
Functional Regulation of the Plasma Protein Histidine-Rich Glycoprotein by Zn2+ in Settings of Tissue Injury



Abstract
:1. Introduction
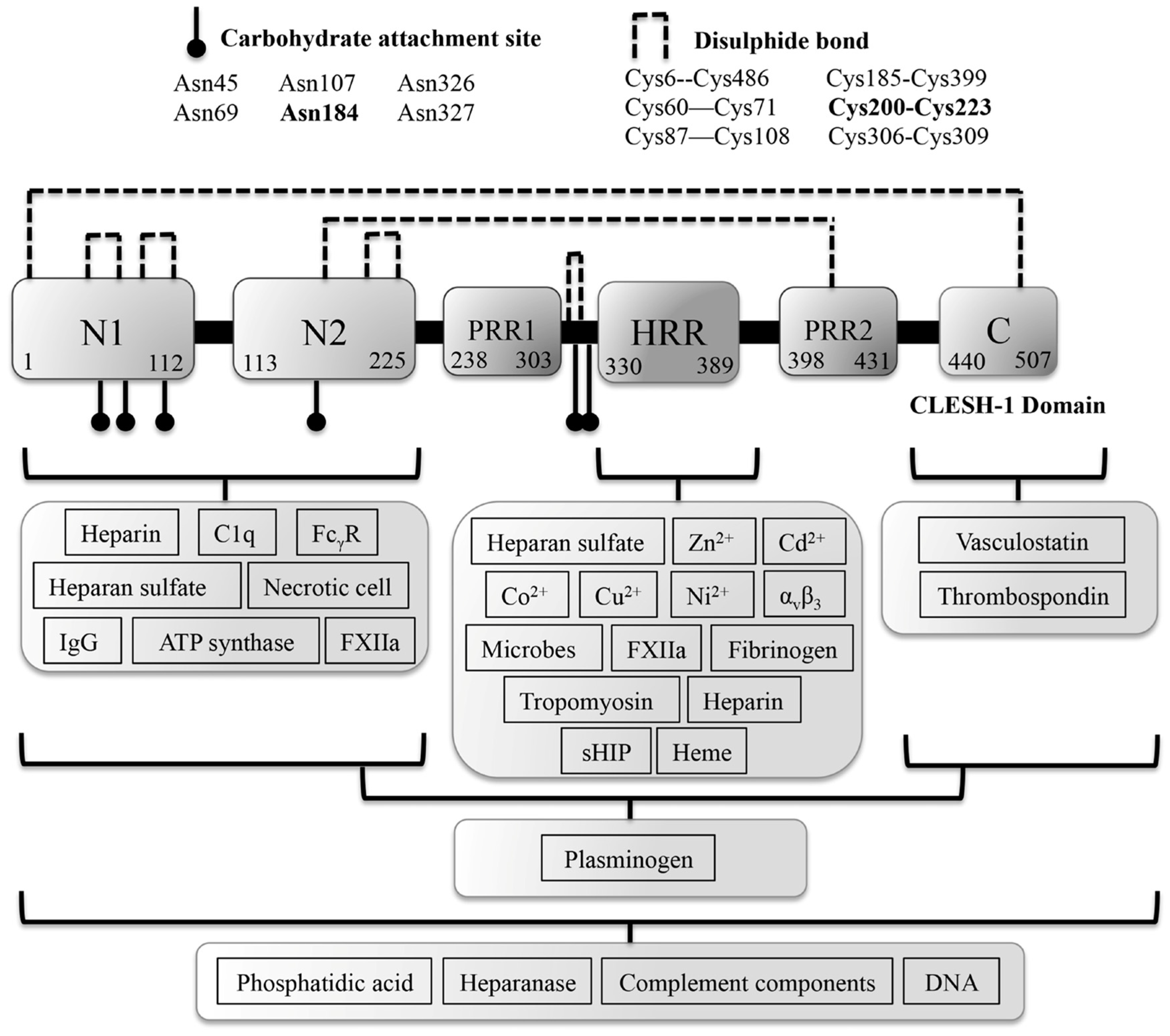
2. The Functional Significance of Zn2+ Coordination Geometry in Histidine-Rich Glycoprotein Function
2.1. HRG Binding to Other Divalent Metal Cations
3. HRG Binds Activated Factor XIIa, Plasminogen, Heparin, Fibrinogen and Fibrin to Regulate Coagulation
3.1. HRG Serves as a Pro-Coagulant Molecule by Neutralizing Heparin
3.2. HRG Binds Activated Factor XIIa to Subdue Its Pro-Coagulant Activities
3.3. HRG Interacts with Fibrin(ogen) to Regulate the Formation of the Platelet Plug
3.4. A Summary of HRG Activities in Context of Hemostasis
4. The Antibacterial and Antifungal Properties of HRG at Sites of Infection
4.1. HRG Central HRR Core Contributes to Multiple Modes of Killing Bacteria and Involves Changes in pH
4.1.1. HRG Antibacterial Properties under Acidic Conditions
4.1.2. HRG Coordinates with Zn2+ Around Physiological pH to Maintain Antibacterial Activity
4.1.3. HRG Central HRR Core is an Obstacle for Invading Microbes
4.2. HRG Exhibits Antifungal Activity by Utilizing Its Histidine-Rich Core
5. Plasmin-Mediated Cleavage of HRG Alters Its Biological Function and Regulates Plasminogen Activation
6. The Role of HRG in the Innate Immunity and Apoptotic/Necrotic Cell Clearance
6.1. HRG Maintains the Formation of Soluble Immune Complexes
6.1.1. Insoluble Immune Complexes and Regulation of Complement System by HRG
6.2. HRG Potentiates Ingestion of Apoptotic and Necrotic Cell Clearance through Phagocytosis
6.2.1. HRG Potentiates Ingestion of Necrotic Cells by Targeting Heparan Sulfate
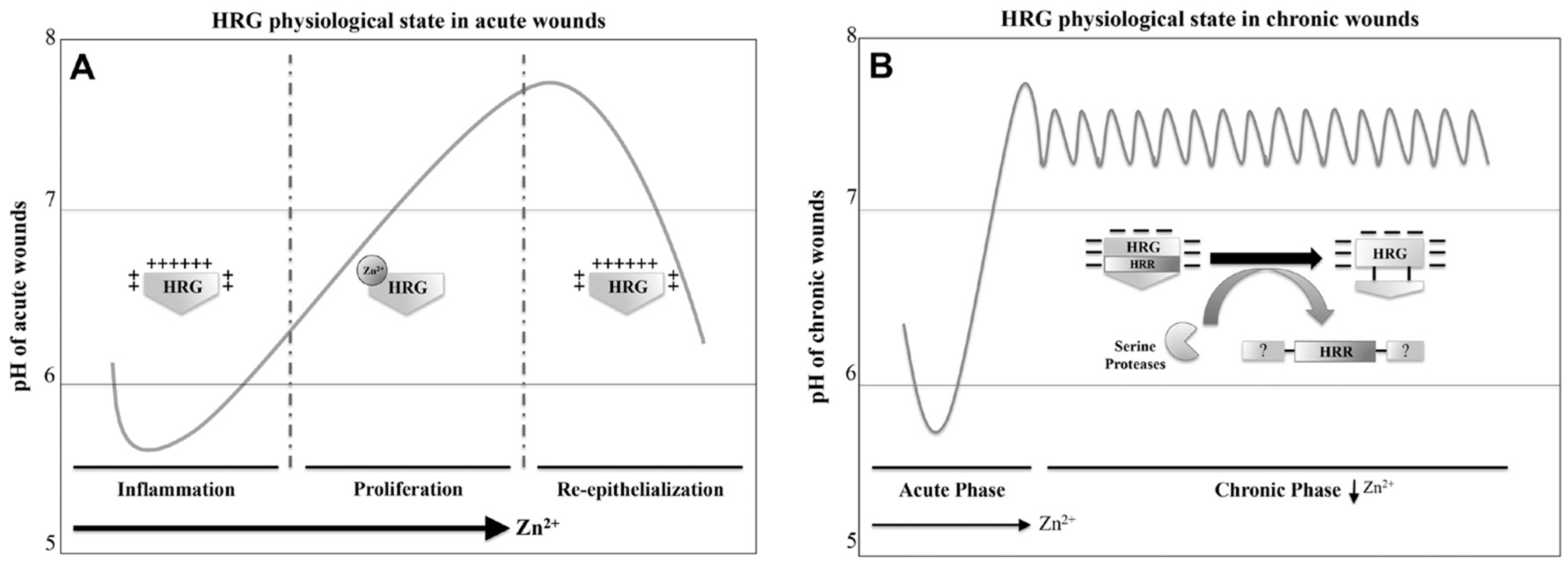
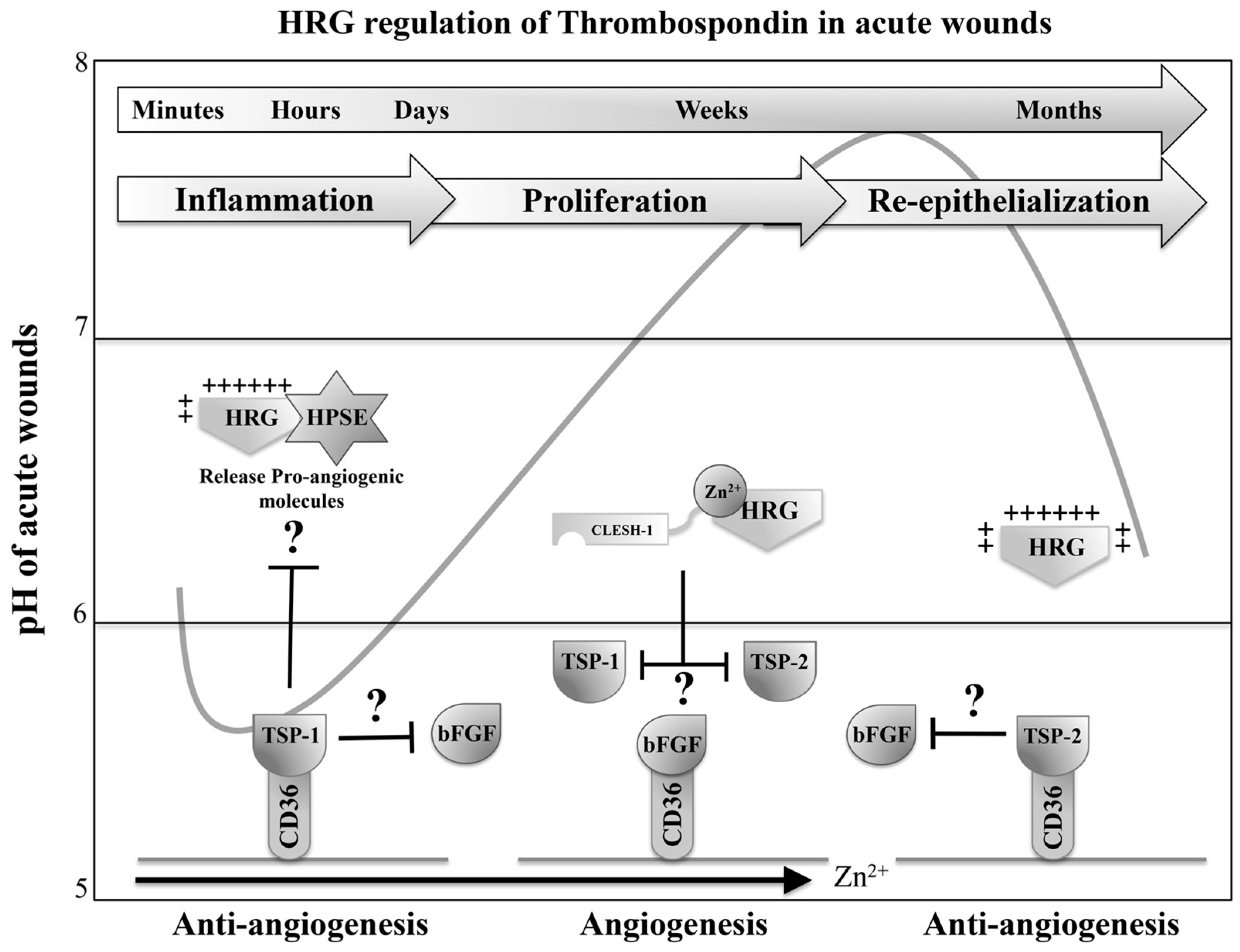
7. HRG Regulates Angiogenesis during Wound Healing
7.1. HRG Potentially Inhibits the Anti-Angiogenic Activity of Thrombospondin 1 and 2 during Acute Wound-Healing Processes
7.2. Chronic Wound Environment and the Potential Role of HRG
7.2.1. HRG Anti-Angiogenic Peptide Suppresses Tumour Growth
8. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Haupt, H.; Heimburger, N. Human serum proteins with high affinity for carboxymethylcellulose. I. Isolation of lysozyme, C1q and 2 hitherto unknown -globulins. Hoppe Seylers Z. Physiol. Chem. 1972, 353, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Heimburger, N.; Haupt, H.; Kranz, T.; Baudner, S. Human serum proteins with high affinity for carboxymethylcellulose. II. Physico-chemical and immunological characterization of a histidine 3, 8S- 2-glycoprotein (CM-Protein I) (in German). Hoppe Seylers Z. Physiol. Chem. 1972, 353, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Poon, I.K.; Patel, K.K.; Davis, D.S.; Parish, C.R.; Hulett, M.D. Histidine-rich glycoprotein: The Swiss Army Knife of mammalian plasma. Blood 2011, 117, 2093–2101. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.L.; Hulett, M.D.; Parish, C.R. Histidine-rich glycoprotein: A novel adaptor protein in plasma that modulates the immune, vascular and coagulation systems. Immunol. Cell Biol. 2005, 83, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Borza, D.-B.; Morgan, W.T. Histidine-Proline-rich Glycoprotein as a Plasma pH Sensor. J. Biol. Chem. 1998, 273, 5492–5499. [Google Scholar] [CrossRef]
- Schneider, L.A.; Korber, A.; Grabbe, S.; Dissemond, J. Influence of pH on wound-healing: A new perspective for wound-therapy? Arch. Dematol. Res. 2007, 298, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Koide, T.; Foster, D.; Yoshitake, S.; Davie, E.W. Amino acid sequence of human histidine-rich glycoprotein derived from the nucleotide sequence of its cDNA. Biochemistry 1986, 25, 2220–2225. [Google Scholar] [CrossRef] [PubMed]
