
Lysosomal Regulation of mTORC1 by Amino Acids in Mammalian Cells


{kind=link}
{kind=link}
Abstract
:1. Overview of Mechanistic Target of Rapamycin Complex 1
2. The Lysosome is the Major Cellular Compartment for mTORC1 Activation
3. Amino Acid-Sensing Signaling to the Lysosomal Membrane
3.1. The Rag GTPase and the Ragulator Complex Form a Super Complex with Vacuolar-ATPase, which Recruits mTORC1 to the Lysosomal Membrane in Response to Amino Acid Availability
3.2. GATOR1 is a GTPase Activating Protein for Rag A/B, while GATOR2 is a Negative Regulator of GATOR1
3.3. SLC38A9 is a Lysosomal Arginine Sensor for mTORC1 Activation
3.4. CASTOR Proteins are Cytosolic Arginine Sensors for mTORC1 Activation
3.5. Sestrin2 is a Leucine Sensor for the mTORC1 Pathway, and Regulates the Activity of Rags through GATORs
3.6. SZT2-Containing KICSTOR Recruits GATOR1 to the Lysosome and Inhibits Amino Acid-Induced mTORC1 Activation
3.7. The FLCN–FNIP Complex Functions as a GAP for Rag C/D
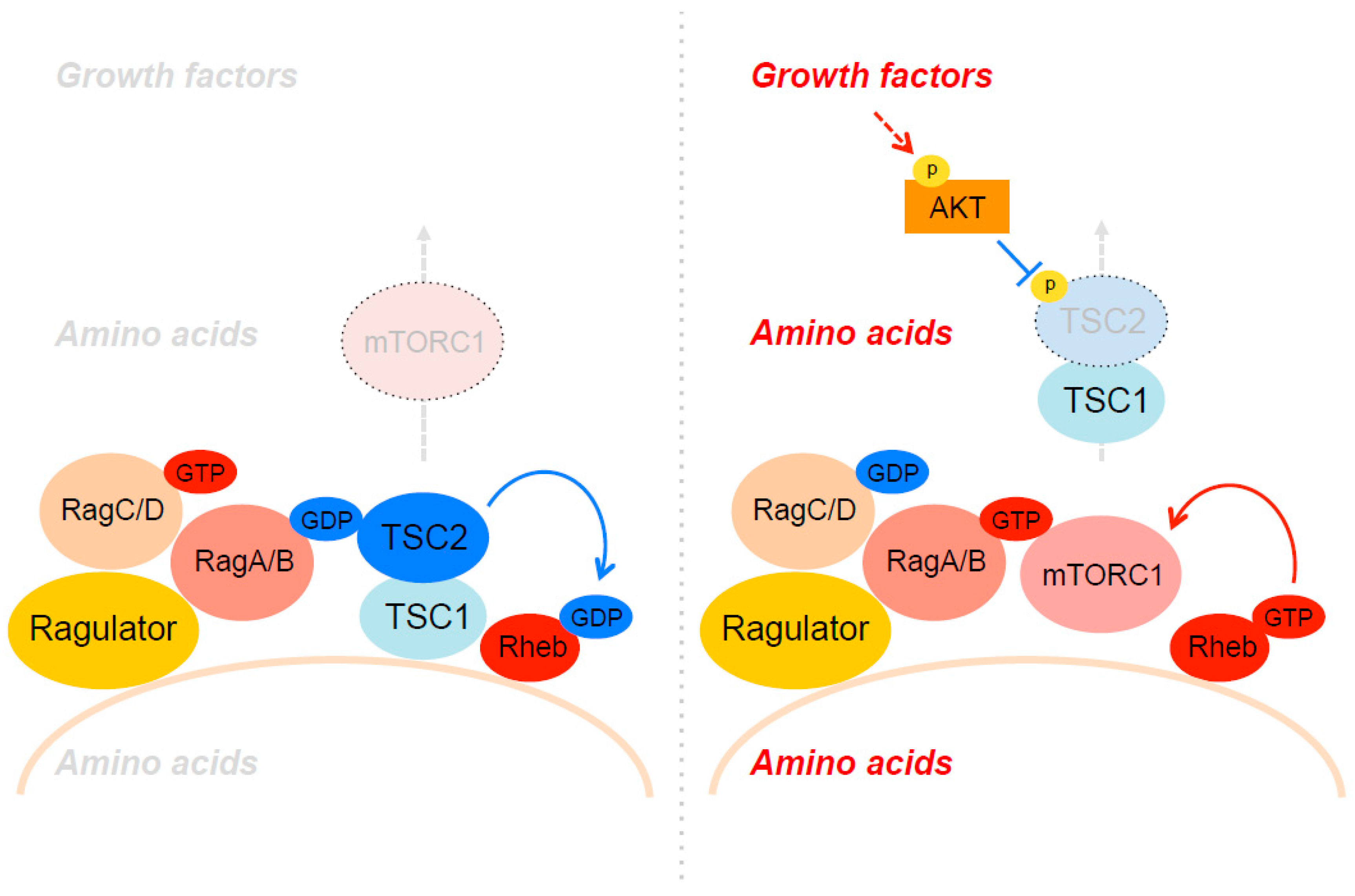
4. The Spatial Regulation of TSC through Akt and Amino Acids
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lempiainen, H.; Halazonetis, T.D. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009, 28, 3067–3073. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates tor action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawlowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem. J. 2007, 405, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. Pras40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Vander Haar, E.; Lee, S.I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Inoki, K.; Ikenoue, T.; Guan, K.L. Identification of SIN1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006, 20, 2820–2832. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. Deptor is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.K.; Murata, K.; Walz, T.; Sabatini, D.M.; Kang, S.A. Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol. Cell 2010, 38, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Aylett, C.H.; Sauer, E.; Imseng, S.; Boehringer, D.; Hall, M.N.; Ban, N.; Maier, T. Architecture of human mTOR complex 1. Science 2016, 351, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Gaubitz, C.; Oliveira, T.M.; Prouteau, M.; Leitner, A.; Karuppasamy, M.; Konstantinidou, G.; Rispal, D.; Eltschinger, S.; Robinson, G.C.; Thore, S.; et al. Molecular basis of the rapamycin insensitivity of target of rapamycin complex 2. Mol. Cell 2015, 58, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Vezina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (ay-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Chen, J.; Schreiber, S.L.; Clardy, J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human frap. Science 1996, 273, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, N.; Yoshino, K.; Hidayat, S.; Tokunaga, C.; Hara, K.; Eguchi, S.; Avruch, J.; Yonezawa, K. Dissociation of Raptor from mTOR is a mechanism of rapamycin-induced inhibition of mTOR function. Genes Cells 2004, 9, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The mammalian target of rapamycin (mTOR) partner, Raptor, binds the mtor substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J. Biol. Chem. 2003, 278, 15461–15464. [Google Scholar] [CrossRef] [PubMed]
