
DNA Methylation Targeting: The DNMT/HMT Crosstalk Challenge

Abstract
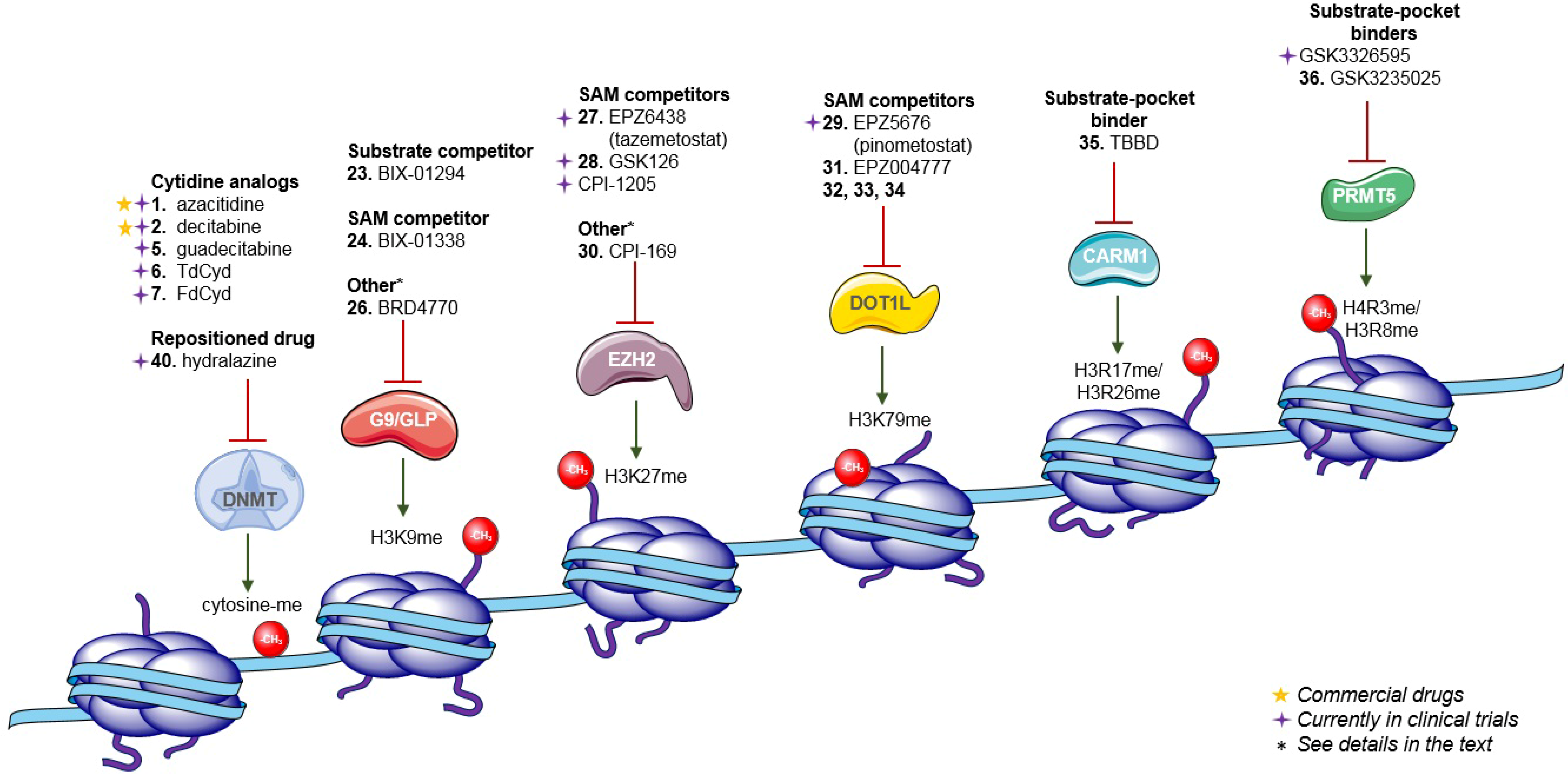
:1. Introduction
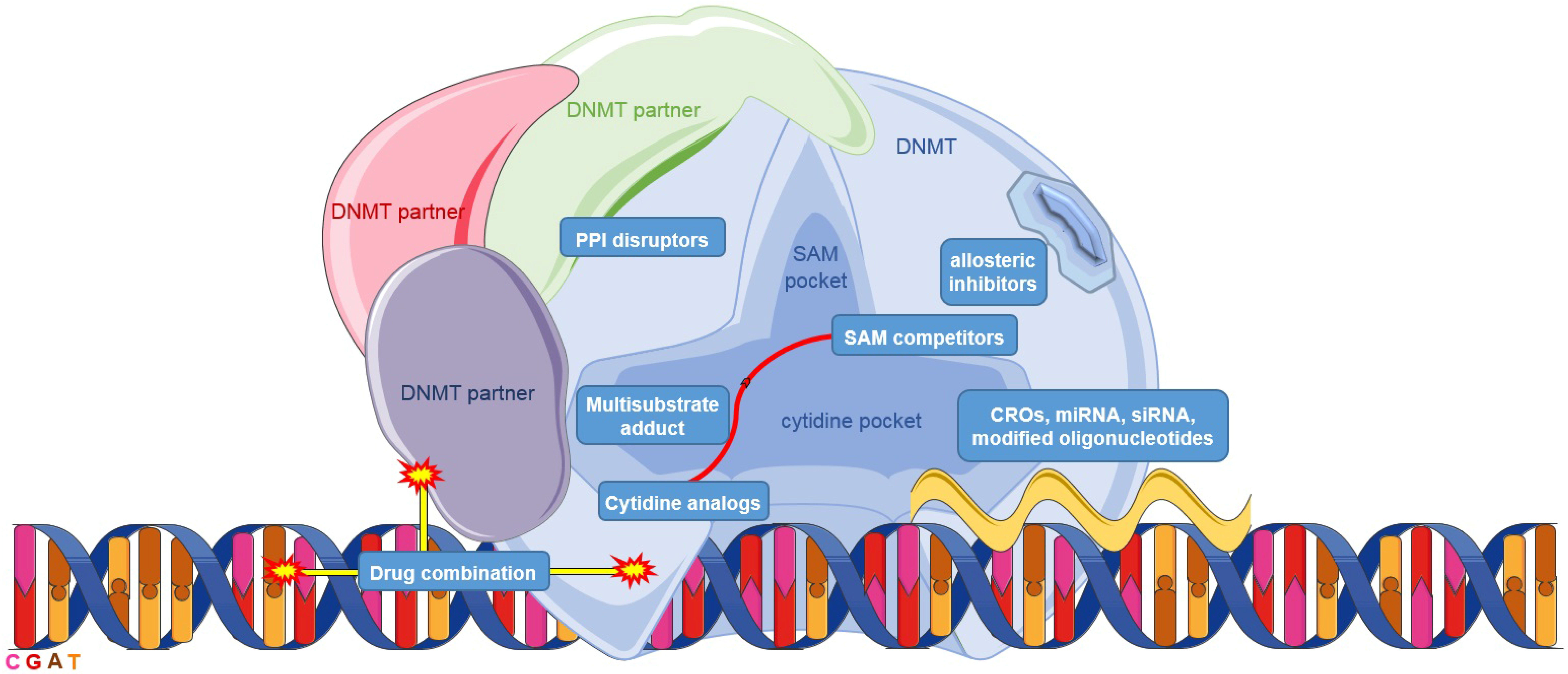
2. Inhibition of DNA Methylation
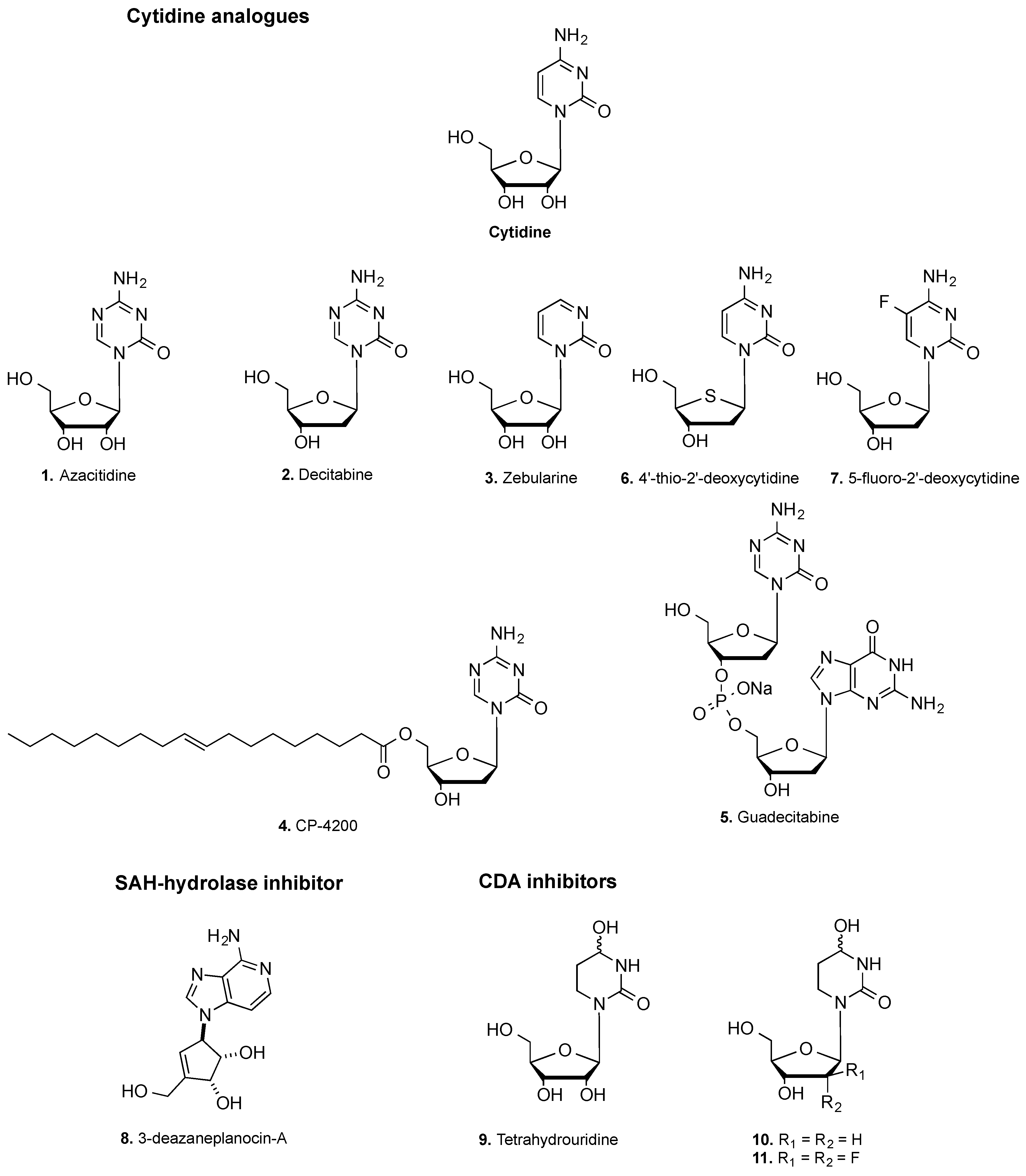
2.1. Cytidine Analogs
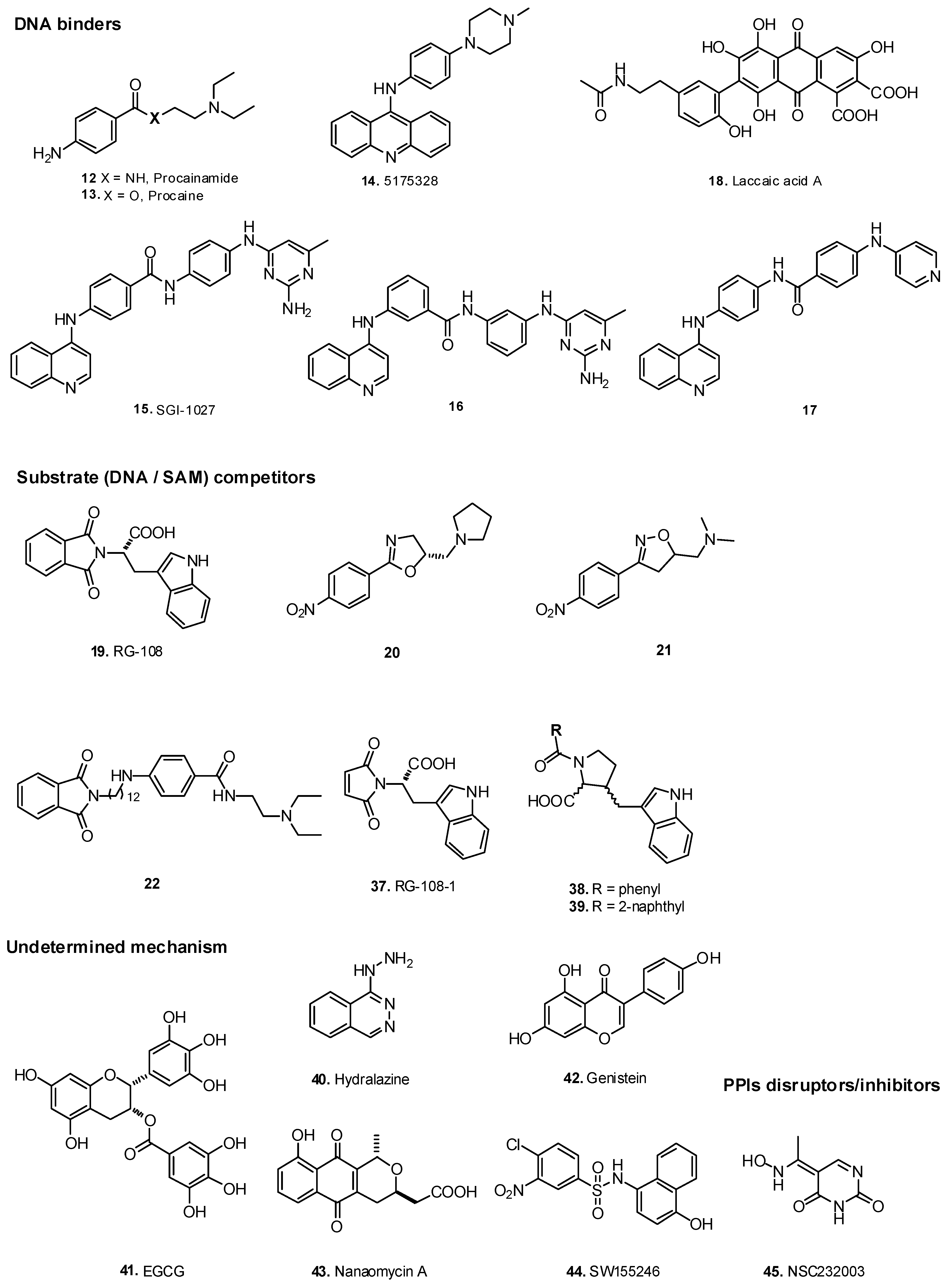
2.2. DNA Binders
2.3. Oligonucleotides
2.4. S-adenosyl-l-methionine Cofactor Competitors
3. Inhibition of Histone Methylation
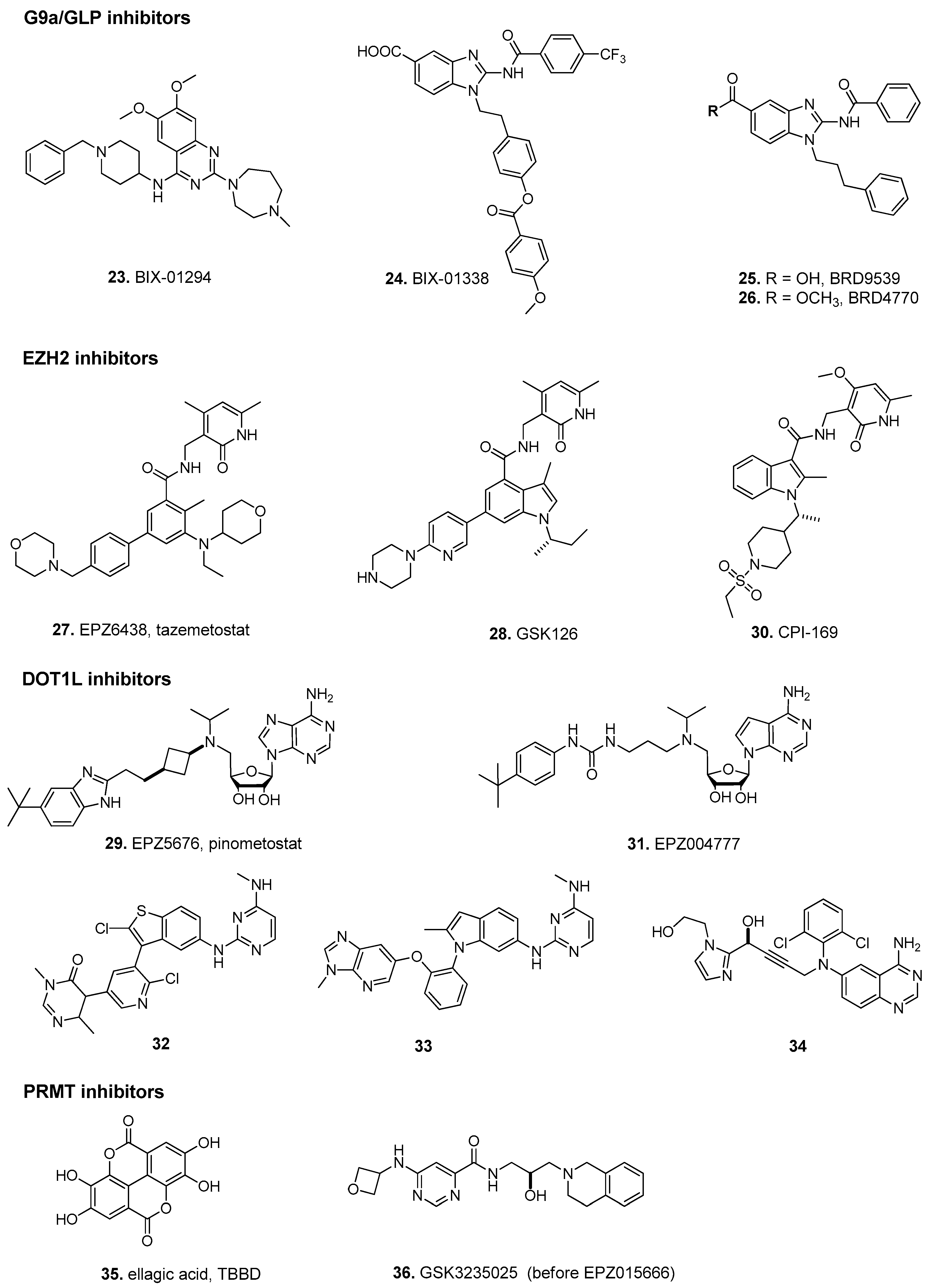
3.1. Histone Lysine Methyltransferases
3.2. Protein Arginine N-Methyltransferases
4. DNA Methyltransferase-Isoform Selectivity
5. Inhibition of DNA Methylation: Other Approaches
5.1. Allosteric and Bisubstrate Approaches
5.2. Repositioned Drugs and Natural Products
5.3. Protein-Protein Disruptors
5.4. Multitarget Inhibitors
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Gros, C.; Fahy, J.; Halby, L.; Dufau, I.; Erdmann, A.; Gregoire, J.-M.; Ausseil, F.; Vispé, S.; Arimondo, P.B. DNA methylation inhibitors in cancer: Recent and future approaches. Biochimie 2012, 94, 2280–2296. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.B.; Jones, P.A. Epigenetic therapy of cancer: Past, present and future. Nat. Rev. Drug Discov. 2006, 5, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Valente, S.; Liu, Y.; Schnekenburger, M.; Zwergel, C.; Cosconati, S.; Gros, C.; Tardugno, M.; Labella, D.; Florean, C.; Minden, S.; et al. Selective Non-nucleoside Inhibitors of Human DNA Methyltransferases Active in Cancer Including in Cancer Stem Cells. J. Med. Chem. 2014, 57, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Duymich, C.E.; Charlet, J.; Yang, X.; Jones, P.A.; Liang, G. DNMT3B isoforms without catalytic activity stimulate gene body methylation as accessory proteins in somatic cells. Nat. Commun. 2016, 7, 11453. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, S.; Van Emburgh, B.O.; Shan, J.; Su, Z.; Fields, C.R.; Vieweg, J.; Hamazaki, T.; Schwartz, P.H.; Terada, N.; Robertson, K.D. A novel DNMT3B splice variant expressed in tumor and pluripotent cells modulates genomic DNA methylation patterns and displays altered DNA binding. Mol. Cancer Res. MCR 2009, 7, 1622–1634. [Google Scholar] [CrossRef] [PubMed]
- Rondelet, G.; Dal Maso, T.; Willems, L.; Wouters, J. Structural basis for recognition of histone H3K36me3 nucleosome by human de novo DNA methyltransferases 3A and 3B. J. Struct. Biol. 2016, 194, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A.; Jurkowska, R.Z. Allosteric control of mammalian DNA methyltransferases—A new regulatory paradigm. Nucleic Acids Res. 2016, 44, 8556–8575. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Hua, S.; He, X.; Zhang, Y. DNA methyltransferases and methyl-binding proteins of mammals. Acta Biochim. Biophys. Sin. 2010, 42, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Terashima, M.; Tange, S.; Ishimura, A. Roles of histone methyl-modifying enzymes in development and progression of cancer. Cancer Sci. 2013, 104, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kikuchi, J. Epigenetic mechanisms of cell adhesion-mediated drug resistance in multiple myeloma. Int. J. Hematol. 2016, 104, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Morera, L.; Lübbert, M.; Jung, M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenet. 2016, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Schwämmle, V.; Sidoli, S.; Ruminowicz, C.; Wu, X.; Lee, C.-F.; Helin, K.; Jensen, O.N. Systems Level Analysis of Histone H3 Post-translational Modifications (PTMs) Reveals Features of PTM Crosstalk in Chromatin Regulation. Mol. Cell. Proteom. MCP 2016, 15, 2715–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baubec, T.; Colombo, D.F.; Wirbelauer, C.; Schmidt, J.; Burger, L.; Krebs, A.R.; Akalin, A.; Schübeler, D. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 2015, 520, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Plass, C.; Pfister, S.M.; Lindroth, A.M.; Bogatyrova, O.; Claus, R.; Lichter, P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat. Rev. Genet. 2013, 14, 765–780. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Lapinska, K.; Snyder, N.; Leary, M.; Rollinson, S.; Sarkar, S. Use of epigenetic drugs in disease: An overview. Genet. Epigenet. 2014, 6, 9–19. [Google Scholar] [PubMed]
- Erdmann, A.; Halby, L.; Fahy, J.; Arimondo, P.B. Targeting DNA Methylation with Small Molecules: What’s next? J. Med. Chem. 2015, 58, 2569–2583. [Google Scholar] [CrossRef] [PubMed]
- Deltour, S.; Chopin, V.; Leprince, D. Modifications épigénétiques et cancer. Med./Sci. 2005, 21, 405–411. (in French). [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Roberts, J.M.; Qi, J.; Bradner, J.E. Inhibitors of emerging epigenetic targets for cancer therapy: A patent review (2010–2014). Pharm. Pat. Anal. 2015, 4, 261–284. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Hu, G.; Luo, C.; Liang, Z. DNA methyltransferase inhibitors: An updated patent review (2012–2015). Expert Opin. Ther. Pat. 2016, 26, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Gelato, K.A.; Shaikhibrahim, Z.; Ocker, M.; Haendler, B. Targeting epigenetic regulators for cancer therapy: Modulation of bromodomain proteins, methyltransferases, demethylases, and microRNAs. Expert Opin. Ther. Targets 2016, 20, 783–799. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.; Hui, C. Synthesis of lysine methyltransferase inhibitors. Front. Chem. 2015, 3, 44. [Google Scholar]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Gabbara, S.; Bhagwat, A.S. The mechanism of inhibition of DNA (cytosine-5-)-methyltransferases by 5-azacytosine is likely to involve methyl transfer to the inhibitor. Biochem. J. 1995, 307 Pt 1, 87–92. [Google Scholar] [CrossRef]