- Koide, T.; Foster, D.; Odani, S. The heparin-binding site(s) of histidine-rich glycoprotein as suggested by sequence homology with antithrombin III. FEBS Lett. 1986, 194, 242–244. [Google Scholar] [CrossRef]
- Lansdown, A.B.G. Calcium: A potential central regulator in wound healing in the skin. Wound Repair Regen. 2002, 10, 271–285. [Google Scholar] [CrossRef]
- Schornack, P.A.; Gillies, R.J. Contributions of cell metabolism and H+ diffusion to the acidic pH of tumors. Neoplasia 2003, 5, 135–145. [Google Scholar] [CrossRef]
- Landsdown, A.B.G.; Mirastschijiski, U.; Stubbs, N.; Scanlon, E.; Agren, M.S. Zinc in wound healing: Theoretical, experimental and clinical aspects. Wound Repair Regen. 2007, 15, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Thulin, Å.; Ringvall, M.; Dimberg, A.; Kårehed, K.; Väisänen, T.; Väisänen, M.-R.; Hamad, O.; Wang, J.; Bjerkvig, R.; Nilsson, B.; et al. Activated Platelets Provide a Functional Microenvironment for the Antiangiogenic Fragment of Histidine-Rich Glycoprotein. Mol. Cancer Res. 2009, 7, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Morgan, W.T. Human serum histidine-rich glycoprotein I. Interactions with heme, metal ions and organic ligands. Biochim. Biophys. Acta 1978, 533, 319–333. [Google Scholar] [CrossRef]
- MacQuarrie, J.L.; Stafford, A.R.; Yau, J.W.; Leslie, B.A.; Vu, T.T.; Fredenburgh, J.C.; Weitz, J.I. Histidine-rich glycoprotein binds factor XIIa with high affinity and inhibits contact initiated coagulation. Blood 2011, 117, 4134–4141. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.K.; Poon, I.K.H.; Talbo, G.H.; Perugini, M.A.; Taylor, N.L.; Ralph, T.J.; Hoogenraad, N.J.; Hulett, M.D. New method for purifying histidine-rich glycoprotein from human plasma redefines its functional properties. IUBMB Life 2013, 65, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Kassaar, O.; Mcmahon, S.A.; Thompson, R.; Botting, C.H.; Naismith, J.H.; Stewart, A.J. Crystal structure of histidine-rich glycoprotein N2 domain reveals redox activity at an interdomain disulphide bridge: Implications for angiogenic regulation. Blood 2014, 123, 1948–1955. [Google Scholar] [CrossRef] [PubMed]
- Arwert, E.N.; Hoste, E.; Watt, F.M. Epithelial stem cells, wound healing and cancer. Nat. Rev. 2012, 12, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Shaw, T.J.; Martin, P. Wound repair at a glance. J. Cell Sci. 2009, 122, 3209–3213. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Singer, A.J.; Clark, R.A.F. Cutaneous wound healing. N. Engl. J. Med. 1999, 341, 738–746. [Google Scholar] [PubMed]
- Whitehouse, R.C.; Prasad, A.S.; Rabbani, P.I.; Cossack, Z.T. Zinc in Plasma, Neutrophils, Lymphocytes, and Erythrocytes as Determined by Flameless Atomic Absorption Spectrophotometry. Clin. Chem. 1982, 28, 478–480. [Google Scholar]
- Harris, W.R.; Keen, C. Calculations of the distribution of zinc in a computer model of human serum. J. Nutr. 1989, 119, 1677–1682. [Google Scholar] [PubMed]
- Sarkar, B. Metal-Protein Interactions in Transport, Accumulation and Excretion of Metals. Biol. Trace Element Res. 1989, 21, 137–144. [Google Scholar] [CrossRef]
- Gorodetsky, R.; Mou, X.; Blankenfeld, A.; Marx, G. Platelet multielemental composition, lability, and subcellular localization. Am. J. Hematol. 1993, 42, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Stewart, A.J.; Sadler, P.J.; Pinheiro, T.J.T.; Blindauer, C.A. Albumin as a zinc carrier: Properties of its high-affinity zinc-binding site. Biochem. Soc. Trans. 2008, 36, 1317–1321. [Google Scholar] [CrossRef] [PubMed]
