Anandamide Revisited: How Cholesterol and Ceramides Control Receptor-Dependent and Receptor-Independent Signal Transmission Pathways of a Lipid Neurotransmitter


 and
and 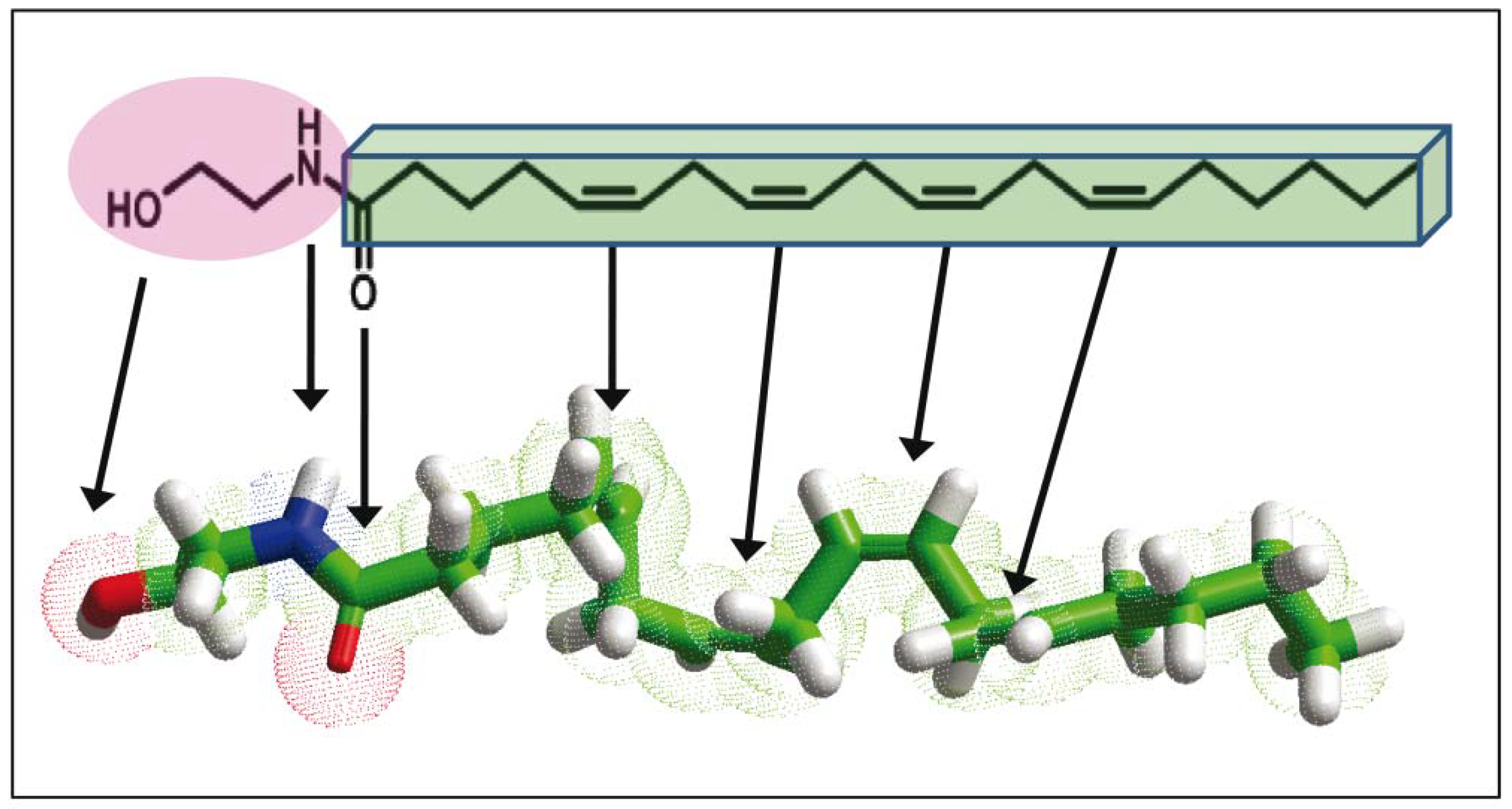{kind=link}
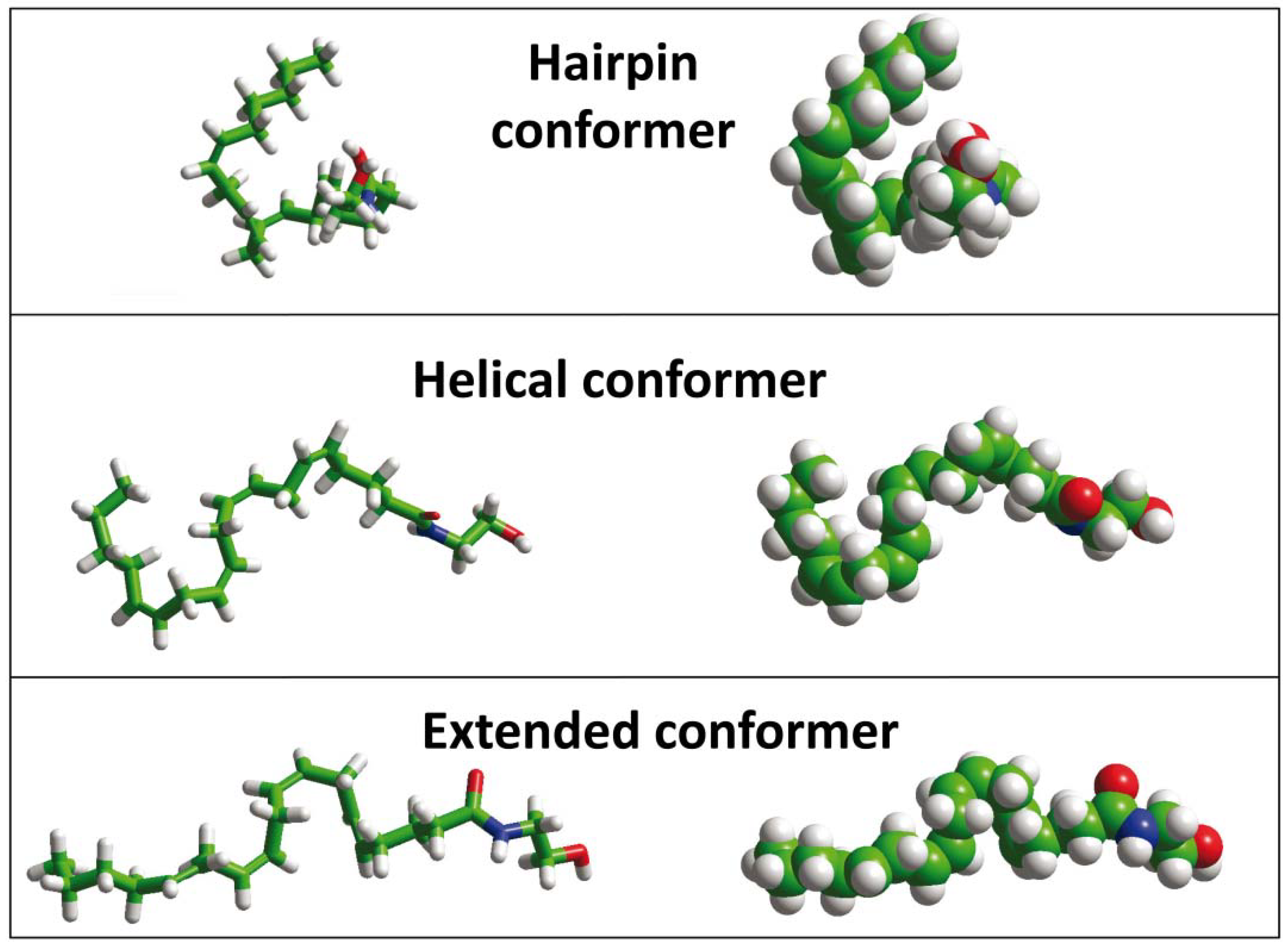{kind=link}
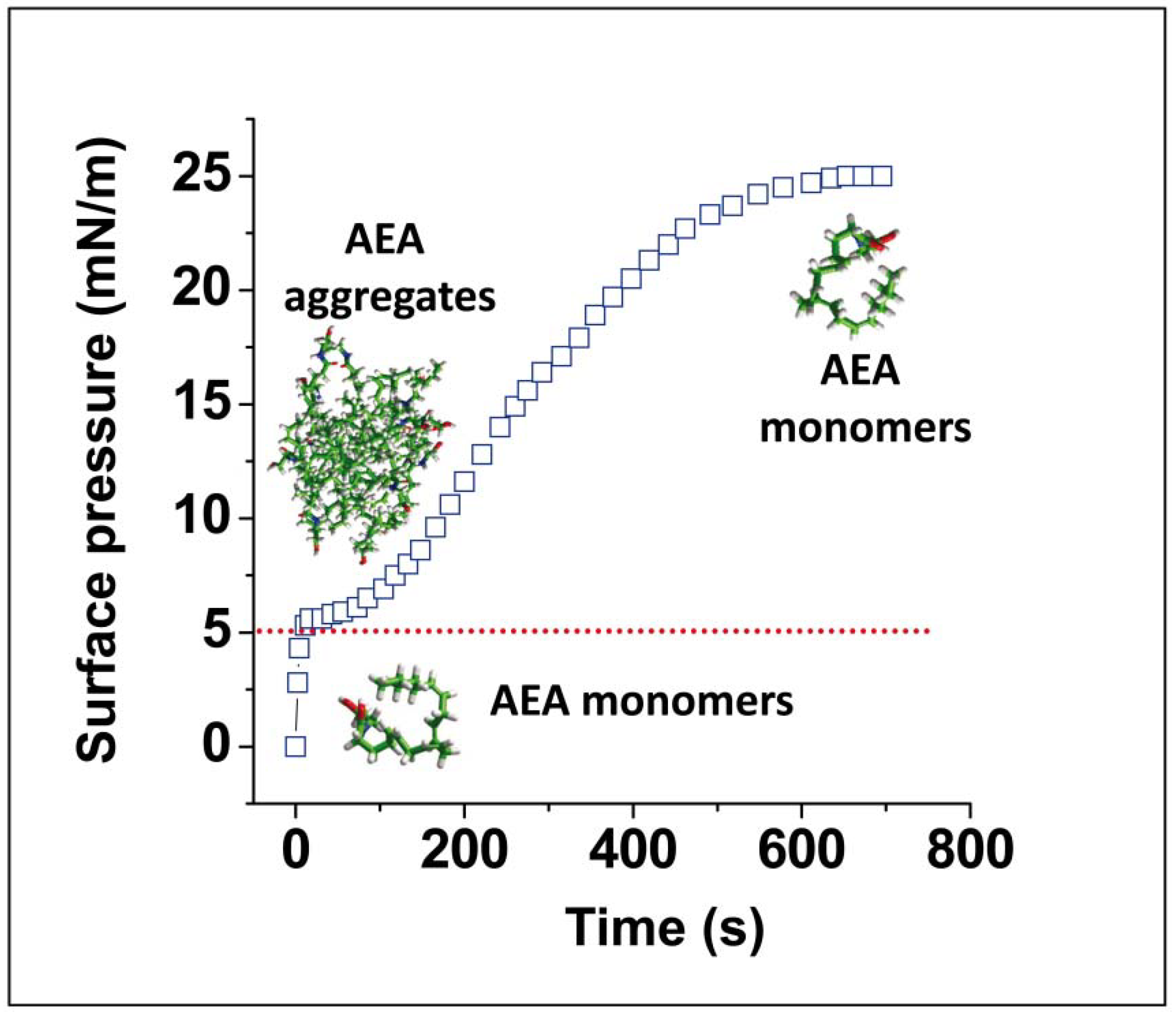{kind=link}