- Schalm, S.S.; Blenis, J. Identification of a conserved motif required for mTOR signaling. Curr. Biol. 2002, 12, 632–639. [Google Scholar] [CrossRef]
- Schalm, S.S.; Fingar, D.C.; Sabatini, D.M.; Blenis, J. TOS motif-mediated Raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol. 2003, 13, 797–806. [Google Scholar] [CrossRef]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind Raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Araki, Y.; Kontani, K.; Nishina, H.; Katada, T. Novel role of the small GTPase Rheb: Its implication in endocytic pathway independent of the activation of mammalian target of rapamycin. J. Biochem. 2005, 137, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Nakagawa, M.; Young, S.G.; Yamanaka, S. Differential membrane localization of ERAS and Rheb, two Ras-related proteins involved in the phosphatidylinositol 3-kinase/mTOR pathway. J. Biol. Chem. 2005, 280, 32768–32774. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Zhang, Y.J.; Wei, Y.H.; Cho, J.H.; Morris, L.E.; Wang, H.Y.; Zheng, X.F. Rab1A is an mTORC1 activator and a colorectal oncogene. Cancer Cell 2014, 26, 754–769. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kim, J.; Alexander, A.; Cai, S.; Tripathi, D.N.; Dere, R.; Tee, A.R.; Tait-Mulder, J.; Di Nardo, A.; Han, J.M.; et al. A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat. Cell Biol. 2013, 15, 1186–1196. [Google Scholar] [CrossRef] [PubMed]
- Melser, S.; Chatelain, E.H.; Lavie, J.; Mahfouf, W.; Jose, C.; Obre, E.; Goorden, S.; Priault, M.; Elgersma, Y.; Rezvani, H.R.; et al. Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab. 2013, 17, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Clark, G.J.; Kinch, M.S.; Rogers-Graham, K.; Sebti, S.M.; Hamilton, A.D.; Der, C.J. The Ras-related protein Rheb is farnesylated and antagonizes Ras signaling and transformation. J. Biol. Chem. 1997, 272, 10608–10615. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 is a third subunit of the TSC1-TSCc2 complex upstream of mTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, X.; Saucedo, L.J.; Ru, B.; Edgar, B.A.; Pan, D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol. 2003, 5, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex gene products, tuberin and hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef]
- Garami, A.; Zwartkruis, F.J.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 gap activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.F.; Rebhun, J.F.; Clark, G.J.; Quilliam, L.A. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J. Biol. Chem. 2003, 278, 32493–32496. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. Rheb fills a gap between TSC and TOR. Trends Biochem. Sci. 2003, 28, 573–576. [Google Scholar] [CrossRef] [PubMed]
- van Slegtenhorst, M.; Nellist, M.; Nagelkerken, B.; Cheadle, J.; Snell, R.; van den Ouweland, A.; Reuser, A.; Sampson, J.; Halley, D.; van der Sluijs, P. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum. Mol. Genet. 1998, 7, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Demetriades, C.; Doumpas, N.; Teleman, A.A. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 2014, 156, 786–799. [Google Scholar] [CrossRef] [PubMed]