- Pasha, Q.; Malik, S.A.; Shah, M.H. Statistical analysis of trace metals in the plasma of cancer patients versus controls. J. Hazardous Mater. 2008, 153, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.J.; Stafford, A.R.; Leslie, B.A.; Kim, P.Y.; Vaezzadeh, N.; Ni, R.; Fredenburgh, J.C.; Weitz, J.I. Zinc delays clot lysis by attenuating plasminogen activation and plasmin-mediated fibrin degradation. J. Thromb. Haemost. 2015, 113, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Poon, I.K.H.; Olsson, A.-K.; Hulett, M.D.; Parish, C.R. Regulation of histidine-rich glycoprotein (HRG) function via plasmin-mediated proteolytic cleavage. Biochem. J. 2009, 424, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Kumar, A.; Durani, S. Analysis of the structural consensus of the zinc coordination centers of metalloprotein structures. Biochim. Biophys. Acta 2007, 1774, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. New perspectives of zinc coordination environments in proteins. J. Inorg. Biochem. 2012, 111, 110–116. [Google Scholar] [CrossRef] [PubMed]
- CJ, W.; TR, Y.; AG, W.; Z, X. CopC Protein from Pseudomonas fluorescens SBW25 Features a Conserved Novel High-Affinity Cu(II) Binding Site. Inorg. Chem. 2015, 54, 2950–2959. [Google Scholar]
- L, Z.; M, K.; MJ, M.; Z, X.; AG, W. Intermolecular Transfer of Copper Ions from the CopC Protein of Pseudomonas syringae. Crystal Structures of Fully Loaded Cu(I)Cu(II) forms. J. Am. Chem. Soc. 2006, 128, 5834–5850. [Google Scholar]
- Lawton, T.J.; Kenney, G.E.; Hurley, J.D.; Rosenzweig, A.C. The CopC Family: Structural and Bioinformatic Insights into a Diverse Group of Periplasmic Copper Binding Proteins. Biochemistry 2016, 55, 2278–2290. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Metals on the move: Zinc ions in cellular regulation and in the coordination dynamics of zinc proteins. Biometals 2011, 24, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Rydengård, V.; Shannon, O.; Lundqvist, K.; Kacprzyk, L.; Chalupka, A.; Olsson, A.-K.; Mörgelin, M.; Jahnen-Dechent, W.; Malmsten, M.; Schmidtchen1, A. Histidine-Rich Glycoprotein Protects from Systemic Candida Infection. PLoS Pathog. 2008, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Morgan, W.T. Interactions of the Histidine-Rich Glycoprotein of Serum with Metals. Biochemistry 1981, 20, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Hutchens, T.W.; Yip, T.-T. Synthetic metal-binding protein surface domains for ion-dependent interaction chromatography. J. Chromatogr. 1992, 604, 133–141. [Google Scholar] [CrossRef]
- Potocki, S.; Valensinb, D.; Kozlowski, H. The specificity of interaction of Zn2+, Ni2+ and Cu2+ ions with the histidine-rich domain of the TjZNT1 ZIP family transporter. Dalton Trans. 2014, 43, 10215–10223. [Google Scholar] [CrossRef] [PubMed]
- Ilari, A.; Alaleona, F.; Petrarca, P.; Battistoni, A.; Chiancone, E. The X-ray Structure of the Zinc Transporter ZnuA from Salmonella enterica Discloses a Unique Triad of Zinc-Coordinating Histidines. J. Mol. Biol. 2011, 409, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.; Zhang, Y.; Sun, X.; Watt, R.M.; He, Q.-Y.; Huang, J.-D.; Wilcox, D.E.; Sun, H. Thermodynamic and Kinetic Aspects of Metal Binding to the Histidine-rich Protein, Hpn. J. Am. Chem. Soc. 2006, 128, 11330–11331. [Google Scholar] [CrossRef] [PubMed]
