair. Free Radic. Biol. Med. 2016, 101, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Ramiro, A.; Ramírez-Ortega, D.; Pérez de la Cruz, V.; Hérnandez-Pedro, N.Y.; González-Esquivel, D.F.; Sotelo, J.; Pineda, B. Role of redox status in development of glioblastoma. Front. Immunol. 2016, 7, 156. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, N.; Jeyasekharan, A.D.; Bernal, J.A.; Venkitaraman, A.R. HP1-β mobilization promotes chromatin changes that initiate the DNA damage response. Nature 2008, 453, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Luijsterburg, M.S.; Dinant, C.; Lans, H.; Stap, J.; Wiernasz, E.; Lagerwerf, S.; Warmerdam, D.O.; Lindh, M.; Brink, M.C.; Dobrucki, J.W.; et al. Heterochromatin protein 1 is recruited to various types of DNA damage. J. Cell Biol. 2009, 185, 577–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldeyron, C.; Soria, G.; Roche, D.; Cook, A.J.; Almouzni, G. HP1α recruitment to DNA damage by p150CAF-1 promotes homologous recombination repair. J. Cell Biol. 2011, 193, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Downs, J.A.; Nussenzweig, M.C.; Nussenzweig, A. Chromatin dynamics and the preservation of genetic information. Nature 2007, 447, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Kruhlak, M.J.; Celeste, A.; Dellaire, G.; Fernandez-Capetillo, O.; Müller, W.G.; McNally, J.G.; Bazett-Jones, D.P.; Nussenzweig, A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell Biol. 2006, 172, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Cowell, I.G.; Sunter, N.J.; Singh, P.B.; Austin, C.A.; Durkacz, B.W.; Tilby, M.J. γH2AX foci form preferentially in euchromatin after ionising-radiation. PLoS ONE 2007, 2, e1057. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Kruhlak, M.; Dotiwala, F.; Nussenzweig, A.; Haber, J.E. Heterochromatin is refractory to γ-H2AX modification in yeast and mammals. J. Cell Biol. 2007, 178, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Ryu, T.; Spatola, B.; Delabaere, L.; Bowlin, K.; Hopp, H.; Kunitake, R.; Karpen, G.H.; Chiolo, I. Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat. Cell Biol. 2015, 17, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.L.; Xiang, J.F.; Kong, N.; Cai, X.J.; Xie, A.Y. Buried territories: Heterochromatic response to DNA double-strand breaks. Acta Biochim. Biophys. Sin. 2016, 48, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Jeggo, P.; Lobrich, M. The influence of heterochromatin on DNA double strand break repair: Getting the strong, silent type to relax. DNA Repair 2010, 9, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, N.; Chen, Y.C.M.; Spector, D.L.; de Lange, T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 2008, 456, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Noon, A.T.; Shibata, A.; Rief, N.; Lobrich, M.; Stewart, G.S.; Jeggo, P.A.; Goodarzi, A.A. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat. Cell Biol. 2010, 12, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Stanley, F.K.; Moore, S.; Goodarzi, A.A. CHD chromatin remodelling enzymes and the DNA damage response. Mutat. Res. 2013, 750, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Kurka, T.; Jeggo, P.A. KAP-1 phosphorylation regulates CHD3 nucleosome remodeling during the DNA double-strand break response. Nat. Struct. Mol. Biol. 2011, 18, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Klement, K.; Luijsterburg, M.S.; Pinder, J.B.; Cena, C.S.; Nero, V.D.; Wintersinger, C.M.; Dellaire, G.; Attikum, H.V.; Goodarzi, A.A. Opposing ISWI- and CHD-class chromatin remodeling activities orchestrate heterochromatic DNA repair. J. Cell Biol. 2014, 207, 717–733. [Google Scholar] [CrossRef] [PubMed]