- Carroll, B.; Maetzel, D.; Maddocks, O.D.; Otten, G.; Ratcliff, M.; Smith, G.R.; Dunlop, E.A.; Passos, J.F.; Davies, O.R.; Jaenisch, R.; et al. Control of TSC2-Rheb signaling axis by arginine regulates mTORC1 activity. Elife 2016, 5, e11058. [Google Scholar] [CrossRef] [PubMed]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Hall, M.N. TSC on the peroxisome controls mTORC1. Nat. Cell Biol. 2013, 15, 1135–1136. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator is a gef for the rag GTPases that signal amino acid levels to mTORC1. Cell 2012, 150, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. MTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.M.; Finn, S.G.; Tee, A.R.; Browne, G.J.; Proud, C.G. The tuberous sclerosis protein TSC2 is not required for the regulation of the mammalian target of rapamycin by amino acids and certain cellular stresses. J. Biol. Chem. 2005, 280, 18717–18727. [Google Scholar] [CrossRef] [PubMed]
- Roccio, M.; Bos, J.L.; Zwartkruis, F.J. Regulation of the small GTPase Rheb by amino acids. Oncogene 2006, 25, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, T.; Hirose, E.; Nakashima, N.; Ii, M.; Nishimoto, T. Novel G proteins, Rag C and Rag D, interact with GTP-binding proteins, Rag A and Rag B. J. Biol. Chem. 2001, 276, 7246–7257. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, N.; Rapley, J.; Avruch, J. Amino acids activate mammalian target of rapamycin (mTOR) complex 1 without changing Rag GTPase guanyl nucleotide charging. J. Biol. Chem. 2014, 289, 2658–2674. [Google Scholar] [CrossRef] [PubMed]
- Nada, S.; Hondo, A.; Kasai, A.; Koike, M.; Saito, K.; Uchiyama, Y.; Okada, M. The novel lipid raft adaptor p18 controls endosome dynamics by anchoring the MEK-ERK pathway to late endosomes. EMBO J. 2009, 28, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Aravind, L. Dynein light chains of the Roadblock/LC7 group belong to an ancient protein superfamily implicated in NTPase regulation. Curr. Biol. 2000, 10, R774–R776. [Google Scholar] [CrossRef]
- Miertzschke, M.; Koerner, C.; Vetter, I.R.; Keilberg, D.; Hot, E.; Leonardy, S.; Sogaard-Andersen, L.; Wittinghofer, A. Structural analysis of the Ras-like G protein MgIA and its cognate GAP MgIB and implications for bacterial polarity. EMBO J. 2011, 30, 4185–4197. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J. Structural basis for activation of arf GTPase: Mechanisms of guanine nucleotide exchange and GTP-myristoyl switching. Cell 1998, 95, 237–248. [Google Scholar] [CrossRef]
- Manifava, M.; Smith, M.; Rotondo, S.; Walker, S.; Niewczas, I.; Zoncu, R.; Clark, J.; Ktistakis, N.T. Dynamics of mTORC1 activation in response to amino acids. Elife 2016, 5, e19960. [Google Scholar] [CrossRef] [PubMed]
- Ogmundsdottir, M.H.; Heublein, S.; Kazi, S.; Reynolds, B.; Visvalingam, S.M.; Shaw, M.K.; Goberdhan, D.C. Proton-assisted amino acid transporter PAT1 complexes with Rag GTPases and activates TORC1 on late endosomal and lysosomal membranes. PLoS ONE 2012, 7, e36616. [Google Scholar] [CrossRef] [PubMed]