- Paksi, Z.; Jancsó, A.; Pacello, F.; Nagy, N.; Battistoni, A.; Gajda, T. Copper and zinc binding properties of the N-terminal histidine-rich sequence of Haemophilus ducreyi Cu,Zn superoxide dismutase. J. Biol. Chem. 2008, 102, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Waite, J.H. Proteins in Load-Bearing Junctions: The Histidine-Rich Metal-Binding Protein of Mussel Byssus. Biochemistry 2006, 45, 14223–14231. [Google Scholar] [CrossRef] [PubMed]
- Borza, D.-B.; Tatum, F.M.; Morgan, W.T. Domain structure and conformation of histidine-proline-rich glycoprotein. Biochemistry 1996, 35, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Hulett, M.D.; Parish, C.R. Murine histidine-rich glycoprotein: Cloning, characterization and cellular origin. Immunol. Cell. Biol. 2000, 78, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Guthans, S.L.; Morgan, W.T. The interaction of Zinc, Nickel and Cadmium with serum albumin and histidine-rich glycoprotein assessed by equilibrium dialysis and immunoadsorbent chromatography. Arch. Biochim. Biophys. 1982, 218, 320–328. [Google Scholar] [CrossRef]
- Waldron, K.J.; Rutherford, J.C.; Ford, D.; Robinson, N.J. Metalloproteins and metal sensing. Nature 2009, 460, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Kassaar, O.; Schwarz-linek, U.; Blindauer, C.A.; Stewart, A.J. Plasma free fatty acid levels influence Zn(2+)-dependent histidine-rich glycoprotein–heparin interactions via an allosteric switch on serum albumin. J. Thromb. Haemost. 2015, 13, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priebatsch, K.M.; Poon, I.K.H.; Patel, K.K.; Kvansakul, M.; Hulett, M.D. Divalent metal binding by histidine-rich glycoprotein differentially regulates higher order oligomerisation and proteolytic processing. FEBS Lett. 2016, 591, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Rydengård, V.; Olsson, A.-K.; Morgelin, M.; Schmidtchen, A. Histidine-rich glycoprotein exerts antibacterial activity. FEBS J. 2007, 274, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Greener, B.; Hughes, A.A.; Bannister, N.P.; Douglass, J. Proteases and pH in chronic wounds. J. Wound Care. 2005, 14, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, H.H.; Heemskerk, J.W.M.; Levi, M.; Reitsma, P.H. New fundamentals in hemostasis. Physiol. Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida-Straeten, N.; Ensslen, S.; Schäfer, C.; Wöltje, M.; Denecke, B.; Moser, M.; Gräber, S.; Wakabayashi, S.; Koide, T.; Jahnen-Dechent, W. Enhanced blood coagulation and fibrinolysis in mice lacking histidine-rich glycoprotein. J. Thromb. Haemost. 2005, 3, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Leung, L.L.K. Interaction of Histidine-rich Glycoprotein with Fibrinogen and Fibrin. J. Clin. Invest. 1986, 77, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.L.; Hulett, M.D.; Altin, J.G.; Hogg, P.; Parish, C.R. Plasminogen Is Tethered with high Affinity to the Cell Surface by the Plasma Protein, Histidine-rich Glycoprotein. J. Biol. Chem. 2004, 279, 38267–38276. [Google Scholar] [CrossRef]
- Kluszynski, B.A.; Kim, C.; Faulk, W.P. Zinc as a Cofactor for Heparin Neutralization by Histidine-rich Glycoprotein. J. Biol. Chem. 1997, 272, 13541–13547. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.L.; Hulett, M.D.; Parish, C.R. Histidine-rich glycoprotein binds to cell surface heparan sulfate via its N-terminal domain following Zn2+ chelation. J. Biol. Chem. 2004, 279, 30114–30122. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.T.; Zhou, J.; Leslie, B.A.; Stafford, A.R.; Fredenburgh, J.C.; Ni, R.; Qiao, S.; Vaezzadeh, N.; Jahnen-Dechent, W.; Monia, B.P.; Gross, P.L.; Weitz, J.I. Arterial thrombosis is accelerated in histidine-rich glycoprotein deficient mice. Blood 2015, 11, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.T.; Leslie, B.A.; Stafford, A.R.; Zhou, J.; Fredenburgh, J.C.; Weitz, J.I. Histidine-rich glycoprotein binds DNA and RNA and attenuates their capacity to activate the intrinsic coagulation pathway. J. Thromb. Haemost. 