- Kim, C.H.; Marquez, V.E.; Mao, D.T.; Haines, D.R.; McCormack, J.J. Synthesis of pyrimidin-2-one nucleosides as acid-stable inhibitors of cytidine deaminase. J. Med. Chem. 1986, 29, 1374–1380. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.C.; Matsen, C.B.; Gonzales, F.A.; Ye, W.; Greer, S.; Marquez, V.E.; Jones, P.A.; Selker, E.U. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J. Natl. Cancer Inst. 2003, 95, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.B.; Jeong, S.; Egger, G.; Liang, G.; Phiasivongsa, P.; Tang, C.; Redkar, S.; Jones, P.A. Delivery of 5-aza-2′-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res. 2007, 67, 6400–6408. [Google Scholar] [CrossRef] [PubMed]
- Brueckner, B.; Rius, M.; Markelova, M.R.; Fichtner, I.; Hals, P.-A.; Sandvold, M.L.; Lyko, F. Delivery of 5-azacytidine to human cancer cells by elaidic acid esterification increases therapeutic drug efficacy. Mol. Cancer Ther. 2010, 9, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Horton, J.R.; Jones, G.D.; Walker, R.T.; Roberts, R.J.; Cheng, X. DNA containing 4′-thio-2′-deoxycytidine inhibits methylation by HhaI methyltransferase. Nucleic Acids Res. 1997, 25, 2773–2783. [Google Scholar] [CrossRef] [PubMed]
- Thottassery, J.V.; Sambandam, V.; Allan, P.W.; Maddry, J.A.; Maxuitenko, Y.Y.; Tiwari, K.; Hollingshead, M.; Parker, W.B. Novel DNA methyltransferase-1 (DNMT1) depleting anticancer nucleosides, 4′-thio-2′-deoxycytidine and 5-aza-4′-thio-2′-deoxycytidine. Cancer Chemother. Pharmacol. 2014, 74, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Newman, E.M.; Morgan, R.J.; Kummar, S.; Beumer, J.H.; Blanchard, M.S.; Ruel, C.; El-Khoueiry, A.B.; Carroll, M.I.; Hou, J.M.; Li, C.; et al. A phase I, pharmacokinetic, and pharmacodynamic evaluation of the DNA methyltransferase inhibitor 5-fluoro-2′-deoxycytidine, administered with tetrahydrouridine. Cancer Chemother. Pharmacol. 2015, 75, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Miranda, T.B.; Cortez, C.C.; Yoo, C.B.; Liang, G.; Abe, M.; Kelly, T.K.; Marquez, V.E.; Jones, P.A. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther. 2009, 8, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Momparler, R.L.; Côté, S. Targeting of cancer stem cells by inhibitors of DNA and histone methylation. Expert Opin. Investig. Drugs 2015, 24, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Cang, S.; Ma, Y.; Petrillo, R.L.; Liu, D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J. Hematol. Oncol. 2010, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, D.; Duvall, B.; Delahanty, G.; Mistry, B.; Alt, J.; Rojas, C.; Rowbottom, C.; Sanders, K.; Schuck, E.; Huang, K.-C.; et al. Design, synthesis, and pharmacological evaluation of fluorinated tetrahydrouridine derivatives as inhibitors of cytidine deaminase. J. Med. Chem. 2014, 57, 2582–2588. [Google Scholar] [CrossRef] [PubMed]
- Segura-Pacheco, B.; Trejo-Becerril, C.; Perez-Cardenas, E.; Taja-Chayeb, L.; Mariscal, I.; Chavez, A.; Acuña, C.; Salazar, A.M.; Lizano, M.; Dueñas-Gonzalez, A. Reactivation of tumor suppressor genes by the cardiovascular drugs hydralazine and procainamide and their potential use in cancer therapy. Clin. Cancer Res. 2003, 9, 1596–1603. [Google Scholar] [PubMed]
- Villar-Garea, A.; Fraga, M.F.; Espada, J.; Esteller, M. Procaine is a DNA-demethylating agent with growth-inhibitory effects in human cancer cells. Cancer Res. 2003, 63, 4984–4989. [Google Scholar] [PubMed]
- Lee, B.H.; Yegnasubramanian, S.; Lin, X.; Nelson, W.G. Procainamide is a specific inhibitor of DNA methyltransferase 1. J. Biol. Chem. 2005, 280, 40749–40756. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.Z.; Healey, M.A.; Lee, C.; Poh, W.; Yerram, S.R.; Patel, K.; Azad, N.S.; Herman, J.G.; Kern, S.E. DNA-intercalators causing rapid re-expression of methylated and silenced genes in cancer cells. Oncotarget 2013, 4, 298–309. [Google Scholar] [CrossRef]
- Gros, C.; Fleury, L.; Nahoum, V.; Faux, C.; Valente, S.; Labella, D.; Cantagrel, F.; Rilova, E.; Bouhlel, M.A.; David-Cordonnier, M.-H.; et al. New insights on the mechanism of quinoline-based DNA Methyltransferase inhibitors. J. Biol. Chem. 2015, 290, 6293–6302. [Google Scholar] [CrossRef] [PubMed]
- Datta, J.; Ghoshal, K.; Denny, W.A.; Gamage, S.A.; Brooke, D.G.; Phiasivongsa, P.; Redkar, S.; Jacob, S.T. A New Class of Quinoline-Based DNA Hypomethylating Agents Reactivates Tumor Suppressor Genes by Blocking DNA Methyltransferase 1 Activity and Inducing Its Degradation. Cancer Res. 2009, 69, 4277–4285. [Google Scholar] [CrossRef] [PubMed]