- Chantranupong, L.; Scaria, S.M.; Saxton, R.A.; Gygi, M.P.; Shen, K.; Wyant, G.A.; Wang, T.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell 2016, 165, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.L. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Lerman, M.I.; Minna, J.D. The 630-kb lung cancer homozygous deletion region on human chromosome 3p21.3: Identification and evaluation of the resident candidate tumor suppressor genes. The international lung cancer chromosome 3p21.3 tumor suppressor gene consortium. Cancer Res. 2000, 60, 6116–6133. [Google Scholar] [PubMed]
- Li, J.; Wang, F.; Haraldson, K.; Protopopov, A.; Duh, F.M.; Geil, L.; Kuzmin, I.; Minna, J.D.; Stanbridge, E.; BRaga, E.; et al. Functional characterization of the candidate tumor suppressor gene NPRL2/G21 located in 3p21.3c. Cancer Res. 2004, 64, 6438–6443. [Google Scholar] [CrossRef] [PubMed]
- Seng, T.J.; Ichimura, K.; Liu, L.; Tingby, O.; Pearson, D.M.; Collins, V.P. Complex chromosome 22 rearrangements in astrocytic tumors identified using microsatellite and chromosome 22 tile path array analysis. Genes Chromosom. Cancer 2005, 43, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tsun, Z.Y.; Wolfson, R.L.; Shen, K.; Wyant, G.A.; Plovanich, M.E.; Yuan, E.D.; Jones, T.D.; Chantranupong, L.; Comb, W.; et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORTORC1. Science 2015, 347, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Rebsamen, M.; Pochini, L.; Stasyk, T.; de Araujo, M.E.; Galluccio, M.; Kandasamy, R.K.; Snijder, B.; Fauster, A.; Rudashevskaya, E.L.; Bruckner, M.; et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 2015, 519, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, B.E.; Waag, E.; Jacobsson, J.A.; Stephansson, O.; Rumaks, J.; Svirskis, S.; Alsio, J.; Roman, E.; Ebendal, T.; Klusa, V.; et al. The evolutionary history and tissue mapping of amino acid transporters belonging to solute carrier families SLC32, SLC36, and SLC38. J. Mol. Neurosci. 2008, 35, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; van der Welle, R.E.; Mydock-McGrane, L.; Jiang, X.; van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Chantranupong, L.; Knockenhauer, K.E.; Schwartz, T.U.; Sabatini, D.M. Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature 2016, 536, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Lang, E.J.; Cross, P.J.; Mittelstadt, G.; Jameson, G.B.; Parker, E.J. Allosteric action: The varied ACT domains regulating enzymes of amino-acid metabolism. Curr. Opin. Struct. Biol. 2014, 29, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Grant, G.A. The ACT domain: A small molecule binding domain and its role as a common regulatory element. J. Biol. Chem. 2006, 281, 33825–33829. [Google Scholar] [CrossRef] [PubMed]
- Chipman, D.M.; Shaanan, B. The ACT domain family. Curr. Opin. Struct. Biol. 2001, 11, 694–700. [Google Scholar] [CrossRef]
- Aravind, L.; Koonin, E.V. Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database searches. J. Mol. Biol. 1999, 287, 1023–1040. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wang, R.; Zhang, T.; Ding, J. Structural insight into the arginine-binding specificity of CASTOR1 in amino acid-dependent mTORC1 signaling. Cell Discov. 2016, 2, 16035. [Google Scholar] [CrossRef] [PubMed]