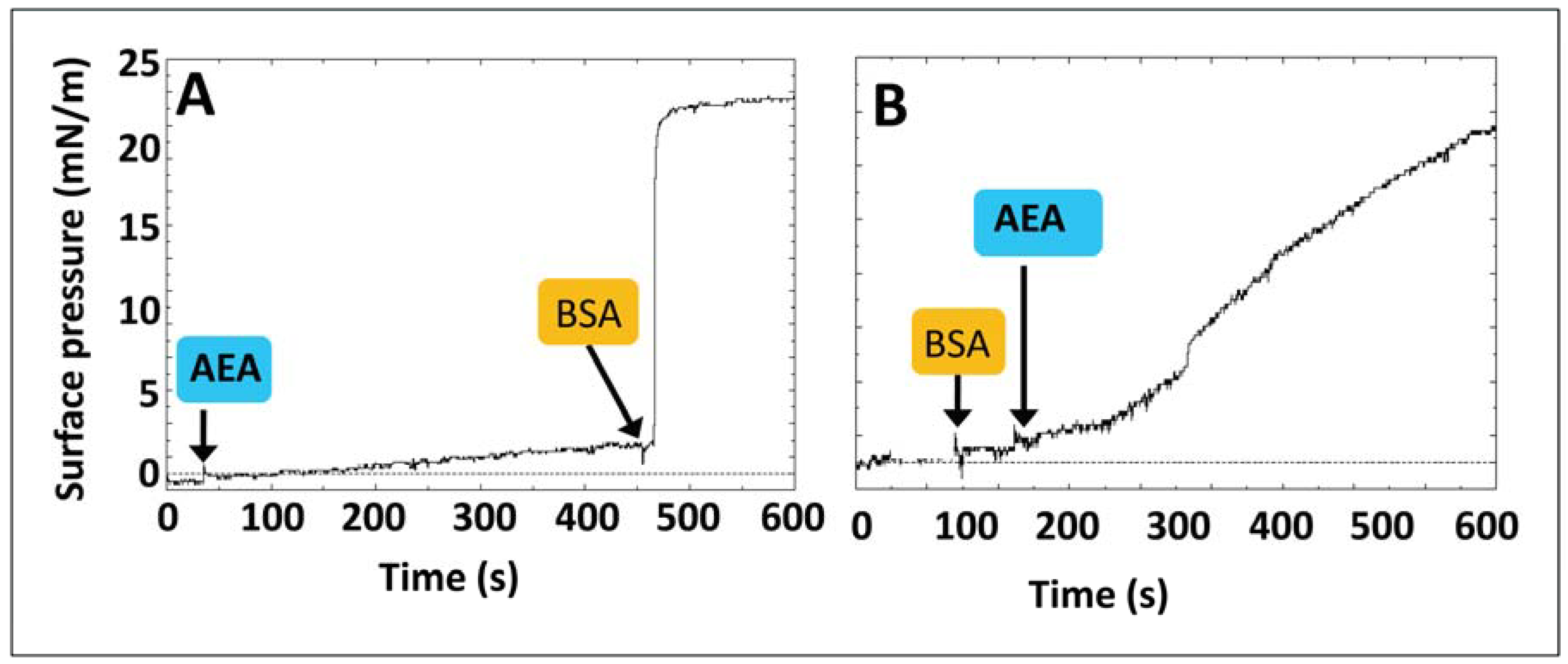{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Membrane Domains and the Synapse: General Aspects
3. Biochemical and Physico-Chemical