- Gamage, S.A.; Brooke, D.G.; Redkar, S.; Datta, J.; Jacob, S.T.; Denny, W.A. Structure–activity relationships for 4-anilinoquinoline derivatives as inhibitors of the DNA methyltransferase enzyme DNMT1. Bioorg. Med. Chem. 2013, 21, 3147–3153. [Google Scholar] [CrossRef] [PubMed]
- Rilova, E.; Erdmann, A.; Gros, C.; Masson, V.; Aussagues, Y.; Poughon-Cassabois, V.; Rajavelu, A.; Jeltsch, A.; Menon, Y.; Novosad, N.; et al. Design, Synthesis and Biological Evaluation of 4-Amino-N-(4-aminophenyl)benzamide Analogues of Quinoline-Based SGI-1027 as Inhibitors of DNA Methylation. ChemMedChem 2014, 9, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Fagan, R.L.; Cryderman, D.E.; Kopelovich, L.; Wallrath, L.L.; Brenner, C. Laccaic acid A is a direct, DNA-competitive inhibitor of DNA methyltransferase 1. J. Biol. Chem. 2013, 288, 23858–23867. [Google Scholar] [CrossRef] [PubMed]
- Halby, L.; Champion, C.; Sénamaud-Beaufort, C.; Ajjan, S.; Drujon, T.; Rajavelu, A.; Ceccaldi, A.; Jurkowska, R.; Lequin, O.; Nelson, W.G.; et al. Rapid Synthesis of New DNMT Inhibitors Derivatives of Procainamide. ChemBioChem 2012, 13, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Asgatay, S.; Champion, C.; Marloie, G.; Drujon, T.; Senamaud-Beaufort, C.; Ceccaldi, A.; Erdmann, A.; Rajavelu, A.; Schambel, P.; Jeltsch, A.; et al. Synthesis and evaluation of analogues of N-Phthaloyl-l-tryptophan (RG108) as Inhibitors of DNA Methyltransferase 1. J. Med. Chem. 2014, 57, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Tanaka, R.; Hamada, S.; Nakagawa, H.; Miyata, N. Design, synthesis, inhibitory activity, and binding mode study of novel DNA methyltransferase 1 inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1124–1127. [Google Scholar] [CrossRef] [PubMed]
- Castellano, S.; Kuck, D.; Viviano, M.; Yoo, J.; López-Vallejo, F.; Conti, P.; Tamborini, L.; Pinto, A.; Medina-Franco, J.L.; Sbardella, G. Synthesis and biochemical evaluation of ∆2-isoxazoline derivatives as DNA methyltransferase 1 inhibitors. J. Med. Chem. 2011, 54, 7663–7677. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-L.; Han, W.-F.; Geng, Y.; Su, J.-S. A genome-wide study of DNA methylation modified by epigallocatechin-3-gallate in the CAL-27 cell line. Mol. Med. Rep. 2015, 12, 5886–5890. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, V.; Vaid, M.; Katiyar, S.K. (−)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis 2011, 32, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, A.; Rajavelu, A.; Champion, C.; Rampon, C.; Jurkowska, R.; Jankevicius, G.; Sénamaud-Beaufort, C.; Ponger, L.; Gagey, N.; Dali Ali, H.; et al. C5-DNA methyltransferase inhibitors: From screening to effects on zebrafish embryo development. ChemBioChem 2011, 12, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Kuck, D.; Caulfield, T.; Lyko, F.; Medina-Franco, J.L. Nanaomycin A selectively inhibits DNMT3B and reactivates silenced tumor suppressor genes in human cancer cells. Mol. Cancer Ther. 2010, 9, 3015–3023. [Google Scholar] [CrossRef] [PubMed]
- Kilgore, J.A.; Du, X.; Melito, L.; Wei, S.; Wang, C.; Chin, H.G.; Posner, B.; Pradhan, S.; Ready, J.M.; Williams, N.S. Identification of DNMT1 selective antagonists using a novel scintillation proximity assay. J. Biol. Chem. 2013, 288, 19673–19684. [Google Scholar] [CrossRef] [PubMed]
- Ruscio, A.D.; Ebralidze, A.K.; Tenen, D.G.; Leone, G. Chimeric RNA Oligonucleotides and Uses Thereof. U.S. Patent 20140171492 A1, 18 October 2012. [Google Scholar]
- Pradhan, S.; Esteve, P.; Zhang, G. Dnmt Inhibitors. Patent WO2015073360, 21 May 2015. [Google Scholar]
- Sledziewski, A.; Devos, T.; Kole, R. Oligonucleotide Inhibitors of DNA Methyltransferases and Their Use in Treating Diseases. Patent WO2014011573, 18 June 2015. [Google Scholar]
- Winquist, E.; Knox, J.; Ayoub, J.-P.; Wood, L.; Wainman, N.; Reid, G.K.; Pearce, L.; Shah, A.; Eisenhauer, E. Phase II trial of DNA methyltransferase 1 inhibition with the antisense oligonucleotide MG98 in patients with metastatic renal carcinoma: A National Cancer Institute of Canada Clinical Trials Group investigational new drug study. Investig. New Drugs 2006, 24, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Amato, R.J.; Stephenson, J.; Hotte, S.; Nemunaitis, J.; Bélanger, K.; Reid, G.; Martell, R.E. MG98, a second-generation DNMT1 inhibitor, in the treatment of advanced renal cell carcinoma. Cancer Investig. 2012, 30, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Heaphy, C.E.A.; Havelange, V.; Fabbri, M.; Volinia, S.; Tsao, T.; Zanesi, N.; Kornblau, S.M.; Marcucci, G.; Calin, G.A.; et al. MicroRNA 29b functions in acute myeloid leukemia. Blood 2009, 114, 5331–5341. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Rossi, M.; Raimondi, L.; Pitari, M.R.; Botta, C.; Tagliaferri, P.; Tassone, P. miR-29s: A family of epi-miRNAs with therapeutic implications in hematologic malignancies. Oncotarget 2015, 6, 12837–12861. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.J.; Donehower, R.C.; Eisenhauer, E.A.; Wainman, N.; Shah, A.K.; Bonfils, C.; MacLeod, A.R.; Besterman, J.M.; Reid, G.K. A phase I pharmacokinetic and pharmacodynamic study of the DNA methyltransferase 1 inhibitor MG98 administered twice weekly. Ann. Oncol. 2003, 14, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Gelmon, K.A.; Siu, L.L.; Moore, M.J.; Britten, C.D.; Mistry, N.; Klamut, H.; D’Aloisio, S.; MacLean, M.; Wainman, N.; et al. Phase I and pharmacologic study of the human DNA methyltransferase antisense oligodeoxynucleotide MG98 given as a 21-day continuous infusion every 4 weeks. Investig. New Drugs 2003, 21, 85–97. [Google Scholar] [CrossRef]