- Gai, Z.; Wang, Q.; Yang, C.; Wang, L.; Deng, W.; Wu, G. Structural mechanism for the arginine sensing and regulation of CASTOR1 in the mTORC1 signaling pathway. Cell Discov. 2016, 2, 16051. [Google Scholar] [CrossRef] [PubMed]
- Chantranupong, L.; Wolfson, R.L.; Orozco, J.M.; Saxton, R.A.; Scaria, S.M.; Bar-Peled, L.; Spooner, E.; Isasa, M.; Gygi, S.P.; Sabatini, D.M. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Parmigiani, A.; Nourbakhsh, A.; Ding, B.; Wang, W.; Kim, Y.C.; Akopiants, K.; Guan, K.L.; Karin, M.; Budanov, A.V. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep. 2014, 9, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Ro, S.H.; Kim, M.; Park, H.W.; Semple, I.A.; Park, H.; Cho, U.S.; Wang, W.; Guan, K.L.; Karin, M.; et al. Sestrin2 inhibits mTORC1 through modulation of GATOR complexes. Sci. Rep. 2015, 5, 9502. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Miguel, S.; Buckbinder, L.; Jean, P.; Gelbert, L.; Talbott, R.; Laidlaw, J.; Seizinger, B.; Kley, N. PA26, a novel target of the p53 tumor suppressor and member of the gadd family of DNA damage and growth arrest inducible genes. Oncogene 1999, 18, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Shoshani, T.; Faerman, A.; Zelin, E.; Kamer, I.; Kalinski, H.; Gorodin, S.; Fishman, A.; Chajut, A.; Einat, P.; et al. Identification of a novel stress-responsive gene HI95 involved in regulation of cell viability. Oncogene 2002, 21, 6017–6031. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Budanov, A.V.; Karin, M. Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab. 2013, 18, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Knockenhauer, K.E.; Wolfson, R.L.; Chantranupong, L.; Pacold, M.E.; Wang, T.; Schwartz, T.U.; Sabatini, D.M. Structural basis for leucine sensing by the Sestrin2-mTORTORC1 pathway. Science 2016, 351, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, R.L.; Chantranupong, L.; Wyant, G.A.; Gu, X.; Orozco, J.M.; Shen, K.; Condon, K.J.; Petri, S.; Kedir, J.; Scaria, S.M.; et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 2017, 543, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Yin, N.; Li, M.O. SZT2 dictates GATOR control of mTORC1 signalling. Nature 2017, 543, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Yin, N.; Li, M.O. Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling. Cell 2014, 159, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; An, S.; Ro, S.H.; Teixeira, F.; Park, G.J.; Kim, C.; Cho, C.S.; Kim, J.S.; Jakob, U.; Lee, J.H.; et al. Janus-faced Sestrin2 controls ROS and mTOR signalling through two separate functional domains. Nat. Commun. 2015, 6, 10025. [Google Scholar] [CrossRef] [PubMed]
- Mc Cormack, A.; Sharpe, C.; Gregersen, N.; Smith, W.; Hayes, I.; George, A.M.; Love, D.R. 12q14 microdeletions: Additional case series with confirmation of a macrocephaly region. Case Rep. Genet. 2015, 192071. [Google Scholar] [CrossRef] [PubMed]
- Pajusalu, S.; Reimand, T.; Ounap, K. Novel homozygous mutation in KPTN gene causing a familial intellectual disability-macrocephaly syndrome. Am. J. Med. Genet. 2015, 167A, 1913–1915. [Google Scholar] [CrossRef] [PubMed]