2015, 115, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Riedel, T.; Suttnar, J.; Brynda, E.; Houska, M.; Medved, L.; Dyr, J.E. Fibrinopeptides A and B release in the process of surface fibrin formation. Blood 2011, 117, 1700–1706. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, S. New insights into the functions of histidine-rich glycoprotein. Int. Rev. Cell Mol. Biol. 2012, 304, 467–493. [Google Scholar]
- Vu, T.T.; Stafford, A.R.; Leslie, B.A.; Kim, P.Y.; Fredenburgh, J.C.; Weitz, J.I. Histidine-rich Glycoprotein Binds Fibrin(ogen) with High Affinity and Competes with Thrombin for Binding to the Υ'-Chain. J. Biol. Chem. 2011, 286, 30314–30323. [Google Scholar] [CrossRef] [PubMed]
- Olsen, H.M.; Parish, C.R.; Altin, J.G. Histidine-rich glycoprotein binds to T-cell lines and its effect on T-cell substratum adhesion is strongly potentiated by zinc. Immunology 1996, 88, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Shannon, O.; Rydengård, V.; Schmidtchen, A.; Mörgelin, M.; Alm, P.; Sørensen, O.E.; Björck, L. Histidine-rich glycoprotein promotes bacterial entrapment in clots and decreases mortality in a mouse model of sepsis. Blood 2010, 116, 2365–2372. [Google Scholar] [CrossRef]
- Vanwildemeersch, M.; Olsson, A.-K.; Gottfridsson, E.; Claesson-Welsh, L.; Lindahl, U.; Spillmann, D. The Anti-angiogenic His/Pro-rich fragment of histidine-rich glycoprotein binds to endothelial cell heparan sulfate in a Zn2+ dependent manner. J. Biol. Chem. 2006, 281, 10298–10304. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.-L.; Horn, M.K., III. Histidine-rich glycoprotein plus zinc to neutralize heparin. J. lab. Clin. Med. 2002, 139, 211–217. [Google Scholar] [CrossRef]
- Lane, D.A.; Pejler, G.; Flynn, A.M.; Thompson, E.A.; Lindahl, U. Neutralization of Heparin-related Saccharides by Histidine-rich Glycoprotein and Platelet Factor 4. J. Biol. Chem. 1986, 261, 3980–3986. [Google Scholar]
- Kacprzyk, L.; Rydengård, V.; Mörgelin, M.; Davoudi, M.; Pasupuleti, M.; Malmsten, M.; Schmidtchen, A. Antimicrobial activity of histidine-rich peptides is dependent on acidic conditions. Biochim. Biophys. Acta 2007, 1768, 2667–2680. [Google Scholar] [CrossRef] [PubMed]
- Brogden, N.K.; Mehalick, L.; Fisher, C.L.; Wertz, P.W.; Brogden, K.A. The emerging role of peptides and lipids as antimicrobial epidermal barriers and modulators of local inflammation. Skin Pharmacol. Physiol. 2012, 25, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Streicher, W.W.; Malmström, J.; Pozdnyakova, M.; Frick, I.-M.; Karlsson, L.B.; Cazzamali, G.; Malmström, I.; Rosenberger, G.; Kahn, C.F.; Varjosalo, M.; Wisniewska, L.M.; Happonen, L. Functional and Structural Properties of a Novel Protein and Virulence Factor (Protein sHIP) in Streptococcus pyogenes. J. Biol. Chem. 2014, 289, 18175–18188. [Google Scholar]
- Netea, M.G.; Brown, G.D.; Kullberg, B.J.; Gow, N.A.R. An integrated model of the recognition of Candida albicans by the innate immune system. Nat. Rev. 2008, 6, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Hasmann, A.; Gewessler, U.; Hulla, E.; Schneider, K.P.; Binder, B.; Francesko, A.; Tzanov, T.; Schintler, M.; Palen, J.V.d.; Guebitz, G.M.; Wehrschuetz-Sigl, E. Sensor materials for the detection of human neutrophil elastase and cathepsin G activity in wound fluid. Exp. Dermatol. 2011, 20, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Parry, M.A.A.; Zhang, X.C.; Bode, W. Molecular mechanisms of plasminogen activation: Bacterial cofactors provide clues. Reviews 2000, 99, 53–59. [Google Scholar] [CrossRef]