- Klisovic, R.B.; Stock, W.; Cataland, S.; Klisovic, M.I.; Liu, S.; Blum, W.; Green, M.; Odenike, O.; Godley, L.; Burgt, J.V.; et al. A phase I biological study of MG98, an oligodeoxynucleotide antisense to DNA methyltransferase 1, in patients with high-risk myelodysplasia and acute myeloid leukemia. Clin. Cancer Res. 2008, 14, 2444–2449. [Google Scholar] [CrossRef] [PubMed]
- Plummer, R.; Vidal, L.; Griffin, M.; Lesley, M.; de Bono, J.; Coulthard, S.; Sludden, J.; Siu, L.L.; Chen, E.X.; Oza, A.M.; et al. Phase I study of MG98, an oligonucleotide antisense inhibitor of human DNA methyltransferase 1, given as a 7-day infusion in patients with advanced solid tumors. Clin. Cancer Res. 2009, 15, 3177–3183. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhang, G.; Wu, J.-H.; Jiang, C.-P. Diverse roles of miR-29 in cancer (review). Oncol. Rep. 2014, 31, 1509–1516. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M.; Pelosi, E.; Castelli, G.; Cerio, A.M.; D’Angiò, A.; Porretti, L.; Rebulla, P.; Pavesi, L.; Russo, G.; Giordano, A.; et al. A miRNA Signature in Human Cord Blood Stem and Progenitor Cells as Potential Biomarker of Specific Acute Myeloid Leukemia Subtypes. J. Cell. Physiol. 2015, 230, 1770–1780. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Guo, Q.; Fu, F.-J.; Wang, Z.; Yin, Z.; Wei, Y.-B.; Yang, J.-R. The role of miR-29b in cancer: Regulation, function, and signaling. OncoTargets Ther. 2015, 8, 539–548. [Google Scholar]
- Guo, Z.S.; Hong, J.A.; Irvine, K.R.; Chen, G.A.; Spiess, P.J.; Liu, Y.; Zeng, G.; Wunderlich, J.R.; Nguyen, D.M.; Restifo, N.P.; et al. De novo induction of a cancer/testis antigen by 5-aza-2′-deoxycytidine augments adoptive immunotherapy in a murine tumor model. Cancer Res. 2006, 66, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Dong, W. Double-chain siRNA Molecule of Silencing DNMT (Desoxvribose Nucleic Acid Methyltransferases) Gene and Application of Double-Chain siRNA Molecule. Patent CN 201410021188, 30 April 2014. [Google Scholar]
- Siedlecki, P.; Boy, R.G.; Musch, T.; Brueckner, B.; Suhai, S.; Lyko, F.; Zielenkiewicz, P. Discovery of Two Novel, Small-Molecule Inhibitors of DNA Methylation. J. Med. Chem. 2006, 49, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Brueckner, B.; Garcia Boy, R.; Siedlecki, P.; Musch, T.; Kliem, H.C.; Zielenkiewicz, P.; Suhai, S.; Wiessler, M.; Lyko, F. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005, 65, 6305–6311. [Google Scholar] [CrossRef] [PubMed]
- Castellano, S.; Kuck, D.; Sala, M.; Novellino, E.; Lyko, F.; Sbardella, G. Constrained analogues of procaine as novel small molecule inhibitors of DNA methyltransferase-1. J. Med. Chem. 2008, 51, 2321–2325. [Google Scholar] [CrossRef] [PubMed]
- Lillico, R.; Stesco, N.; Khorshid Amhad, T.; Cortes, C.; Namaka, M.P.; Lakowski, T.M. Inhibitors of enzymes catalyzing modifications to histone lysine residues: Structure, function and activity. Future Med. Chem. 2016, 8, 879–897. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.T.; Weisenberger, D.J.; Velicescu, M.; Gonzales, F.A.; Lin, J.C.Y.; Liang, G.; Jones, P.A. Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2′-deoxycytidine. Cancer Res. 2002, 62, 6456–6461. [Google Scholar] [PubMed]
- Andresini, O.; Ciotti, A.; Rossi, M.N.; Battistelli, C.; Carbone, M.; Maione, R. A cross-talk between DNA methylation and H3 lysine 9 dimethylation at the KvDMR1 region controls the induction of Cdkn1c in muscle cells. Epigenetics 2016, 11, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Chadwick, B.P. Influence of Repressive Histone and DNA Methylation upon D4Z4 Transcription in Non-Myogenic Cells. PLoS ONE 2016, 11, e0160022. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Matsumura, Y.; Fukuda, M.; Kimura, H.; Shinkai, Y. G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J. 2008, 27, 2681–2690. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Ueda, J.; Fukuda, M.; Takeda, N.; Ohta, T.; Iwanari, H.; Sakihama, T.; Kodama, T.; Hamakubo, T.; Shinkai, Y. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 2005, 19, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Casciello, F.; Windloch, K.; Gannon, F.; Lee, J.S. Functional Role of G9a Histone Methyltransferase in Cancer. Front. Immunol. 2015, 6, 487. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Li, T.; Wang, X.; Zhao, E.; Choi, J.-H.; Yang, L.; Zha, Y.; Dong, Z.; Huang, S.; Asara, J.M.; et al. The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab. 2013, 18, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Kubicek, S.; O’Sullivan, R.J.; August, E.M.; Hickey, E.R.; Zhang, Q.; Teodoro, M.L.; Rea, S.; Mechtler, K.; Kowalski, J.A.; Homon, C.A.; et al. Reversal of H3K9me2 by a Small-Molecule Inhibitor for the G9a Histone Methyltransferase. Mol. Cell 2007, 25, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Soumyanarayanan, U.; Dymock, B.W. Recently discovered EZH2 and EHMT2 (G9a) inhibitors. Future Med. Chem. 2016, 8, 1635–1654. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wang, Q.; Paulk, J.; Kubicek, S.; Kemp, M.M.; Adams, D.J.; Shamji, A.F.; Wagner, B.K.; Schreiber, S.L. A Small-Molecule Probe of the Histone Methyltransferase G9a Induces Cellular Senescence in Pancreatic Adenocarcinoma. ACS Chem. Biol. 2012, 7, 1152–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotili, D.; Tarantino, D.; Marrocco, B.; Gros, C.; Masson, V.; Poughon, V.; Ausseil, F.; Chang, Y.; Labella, D.; Cosconati, S.; et al. Properly Substituted Analogues of BIX-01294 Lose Inhibition of G9a Histone Methyltransferase and Gain Selective Anti-DNA Methyltransferase 3A Activity. PLoS ONE 2014, 9, e96941. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.B.; Maksakova, I.A.; Mohn, F.; Leung, D.; Appanah, R.; Lee, S.; Yang, H.W.; Lam, L.L.; Mager, D.L.; Schübeler, D.; et al. DNA methylation in ES cells requires the lysine methyltransferase G9a but not its catalytic activity. EMBO J. 2008, 27, 2691–2701. [Google Scholar] [CrossRef] [PubMed]
- Epsztejn-Litman, S.; Feldman, N.; Abu-Remaileh, M.; Shufaro, Y.; Gerson, A.; Ueda, J.; Deplus, R.; Fuks, F.; Shinkai, Y.; Cedar, H.; et al. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat. Struct. Mol. Biol. 2008, 15, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Sun, L.; Kokura, K.; Horton, J.R.; Fukuda, M.; Espejo, A.; Izumi, V.; Koomen, J.M.; Bedford, M.T.; Zhang, X.; et al. MPP8 mediates the interactions between DNA methyltransferase Dnmt3a and H3K9 methyltransferase GLP/G9a. Nat. Commun. 2011, 2, 533. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Termanis, A.; Özkan, B.; Bao, X.X.; Culley, J.; de Lima Alves, F.; Rappsilber, J.; Ramsahoye, B.; Stancheva, I. G9a/GLP Complex maintains imprinted DNA methylation in embryonic stem cells. Cell Rep. 2016, 15, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, K.; Chesworth, R.; Duncan, K.; Keilhack, H.; Warholic, N.; Klaus, C.; Seki, M.; Shirotori, S.; Kawano, S.; Wigle, T.J.N. Aryl-or Heteroaryl-Substituted Benzene Compounds. WO/2012/142504, 2012. [Google Scholar]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A.; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Bradley, W.D.; Arora, S.; Busby, J.; Balasubramanian, S.; Gehling, V.S.; Nasveschuk, C.G.; Vaswani, R.G.; Yuan, C.-C.; Hatton, C.; Zhao, F.; et al. EZH2 inhibitor efficacy in non-Hodgkin’s lymphoma does not require suppression of H3K27 monomethylation. Chem. Biol. 2014, 21, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- Olhava, E.J.; Chesworth, R.; Kuntz, K. Carbocycle-Substituted Purine and 7-Deazapurine Compounds. WO/2012/075492, 2012. [Google Scholar]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Lipka, D.B.; Kuck, D.; Kliem, C.; Gerhauser, C. Substituted purine and 7-deazapurine compounds as modulators of epigenetic enzymes: A patent evaluation (WO2012075381). Expert Opin. Ther. Pat. 2013, 23, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.-M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Moison, C.; Senamaud-Beaufort, C.; Fourrière, L.; Champion, C.; Ceccaldi, A.; Lacomme, S.; Daunay, A.; Tost, J.; Arimondo, P.B.; Guieysse-Peugeot, A.-L. DNA methylation associated with polycomb repression in retinoic acid receptor β silencing. FASEB J. 2013, 27, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Moison, C.; Assemat, F.; Daunay, A.; Tost, J.; Guieysse-Peugeot, A.-L.; Arimondo, P.B. Synergistic chromatin repression of the tumor suppressor gene RARB in human prostate cancers. Epigenetics 2014, 9, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.; Shi, Z.; Liu, X.; Zhang, A.; Han, L.; Jiang, K.; Kang, C.; Zhang, Q. DNMT1 and EZH2 mediated methylation silences the microRNA-200b/a/429 gene and promotes tumor progression. Cancer Lett. 2015, 359, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Nichol, J.N.; Dupéré-Richer, D.; Ezponda, T.; Licht, J.D.; Miller, W.H. H3K27 Methylation: A Focal Point of Epigenetic Deregulation in Cancer. Adv. Cancer Res. 2016, 131, 59–95. [Google Scholar] [PubMed]
- Balasubramanian, V.; Iyer, P.; Arora, S.; Troyer, P.; Normant, E. Abstract 1697: CPI-169, a novel and potent EZH2 inhibitor, synergizes with CHOP in vivo and achieves complete regression in lymphoma xenograft models. Cancer Res. 2014, 74, 1697. [Google Scholar] [CrossRef]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective Killing of Mixed Lineage Leukemia Cells by a Potent Small-Molecule DOT1L Inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Zhang, Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011, 25, 1345–1358. [Google Scholar] [CrossRef] [PubMed]
- Anglin, J.L.; Song, Y. A medicinal chemistry perspective for targeting histone H3 lysine-79 methyltransferase DOT1L. J. Med. Chem. 2013, 56, 8972–8983. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Polly, P.; Liu, T. The histone methyltransferase DOT1L: Regulatory functions and a cancer therapy target. Am. J. Cancer Res. 2015, 5, 2823–2837. [Google Scholar] [PubMed]