- Baple, E.L.; Maroofian, R.; Chioza, B.A.; Izadi, M.; Cross, H.E.; Al-Turki, S.; Barwick, K.; Skrzypiec, A.; Pawlak, R.; Wagner, K.; et al. Mutations in KPTN cause macrocephaly, neurodevelopmental delay, and seizures. Am. J. Hum. Genet. 2014, 94, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, C.; Angle, B.; Millichap, J.J. Early-life epileptic encephalopathy secondary to SZT2 pathogenic recessive variants. Epileptic Disord. Int. Epilepsy J. Videotape 2016, 18, 195–200. [Google Scholar]
- Basel-Vanagaite, L.; Hershkovitz, T.; Heyman, E.; Raspall-Chaure, M.; Kakar, N.; Smirin-Yosef, P.; Vila-Pueyo, M.; Kornreich, L.; Thiele, H.; Bode, H.; et al. Biallelic SZT2 mutations cause infantile encephalopathy with epilepsy and dysmorphic corpus callosum. Am. J. Hum. Genet. 2013, 93, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Krueger, D.A.; Wilfong, A.A.; Holland-Bouley, K.; Anderson, A.E.; Agricola, K.; Tudor, C.; Mays, M.; Lopez, C.M.; Kim, M.O.; Franz, D.N. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann. Neurol. 2013, 74, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Goffin, A.; Hoefsloot, L.H.; Bosgoed, E.; Swillen, A.; Fryns, J.P. PTEN mutation in a family with Cowden Syndrome and autism. Am. J. Med. Genet. 2001, 105, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Tsun, Z.Y.; Bar-Peled, L.; Chantranupong, L.; Zoncu, R.; Wang, T.; Kim, C.; Spooner, E.; Sabatini, D.M. The folliculin tumor suppressor is a GAP for the Ragc/d GTPases that signal amino acid levels to mTORTORC1. Mol. Cell 2013, 52, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Peli-Gulli, M.P.; Sardu, A.; Panchaud, N.; Raucci, S.; De Virgilio, C. Amino acids stimulate TORC1 through LST4-LST7, a GTPase-activating protein complex for the Rag family GTPase GTR2. Cell Rep. 2015, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Birt, A.R.; Hogg, G.R.; Dube, W.J. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch. Dermatol. 1977, 113, 1674–1677. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, M.L.; Warren, M.B.; Toro, J.R.; Matrosova, V.; Glenn, G.; Turner, M.L.; Duray, P.; Merino, M.; Choyke, P.; Pavlovich, C.P.; et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell 2002, 2, 157–164. [Google Scholar] [CrossRef]
- Petit, C.S.; Roczniak-Ferguson, A.; Ferguson, S.M. Recruitment of Folliculin to lysosomes supports the amino acid-dependent activation of Rag GTPases. J. Cell Biol. 2013, 202, 1107–1122. [Google Scholar] [CrossRef] [PubMed]
- Hartman, T.R.; Nicolas, E.; Klein-Szanto, A.; Al-Saleem, T.; Cash, T.P.; Simon, M.C.; Henske, E.P. The role of the Birt-Hogg-Dubé protein in mTOR activation and renal tumorigenesis. Oncogene 2009, 28, 1594–1604. [Google Scholar] [CrossRef] [PubMed]
- Hudon, V.; Sabourin, S.; Dydensborg, A.B.; Kottis, V.; Ghazi, A.; Paquet, M.; Crosby, K.; Pomerleau, V.; Uetani, N.; Pause, A. Renal tumour suppressor function of the Birt-Hogg-Dubé syndrome gene product Folliculin. J. Med. Genet. 2010, 47, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Takagi, Y.; Kobayashi, T.; Shiono, M.; Wang, L.; Piao, X.; Sun, G.; Zhang, D.; Abe, M.; Hagiwara, Y.; Takahashi, K.; et al. Interaction of Folliculin (Birt-Hogg-Dubé gene product) with a novel FNIP1-like (FNIPl/FNIP2) protein. Oncogene 2008, 27, 5339–5347. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Hong, S.B.; Sharma, N.; Warren, M.B.; Nickerson, M.L.; Iwamatsu, A.; Esposito, D.; Gillette, W.K.; Hopkins, R.F., 3rd; Hartley, J.L.; et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 15552–15557. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Keller, J.R.; Sun, H.W.; Resch, W.; Kuchen, S.; Suh, H.C.; Hasumi, H.; Hasumi, Y.; Kieffer-Kwon, K.R.; Gonzalez, C.G.; et al. The Folliculin-FNIP1 pathway deleted in human Birt-Hogg-Dubé syndrome is required for murine B-cell development. Blood 2012, 120, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Futami, K.; Petillo, D.; Peng, J.; Wang, P.; Knol, J.; Li, Y.; Khoo, S.K.; Huang, D.; Qian, C.N.; et al. Deficiency of FLCN in mouse kidney led to development of polycystic kidneys and renal neoplasia. PLoS ONE 2008, 3, e3581. [Google Scholar] [CrossRef]
- Hasumi, Y.; Baba, M.; Ajima, R.; Hasumi, H.; Valera, V.A.; Klein, M.E.; Haines, D.C.; Merino, M.J.; Hong, S.B.; Yamaguchi, T.P.; et al. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc. Natl. Acad. Sci. USA 2009, 106, 18722–18727. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Jeong, S.J.; Park, M.C.; Kim, G.; Kwon, N.H.; Kim, H.K.; Ha, S.H.; Ryu, S.H.; Kim, S. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 2012, 149, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.S.; Son, K.; Arauz, E.; Han, J.M.; Kim, S.; Chen, J. Leucyl-tRNA synthetase activates Vps34 in amino acid-sensing mTORTORC1 signaling. Cell Rep. 2016, 16, 1510–1517. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product Tuberin as a target of the phosphoinositide 3-kinase/Akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Duran, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Thedieck, K.; Holzwarth, B.; Prentzell, M.T.; Boehlke, C.; Klasener, K.; Ruf, S.; Sonntag, A.G.; Maerz, L.; Grellscheid, S.N.; Kremmer, E.; et al. Inhibition of mTORC1 by Astrin and stress granules prevents apoptosis in cancer cells. Cell 2013, 154, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Wippich, F.; Bodenmiller, B.; Trajkovska, M.G.; Wanka, S.; Aebersold, R.; Pelkmans, L. Dual specificity kinase DURK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell 2013, 152, 791–805. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Clister, T.L.; Lowry, P.R.; Seldin, M.M.; Wong, G.W.; Zhang, J. Dynamic visualization of mTORC1 activity in living cells. Cell Rep. 2015. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, Y.; Jones, E.; Inoki, K. Lysosomal Regulation of mTORC1 by Amino Acids in Mammalian Cells. Biomolecules 2017, 7, 51. https://doi.org/10.3390/biom7030051
Yao Y, Jones E, Inoki K. Lysosomal Regulation of mTORC1 by Amino Acids in Mammalian Cells. Biomolecules. 2017; 7(3):51. https://doi.org/10.3390/biom7030051
Chicago/Turabian StyleYao, Yao, Edith Jones, and Ken Inoki. 2017. "Lysosomal Regulation of mTORC1 by Amino Acids in Mammalian Cells" Biomolecules 7, no. 3: 51. https://doi.org/10.3390/biom7030051
APA StyleYao, Y., Jones, E., & Inoki, K. (2017). Lysosomal Regulation of mTORC1 by Amino Acids in Mammalian Cells. Biomolecules, 7(3), 51. https://doi.org/10.3390/biom7030051