Singularities of the Anandamide Synapse
4. Anandamide Diffusion through the Synaptic Cleft: Alone or Accompanied?
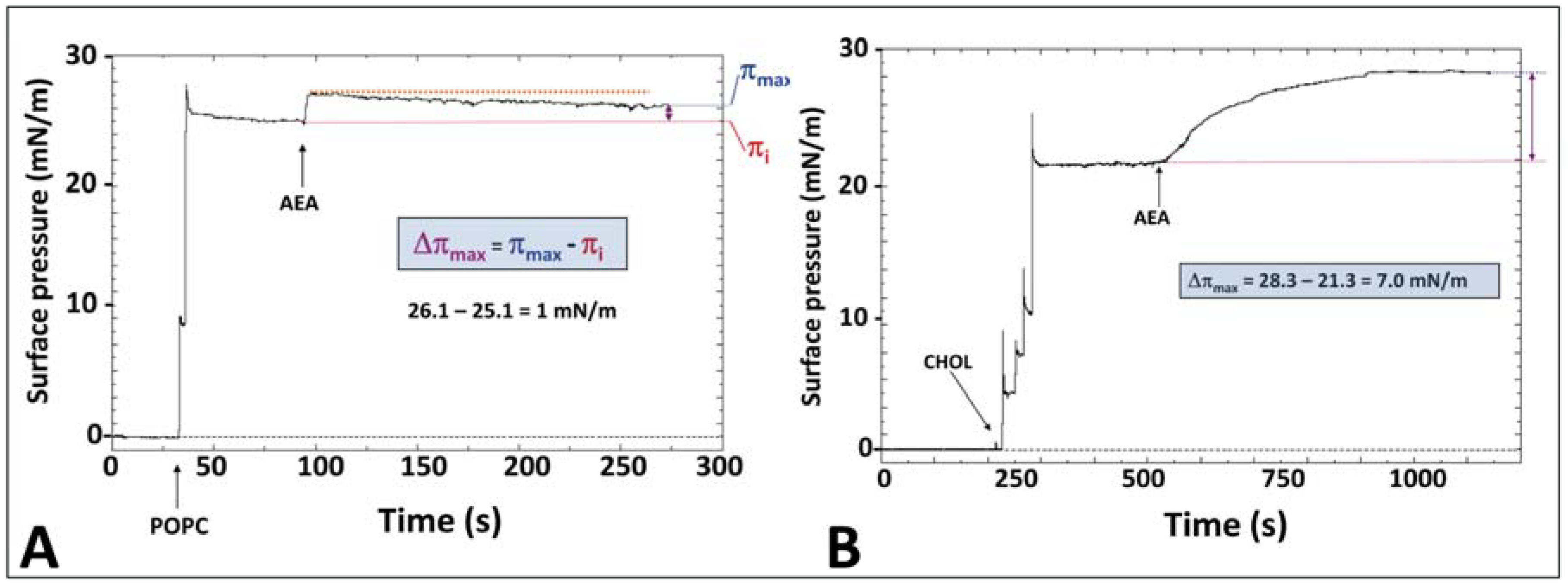