- Wang, X.; Chen, C.-W.; Armstrong, S.A. The role of DOT1L in the maintenance of leukemia gene expression. Curr. Opin. Genet. Dev. 2016, 36, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Farooq, Z.; Banday, S.; Pandita, T.K.; Altaf, M. The many faces of histone H3K79 methylation. Mutat. Res. Mutat. Res. 2016, 768, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Vlaming, H.; van Leeuwen, F. The upstreams and downstreams of H3K79 methylation by DOT1L. Chromosoma 2016, 125, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, D. Recent progress in the treatment of infant acute lymphoblastic leukemia. Pediatr. Int. 2015, 57, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Spurr, S.S.; Bayle, E.D.; Yu, W.; Li, F.; Tempel, W.; Vedadi, M.; Schapira, M.; Fish, P.V. New small molecule inhibitors of histone methyl transferase DOT1L with a nitrile as a non-traditional replacement for heavy halogen atoms. Bioorg. Med. Chem. Lett. 2016, 26, 4518–4522. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Zhang, L.; Yao, Y.; Wang, C.; Redell, M.S.; Dong, S.; Song, Y. Synthesis, activity and metabolic stability of non-ribose containing inhibitors of histone methyltransferase DOT1L. MedChemComm 2013, 4, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhu, H.; Stauffer, F.; Caravatti, G.; Vollmer, S.; Machauer, R.; Holzer, P.; Möbitz, H.; Scheufler, C.; Klumpp, M.; et al. Discovery of Novel Dot1L Inhibitors through a Structure-Based Fragmentation Approach. ACS Med. Chem. Lett. 2016, 7, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Scheufler, C.; Möbitz, H.; Gaul, C.; Ragot, C.; Be, C.; Fernández, C.; Beyer, K.S.; Tiedt, R.; Stauffer, F. Optimization of a Fragment-Based Screening Hit toward Potent DOT1L Inhibitors Interacting in an Induced Binding Pocket. ACS Med. Chem. Lett. 2016, 7, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Wang, H.; Zou, Y.; Zhang, S.; Xiao, J.; Jiang, G.; Zhang, Y.; Lai, Y. Identification of phenoxyacetamide derivatives as novel DOT1L inhibitors via docking screening and molecular dynamics simulation. J. Mol. Graph. Model. 2016, 68, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, L.; Chen, Y.; Hu, J.; Liu, J.; Liu, Y.-C.; Liu, R.; Zhang, Y.; Meng, F.; Zhu, K.; et al. Identification of Novel Disruptor of Telomeric Silencing 1-like (DOT1L) Inhibitors through Structure-Based Virtual Screening and Biological Assays. J. Chem. Inf. Model. 2016, 56, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Warholic, N.M.; Wigle, T.J.; Klaus, C.R.; Allain, C.J.; Raimondi, A.; Porter Scott, M.; Chesworth, R.; Moyer, M.P.; Copeland, R.A.; et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2013, 110, 7922–7927. [Google Scholar] [CrossRef] [PubMed]
- Olhava, E.; Chesworth, R.; Kuntz, K.; Richon, V.; Pollock, R.; Daigle, S. Method of Treating Leukemia. WO2014039839, 13 March 2014. [Google Scholar]
- Selvi, B.R.; Batta, K.; Kishore, A.H.; Mantelingu, K.; Varier, R.A.; Balasubramanyam, K.; Pradhan, S.K.; Dasgupta, D.; Sriram, S.; Agrawal, S.; et al. Identification of a novel inhibitor of coactivator-associated arginine methyltransferase 1 (CARM1)-mediated methylation of histone H3 Arg-17. J. Biol. Chem. 2010, 285, 7143–7152. [Google Scholar] [CrossRef] [PubMed]
- Chan-Penebre, E.; Kuplast, K.G.; Majer, C.R.; Boriack-Sjodin, P.A.; Wigle, T.J.; Johnston, L.D.; Rioux, N.; Munchhof, M.J.; Jin, L.; Jacques, S.L.; et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Yi, P.; Wong, J.; O’Malley, B.W. Signaling within a coactivator complex: Methylation of SRC-3/AIB1 is a molecular switch for complex disassembly. Mol. Cell. Biol. 2006, 26, 7846–7857. [Google Scholar] [CrossRef] [PubMed]
- Van Haren, M.; van Ufford, L.Q.; Moret, E.E.; Martin, N.I. Synthesis and evaluation of protein arginine N-methyltransferase inhibitors designed to simultaneously occupy both substrate binding sites. Org. Biomol. Chem. 2015, 13, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Jahan, S.; Davie, J.R. Protein arginine methyltransferases (PRMTs): Role in chromatin organization. Adv. Biol. Regul. 2015, 57, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Wang, Z. Protein arginine methyltransferase 5 regulates multiple signaling pathways to promote lung cancer cell proliferation. BMC Cancer 2016, 16, 567. [Google Scholar] [CrossRef] [PubMed]
- Kanda, M.; Shimizu, D.; Fujii, T.; Tanaka, H.; Shibata, M.; Iwata, N.; Hayashi, M.; Kobayashi, D.; Tanaka, C.; Yamada, S.; et al. Protein arginine methyltransferase 5 is associated with malignant phenotype and peritoneal metastasis in gastric cancer. Int. J. Oncol. 2016, 49, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Rioux, N.; Duncan, K.W.; Lantz, R.J.; Miao, X.; Chan-Penebre, E.; Moyer, M.P.; Munchhof, M.J.; Copeland, R.A.; Chesworth, R.; Waters, N.J. Species differences in metabolism of EPZ015666, an oxetane-containing protein arginine methyltransferase-5 (PRMT5) inhibitor. Xenobiotica Fate Foreign Compd. Biol. Syst. 2016, 46, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Epizyme Inc. Epizyme Earns $6 Million Milestone Payment from GlaxoSmithKline for Initiation of Clinical Development with First-in-Class PRMT5 Inhibitor. Available online: http://globenewswire.com/news-release/2016/09/15/872059/0/en/Epizyme-Earns-6-Million-Milestone-Payment-from-GlaxoSmithKline-for-Initiation-of-Clinical-Development-with-First-in-Class-PRMT5-Inhibitor.html (accessed on 15 September 2016).