- Baldock, R.A.; Day, M.; Wilkinson, O.J.; Cloney, R.; Jeggo, P.A.; Oliver, A.W.; Watts, F.Z.; Pearl, L.H. ATM localization and heterochromatin repair depend on direct interaction of the 53BP1-BRCT2 domain with γH2AX. Cell Rep. 2015, 13, 2081–2089. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Noon, A.T.; Deckbar, D.; Ziv, Y.; Shiloh, Y.; Lobrich, M.; Jeggo, P.A. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 2008, 31, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Alagoz, M.; Katsuki, Y.; Ogiwara, H.; Ogi, T.; Shibata, A.; Kakarougkas, A.; Jeggo, P. SETDB1, HP1 and SUV39 promote repositioning of 53BP1 to extend resection during homologous recombination in G2 cells. Nucleic Acids Res. 2015, 18, 7931–7944. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Matsuoka, S.; Ballif, B.A.; Zhang, D.; Smogorzewska, A.; Gygi, S.P.; Elledge, S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007, 316, 1194–1198. [Google Scholar] [CrossRef] [PubMed]
- Huen, M.S.; Sy, S.M.; Chen, J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol. 2009, 11, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Brzovic, P.S.; Rajagopal, P.; Hoyt, D.W.; King, M.C.; Klevit, R.E. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol. 2001, 8, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yu, X. Function of BRCA1 in the DNA Damage Response Is Mediated by ADP-Ribosylation. Cancer Cell 2013, 23, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Tsuruga, T.; Kuroda, T.; Takeuchi, J.; Wu, W.; Ohta, T. The BARD1/HP1 interaction: Another clue to heterochromatin involvement in homologous recombination. Mol. Cell. Oncol. 2016, 3, e1030535. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kuo, C.Y.; Stark, J.M.; Shih, H.M.; Ann, D.K. HP1 promotes tumor suppressor BRCA1 functions during the DNA damage response. Nucleic Acids Res. 2013, 41, 5784–5798. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Nishikawa, H.; Fukuda, T.; Vittal, V.; Asano, M.; Miyoshi, Y.; Klevit, R.E.; Ohta, T. Interaction of BARD1 and HP1 Is Required for BRCA1 Retention at Sites of DNA Damage. Cancer Res. 2015, 75, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Densham, R.M.; Garvin, A.J.; Stone, H.R.; Strachan, J.; Baldock, R.A.; Daza-Martin, M.; Fletcher, A.; Blair-Reid, S.; Beesley, J.; Johal, B.; et al. Human BRCA1–BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct. Mol. Biol. 2016, 23, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Costelloe, T.; Louge, R.; Tomimatsu, N.; Mukherjee, B.; Martini, E.; Khadaroo, B.; Dubois, K.; Wiegant, W.W.; Thierry, A.; Burma, S.; et al. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 2012, 489, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Tsouroula, K.; Furst, A.; Rogier, M.; Heyer, V.; Maglott-Roth, A.; Ferrand, A.; Reina-San-Martin, B.; Soutoglou, E. Temporal and spatial uncoupling of DNA double strand break repair pathways within mammalian heterochromatin. Mol. Cell 2016, 63, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Foltánková, V.; Legartová, S.; Kozubek, S.; Hofer, M.; Bártová, E. DNA-damage response in chromatin of ribosomal genes and the surrounding genome. Gene 2013, 522, 156–167. [Google Scholar] [CrossRef] [PubMed]




© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watts, F.Z. Repair of DNA Double-Strand Breaks in Heterochromatin. Biomolecules 2016, 6, 47. https://doi.org/10.3390/biom6040047
Watts FZ. Repair of DNA Double-Strand Breaks in Heterochromatin. Biomolecules. 2016; 6(4):47. https://doi.org/10.3390/biom6040047
Chicago/Turabian StyleWatts, Felicity Z. 2016. "Repair of DNA Double-Strand Breaks in Heterochromatin" Biomolecules 6, no. 4: 47. https://doi.org/10.3390/biom6040047
APA StyleWatts, F. Z. (2016). Repair of DNA Double-Strand Breaks in Heterochromatin. Biomolecules, 6(4), 47. https://doi.org/10.3390/biom6040047