- Fay, W.P.; Garg, N.; Sunkar, M. Vascular functions of the plasminogen activation system. Arterioscler Thromb. Vasc. Biol. 2007, 27, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Juarez, J.C.; Guan, X.; Shipulina, N.V. Histidine-Proline-rich Glycoprotein has Potent Antiangiogenic Activity Mediated Through the Histidine-Proline-rich Domain. Cancer Res. 2002, 62, 5344–5350. [Google Scholar] [PubMed]
- Bosshart, H.; Heinzelmann, M. Endotoxin-neutralizing effects of histidine-rich peptides. FEBS Lett. 2003, 553, 135–140. [Google Scholar] [CrossRef]
- Doñate, F.; Juarez, J.C.; Guan, X. Peptides derived from the histidine-proline domain of histidine-proline-rich glycoprotein bind to tropomyosin and have antiangiogenic and antitumor activities. Cancer Res. 2004, 64, 5812–5817. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.-K.; Larsson, H.; Dixelius, J.; Johansson, I.; Lee, C.; Oellig, C.; Bjork, I.; Claesson-Welsh, L. A Fragment of Histidine-Rich Glycoprotein Is a Potent Inhibitor of Tumor Vascularization. Cancer Res. 2004, 64, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Dixelius, J.; Olsson, A.-K.; Thulin, A.s.; Lee, C.; Johansson, I.; Claesson-Welsh, L. Minimal active domain and mechanism of action of the angiogenesis inhibiton histidine rich glycoprotein. Cancer Res. 2006, 66, 2089–2097. [Google Scholar] [CrossRef]
- Nowak, P.; Zgirski, A. Effects of metal ions on activity of plasmin. Biol. Trace Element Res. 2003, 93, 87–94. [Google Scholar] [CrossRef]
- Bjarnsholt, T.; Kirketerp-Mller, K.; Jensen, P.Ø.; Madsen, K.G.; Phipps, R.; Krogfelt, K.; Hiby, N.; Givskov, M. Why chronic wounds will not heal: A novel hypothesis. Wound Repair Regen. 2008, 16, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, R.L.; Leung, L.L.K.; Harpel, P.C.; Nachman, R.L. Platelet Thrombospondin Forms a Trimolecular Complex with Plasminogen and Histidine-rich Glycoprotein. J. Clin. Investig. 1985, 75, 2065–2073. [Google Scholar] [CrossRef]
- Simantov, R.; Febbraio, M.; Crombie, R.; Asch, A.S.; Nachman, R.L.; Silverstein, R.L. Histidine-rich glycoprotein inhibits the antiangiogenic effect of thrombospondin-1. J. Clin. Investig. 2001, 107, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Walport, M.J. Complement: First of Two Parts. N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [PubMed]
- Sjöberg, A.P.; Trouw, L.A.; Blom, A.M. Complement activation and inhibition: A delicate balance. Trends Immunol. 2009, 30, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Gorgani, N.N.; Smith, B.A.; Kono, D.H.; Theofilopoulos2, A.N. Histidine-Rich Glycoprotein Binds to DNA and FcγRI and Potentiates the Ingestion of Apoptotic Cells by Macrophages. J. Immunol. 2002, 169, 4745–4751. [Google Scholar] [CrossRef] [PubMed]
- Gorgani, N.N.; Parish, C.R.; Smith, S.B.E.; Altin, J.G. Histidine-Rich Glycoprotein Binds to Human IgG and C1q and Inhibits the Formation of Insoluble Immune Complexes. Biochemistry 1997, 36, 6653–6662. [Google Scholar] [CrossRef] [PubMed]
- Poon, I.K.; Hulett, M.D.; Parish, C.R. Histidine-rich glycoprotein is a novel plasma pattern recognition molecule that recruits IgG to facilitate necrotic cell clearance via FcγRI on phagocytes. Blood 2010, 115, 2473–2482. [Google Scholar] [CrossRef] [PubMed]