- Erdmann, A.; Menon, Y.; Gros, C.; Masson, V.; Aussagues, Y.; Ausseil, F.; Novosad, N.; Schambel, P.; Baltas, M.; Arimondo, P.B. Identification and optimization of hydrazone-gallate derivatives as specific inhibitors of DNA methyltransferase 3A. Future Med. Chem. 2016, 8, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Aldawsari, F.S.; Aguayo-Ortiz, R.; Kapilashrami, K.; Yoo, J.; Luo, M.; Medina-Franco, J.L.; Velázquez-Martínez, C.A. Resveratrol-salicylate derivatives as selective DNMT3 inhibitors and anticancer agents. J. Enzyme Inhib. Med. Chem. 2016, 31, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Halby, L.; Arimondo, P.B. Quinazoline Derivatives and Their Use as DNA Methyltransferase Inhibitors. Patent WO2015040169, 26 March 2015. [Google Scholar]
- Yost, J.M.; Korboukh, I.; Liu, F.; Gao, C.; Jin, J. Targets in epigenetics: Inhibiting the methyl writers of the histone code. Curr. Chem. Genom. 2011, 5, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chory, E.J.; Wernimont, A.K.; Tempel, W.; Scopton, A.; Federation, A.; Marineau, J.J.; Qi, J.; Barsyte-Lovejoy, D.; Yi, J.; et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 2012, 3, 1288. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Magaña, M.J.; Martínez-Aguilar, R.; Lucendo, E.; Campillo-Davo, D.; Schulze-Osthoff, K.; Ruiz-Ruiz, C. The antihypertensive drug hydralazine activates the intrinsic pathway of apoptosis and causes DNA damage in leukemic T cells. Oncotarget 2016, 7, 21875–21886. [Google Scholar] [PubMed]
- Deng, C.; Lu, Q.; Zhang, Z.; Rao, T.; Attwood, J.; Yung, R.; Richardson, B. Hydralazine may induce autoimmunity by inhibiting extracellular signal-regulated kinase pathway signaling. Arthritis Rheum. 2003, 48, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Dueñas-González, A.; Lyko, F.; Medina-Franco, J.L. Molecular modeling and molecular dynamics studies of hydralazine with human DNA methyltransferase 1. ChemMedChem 2009, 4, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Dueñas-Gonzalez, A.; Coronel, J.; Cetina, L.; González-Fierro, A.; Chavez-Blanco, A.; Taja-Chayeb, L. Hydralazine-valproate: A repositioned drug combination for the epigenetic therapy of cancer. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1433–1444. [Google Scholar] [CrossRef] [PubMed]
- Candelaria, M.; Herrera, A.; Labardini, J.; González-Fierro, A.; Trejo-Becerril, C.; Taja-Chayeb, L.; Pérez-Cárdenas, E.; de la Cruz-Hernández, E.; Arias-Bofill, D.; Vidal, S.; et al. Hydralazine and magnesium valproate as epigenetic treatment for myelodysplastic syndrome. Preliminary results of a phase-II trial. Ann. Hematol. 2011, 90, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.Z.; Wang, Y.; Ai, N.; Hou, Z.; Sun, Y.; Lu, H.; Welsh, W.; Yang, C.S. Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003, 63, 7563–7570. [Google Scholar] [PubMed]
- Lee, W.J.; Shim, J.-Y.; Zhu, B.T. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol. Pharmacol. 2005, 68, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Shilpi, A.; Parbin, S.; Sengupta, D.; Kar, S.; Deb, M.; Rath, S.K.; Pradhan, N.; Rakshit, M.; Patra, S.K. Mechanisms of DNA methyltransferase–inhibitor interactions: Procyanidin B2 shows new promise for therapeutic intervention of cancer. Chem. Biol. Interact. 2015, 233, 122–138. [Google Scholar] [CrossRef] [PubMed]
- Moseley, V.R.; Morris, J.; Knackstedt, R.W.; Wargovich, M.J. Green tea polyphenol epigallocatechin 3-gallate, contributes to the degradation of DNMT3A and HDAC3 in HCT 116 human colon cancer cells. Anticancer Res. 2013, 33, 5325–5333. [Google Scholar] [PubMed]
- Forester, S.C.; Lambert, J.D. Synergistic inhibition of lung cancer cell lines by (−)-epigallocatechin-3-gallate in combination with clinically used nitrocatechol inhibitors of catechol-O-methyltransferase. Carcinogenesis 2014, 35, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Busch, C.; Burkard, M.; Leischner, C.; Lauer, U.M.; Frank, J.; Venturelli, S. Epigenetic activities of flavonoids in the prevention and treatment of cancer. Clin. Epigenet. 2015, 7, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, T.; Medina-Franco, J.L. Molecular dynamics simulations of human DNA methyltransferase 3B with selective inhibitor nanaomycin A. J. Struct. Biol. 2011, 176, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Medina-Franco, J.L.; Méndez-Lucio, O.; Yoo, J. Rationalization of activity cliffs of a sulfonamide inhibitor of DNA methyltransferases with induced-fit docking. Int. J. Mol. Sci. 2014, 15, 3253–3261. [Google Scholar] [CrossRef] [PubMed]
- Rogawski, D.S.; Grembecka, J.; Cierpicki, T. H3K36 methyltransferases as cancer drug targets: Rationale and perspectives for inhibitor development. Future Med. Chem. 2016, 8, 1589–1607. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jurkowska, R.; Soeroes, S.; Rajavelu, A.; Dhayalan, A.; Bock, I.; Rathert, P.; Brandt, O.; Reinhardt, R.; Fischle, W.; et al. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 2010, 38, 4246–4253. [Google Scholar] [CrossRef] [PubMed]
- Bashtrykov, P.; Jankevicius, G.; Jurkowska, R.Z.; Ragozin, S.; Jeltsch, A. The UHRF1 protein stimulates the activity and specificity of the maintenance DNA methyltransferase DNMT1 by an allosteric mechanism. J. Biol. Chem. 2014, 289, 4106–4115. [Google Scholar] [CrossRef] [PubMed]
- Rottach, A.; Frauer, C.; Pichler, G.; Bonapace, I.M.; Spada, F.; Leonhardt, H. The multi-domain protein Np95 connects DNA methylation and histone modification. Nucleic Acids Res. 2010, 38, 1796–1804. [Google Scholar] [CrossRef] [PubMed]
- Karagianni, P.; Amazit, L.; Qin, J.; Wong, J. ICBP90, a novel methyl K9 H3 binding protein linking protein ubiquitination with heterochromatin formation. Mol. Cell. Biol. 2008, 28, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Cheray, M.; Nadaradjane, A.; Bonnet, P.; Routier, S.; Vallette, F.M.; Cartron, P.-F. Specific inhibition of DNMT1/CFP1 reduces cancer phenotypes and enhances chemotherapy effectiveness. Epigenomics 2014, 6, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Myrianthopoulos, V.; Cartron, P.F.; Liutkevičiūtė, Z.; Klimašauskas, S.; Matulis, D.; Bronner, C.; Martinet, N.; Mikros, E. Tandem virtual screening targeting the SRA domain of UHRF1 identifies a novel chemical tool modulating DNA methylation. Eur. J. Med. Chem. 2016, 114, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, K. Take two: Combining immunotherapy with epigenetic drugs to tackle cancer. Nat. Med. 2016, 22, 8–10. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | IC50 (or EC50) a, µM | Reference | ||
|---|---|---|---|---|
| DNMT1 | DNMT3a | DNMT3b | ||
| 12 (procainamide) | >500 | >300 a | ND | [38] |
| 15 (SGI-1027) | 6 | 8 | 7.5 | [40] |
| 16 | 9 | 2.8 b | ND | [4] |
| 17 | (15) | (0.9) | ND | [43] |
| 18 (laccaic acid A) | 19 | 50 | ND | [44] |
| 19 (RG-108) | 390 | 315 b | ND | [45,46] |
| 37 (RG108-1) | 20 | ND | ND | [47] |
| 38a (2S, 3R) | 98 | ND | ND | [46] |
| 38b (2R, 3S) | 73 | ND | ND | |
| 39a (2S, 3R) | 128 | ND | ND | [46] |
| 39b (2R, 3S) | 50 | ND | ND | |
| 21 | 150 | ND | ND | [48] |
| 22 | 4 | 21 b | ND | [45] |
| 41 (EGCG) | 0.5 | ND | ND | [49] |
| 42 (genistein) | 30 | >100 | ND | [51] |
| 43 (nanaomycin A) | inactive | ND | 0.5 | [52] |
| 44 (SW155246) | 1.2 | 38 | ND | [53] |
| Entry | Inhibitor | Sequence (5’ to 3’) | IC50 (or Ki) a, μM | Reference | ||
|---|---|---|---|---|---|---|
| (DNMT1) | (DNMT3a) | (DNMT3b) | ||||
| 1 | asCEBPα-2 | GCCAGUGGCGAGGGGCGGCGCGG | (0.4341) | ND | ND | [55] |
| 2 | asCEBPα-2HPE | GACAGUGGAGAGGGGCGGAGCGG | (0.1352) | ND | ND | [55] |
| 3 | miR-155-5p | UUAAUGCUAAUCGUGAUAGGGGU | (0.02788) | ND | ND | [55] |
| 4 | MTC-423 | CCTATGCGATCGAGTTTTCT[z]GAT[z]GCATAGG z = zebularine | 0.363 | 1.60 | 17.5 | [56] |
| 5 | MTC-427 | CCTATG[M]GAT[M]GAGTTTTCT[z]GAT[z]GCATAGG′ M = methylcytosine; z = zebularine | 0.295 | 1.52 | 6.20 | [56] |
| 6 | MTC-433 | CCTATG[M]GAT[M]GAGTTTTCT[dz]GAT[dz]GCATAGG M = 5-methylcytosine; dz = deoxyzebularine | 0.00422 | ND | ND | [56] |
| 7 | MG98 | TTCATGTCAGCCAAGGCCAC | ND | ND | ND | [57,58] |
| 8 | miR29b | UAGCACCAUUUGAAAUCAGUGUU | ND | ND | ND | [59] |
| Inhibitor | IC50 a, µM | Reference | |||||
|---|---|---|---|---|---|---|---|
| Suv39H1 | G9a | EZH2 | DOT1L | CARM1 (PRMT4) | PRMT5 | ||
| 23 (BIX-01294) | >10 | 2.7 | ND | ND | ND | ND | [81] |
| 24 (BIX-01338) | 1.1 | 4.7 | ND | ND | ND | ND | [81] |
| 25 (BRD9539) | ND | 6.3 | ND | ND | ND | ND | [83] |
| 27 (EPZ6438, tazemetostat, E7438) | ND | ND | 0.012 | >100 | >100 | >100 | [115] |
| 28 (GSK2816126, GSK126) | >100 | >100 | 0.009 | >100 | >100 | >100 | [90] |
| 29 (EPZ5676, pinometostat) | ND | ND | ND | 0.0008 | >50 | 30 | [116] |
| 30 (CPI-169) | ND | ND | <0.001 b | ND | ND | ND | [91] |
| 31 (EPZ004777) | ND | ND | >50 b | 0.0004 | >50 | 0.521 | [101] |
| 32 | ND | ND | ND | 0.0014 | ND | ND | [111] |
| 33 | ND | ND | ND | 0.0004 | ND | ND | [111] |
| 34 | ND | ND | ND | 0.014 | ND | ND | [112] |
| 35 (ellagic acid, TBBD) | ND | ND | ND | ND | 25 | ND | [117] |
| 36 (GSK3235025, EPZ015666) | ND | ND | ND | ND | ND | 0.022 | [118] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castillo-Aguilera, O.; Depreux, P.; Halby, L.; Arimondo, P.B.; Goossens, L. DNA Methylation Targeting: The DNMT/HMT Crosstalk Challenge. Biomolecules 2017, 7, 3. https://doi.org/10.3390/biom7010003
Castillo-Aguilera O, Depreux P, Halby L, Arimondo PB, Goossens L. DNA Methylation Targeting: The DNMT/HMT Crosstalk Challenge. Biomolecules. 2017; 7(1):3. https://doi.org/10.3390/biom7010003
Chicago/Turabian StyleCastillo-Aguilera, Omar, Patrick Depreux, Ludovic Halby, Paola B. Arimondo, and Laurence Goossens. 2017. "DNA Methylation Targeting: The DNMT/HMT Crosstalk Challenge" Biomolecules 7, no. 1: 3. https://doi.org/10.3390/biom7010003
APA StyleCastillo-Aguilera, O., Depreux, P., Halby, L., Arimondo, P. B., & Goossens, L. (2017). DNA Methylation Targeting: The DNMT/HMT Crosstalk Challenge. Biomolecules, 7(1), 3. https://doi.org/10.3390/biom7010003