- Gorgani, N.N.; Altin, J.G.; parish, C.R. Histidine-rich glycoprotein regulates the binding of monomeric IgG and immune complexes to monocytes. Int. Immunol. 1999, 11, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Manderson, G.A.; Martin, M.; Önnerfjord, P.; Saxne, T.; Schmidtchen, A.; Mollnes, T.E.; Heinegård, D.; Blom, A.M. Interactions of histidine-rich glycoprotein with immunoglobulins and proteins of the complement system. Mol. Immunol. 2009, 46, 3388–3398. [Google Scholar] [CrossRef] [PubMed]
- Gorgani, N.N.; Altin, J.G.; Parish, C.R. Histidine-rich glycoprotein prevents the formation of insoluble immune complexes by rheumatoid factor. Immunology 1999, 98, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Gorgani, N.N.; Parish, C.R.; Altin, J.G. Differential Binding of Histidine-rich Glycoprotein (HRG) to Human IgG Subclasses and IgG Molecules Containing k and l Light Chains. J. Biol. Chem. 1999, 274, 29633–29640. [Google Scholar] [CrossRef] [PubMed]
- Blom, A.M.; Kask, L.; Ramesh, B.; Hillarp, A. Effects of zinc on factor I cofactor activity of C4b-binding protein and factor H. Arch. Biochim. Biophys. 2003, 418, 108–118. [Google Scholar] [CrossRef]
- Marx, G.; Korner, G.; Mou, X.; Gorodetsky, R. Packagin Zinc, Fibrinogen, and Factor XIII in Platelet α-Granules. J. Cell. Physiol. 1993, 156, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Poon, I.K.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. 2014, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Atkin-Smith, G.K.; Tixeira, R.; Paone, S.; Mathivanan, S.; Collins, C.; Liem, M.; Goodall, K.J.; Ravichandran, K.S.; Hulett, M.D.; Poon, I.K.H. A novel mechanism of generating extracellular vesicles during apoptosis via a beads-on-a-string membrane structure. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Poon, I.; Hulett, M.; Parish, C. Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell Death Diff. 2010, 17, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Poon, I.K.; Parish, C.R.; Hulett, M.D. Histidine-rich glycoprotein functions cooperatively with cell surface heparan sulfate on phagocytes to promote necrotic cell uptake. J. Leukoc. Biol. 2010, 88, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.L.; Poon, I.K.H.; Hulett, M.D.; Parish, C.R. Histidine-rich Glycoprotein Specifically Binds to Necrotic Cells via Its Amino-terminal Domain and Facilitates Necrotic Cell Phagocytosis. J. Biol. Chem. 2005, 280, 35733–35741. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.A.S.; Parish, C.R. Heparan sulfate: A ubiquitous glycosaminoglycan with multiple roles in immunity. Front. Immunol. 2013, 4, 1–7. [Google Scholar]
- Simantov, R.; Febbraio, M.; Silverstein, R.L. The antiangiogenic effect of thrombospondin-2 is mediated by CD36 and modulated by histidine-rich glycoprotein. Matrix Biol. 2005, 24, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Bielefeld, K.A.; Amini-Nik, S.; Alman, B.A. Cutaneous wound healing: Recruiting developmental pathways for regeneration. Cell. Mol. Life Sci. 2013, 70, 2059–2081. [Google Scholar] [CrossRef] [PubMed]
- Hale, J.S.; Li, M.; Sinyuk, M.; Jahnen-Dechent, W.; Lathia, J.D.; Silverstein, R.L. Context Dependent Role of the CD36 - Thrombospodin - Histidine-Rich Glycoprotein Axis in Tumor Angiogenesis and Growth. PLoS ONE 2012, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Poon, I.K.; Yee, D.Y.; Jones, A.L.; Wood, R.J.; Davis, D.S.; Freeman, C.; Parish, C.R.; Hulett, M.D. Histidine-rich glycoprotein binds heparanase and regulates its enzymatic activity and cell surface interactions. Int. J. Biochem. Cell Biol. 2010, 42, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, J.; Kirsner, R. Pathophysiology of acute wound healing. Clin. Dermatology. 2007, 25, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.; Nigam, Y. Naturally derived factors and their role in the promotion of angiogenesis for the healing of chronic wounds. Angiogenesis 2013, 16, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Martins, V.L.; Caley, M.; O’Toole, E.A. Matrix metalloproteinases and epidermal wound repair. Cell Tissue Res. 2013, 351, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Harding, K.G.; Morris, H.L.; Patel, G.K. Science, medicine, and the future Healing chronic wounds. Clin. Rev. 2002, 324, 160–163. [Google Scholar] [CrossRef]
- Schäfer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. 2008, 9, 628–638. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Matrisian, L.M. Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 2007, 7, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Ikemoto, Y.; Usami, A.; Koide, T.; Wakabayashi, S. High affinity interactions between histidine-rich glycoprotein and the cell surface type ATP synthase on T-cells. Biochim. Biophys. Acta 2009, 1788, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Klenotic, P.A.; Huang, P.; Palomo, J.; Kaur, B.; Meir, E.G.V.; Vogelbaum, M.A.; Febbraio, M.; Gladson, C.L.; Silverstein, R.L. Histidine-Rich Glycoprotein Modulates the Anti-Angiogenic Effects of Vasculostatin. Am. J. Pathol. 2010, 176, 2039–2049. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HRG Ligand Interactions | ||||||
|---|---|---|---|---|---|---|
| Zn2+ regulated interactions | pH changes | References | ||||
| HRG Ligands | Promoted by Zn2+ | Inhibited by Zn2+/or reduced affinity | Acidic (pH < 7.4) | pH ≥ 7.4 | ||
| Immunological Processes | Heparin | ✔ | ✔ | ✔ | [55,65,66] | |
| Heparan Sulfate | ✔ | ✔ | ✔ | [56,64] | ||
| C1q | ✔ | N/D | ✔ | [87,90] | ||
| Microbes | ✔ | ✔ | ✔ | [35,49,67] | ||
| IgG1κ | ✔ | N/D | ✔ | [92] | ||
| IgGλ | ✔ | N/D | ✔ | [92] | ||
| ATP Synthase | N/D | N/D | ✔ | [111] | ||
| Phosphatidic acid | N/D | N/D | ✔ | [15] | ||
| Heparanase | N/C | ✔ | ✔ | [104] | ||
| Vasculature Processes | Vasculostatin | N/D | N/D | ✔ | [112] | |
| Thrombospondin-1 | N/D | N/D | ✔ | [83] | ||
| Thrombospondin-2 | N/D | N/D | ✔ | [101] | ||
| Plasminogen | N/C | ✔ | ✔ | [28,54] | ||
| Plasmin | ✔ | ✖ | ✔ | [28] | ||
| Fibrinogen | ✔ | ✖ | ✖ | [53,61] | ||
| Fibrin | ✔ | ✖ | ✖ | [53,61] | ||
| Tropomyosin | ✔ | N/D | ✖ | [77] | ||
| FXIIa | ✔ | N/D | ✔ | [14] | ||
| αvβ3 | ✔ | N/D | ✔ | [79] | ||
| Heparanase | N/C | ✔ | ✔ | [104] | ||
| Heparin | ✔ | ✔ | ✔ | [55,65,66] | ||
| DNA/RNA | N/D | N/D | ✔ | [58,86] | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Priebatsch, K.M.; Kvansakul, M.; Poon, I.K.H.; Hulett, M.D. Functional Regulation of the Plasma Protein Histidine-Rich Glycoprotein by Zn2+ in Settings of Tissue Injury. Biomolecules 2017, 7, 22. https://doi.org/10.3390/biom7010022
Priebatsch KM, Kvansakul M, Poon IKH, Hulett MD. Functional Regulation of the Plasma Protein Histidine-Rich Glycoprotein by Zn2+ in Settings of Tissue Injury. Biomolecules. 2017; 7(1):22. https://doi.org/10.3390/biom7010022
Chicago/Turabian StylePriebatsch, Kristin M., Marc Kvansakul, Ivan K. H. Poon, and Mark D. Hulett. 2017. "Functional Regulation of the Plasma Protein Histidine-Rich Glycoprotein by Zn2+ in Settings of Tissue Injury" Biomolecules 7, no. 1: 22. https://doi.org/10.3390/biom7010022
APA StylePriebatsch, K. M., Kvansakul, M., Poon, I. K. H., & Hulett, M. D. (2017). Functional Regulation of the Plasma Protein Histidine-Rich Glycoprotein by Zn2+ in Settings of Tissue Injury. Biomolecules, 7(1), 22. https://doi.org/10.3390/biom7010022