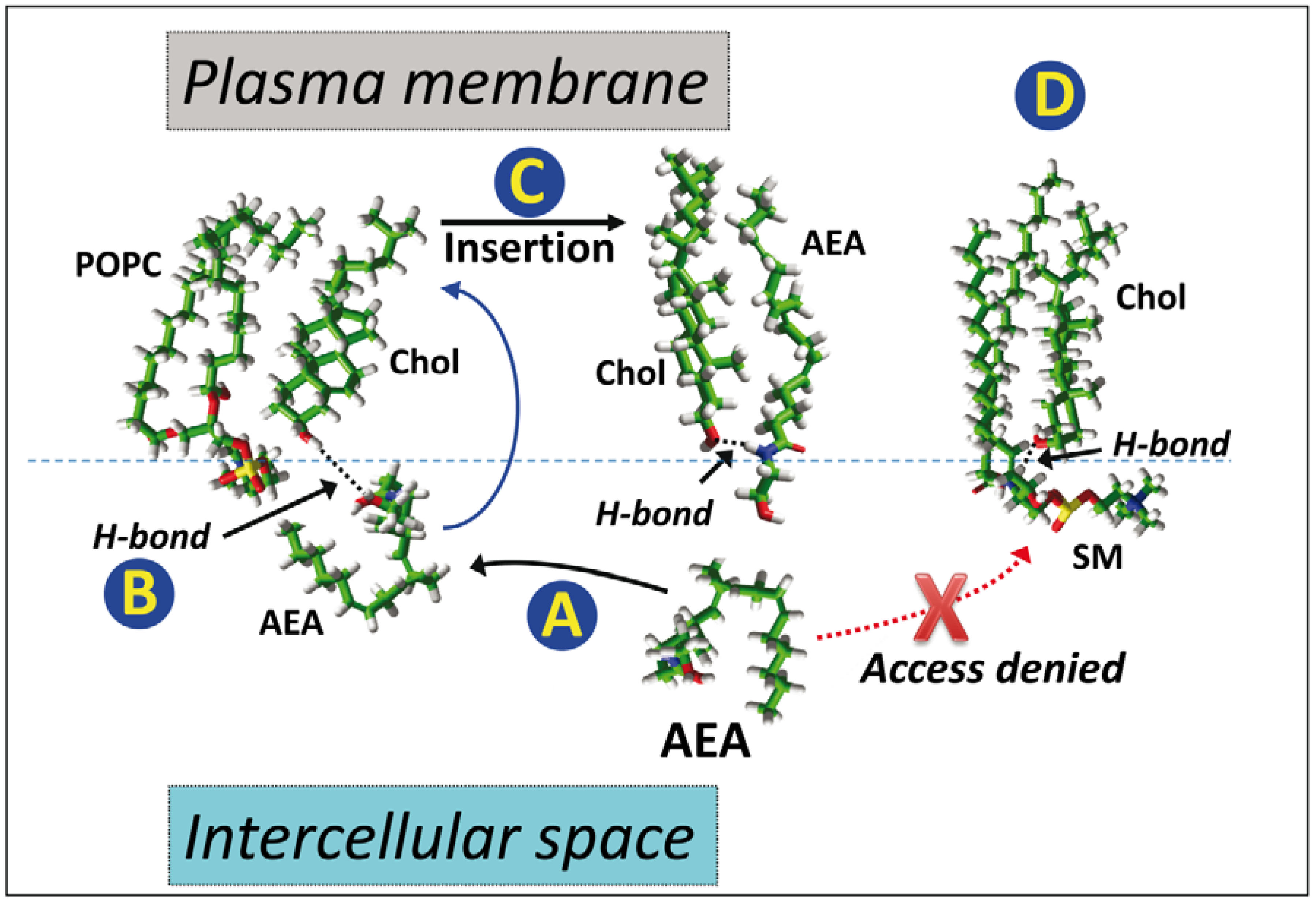
5. Anandamide Insertion in the Plasma Membrane: A Cholesterol-Dependent Process
6. Cholesterol-Dependent Anandamide Membrane Crossing
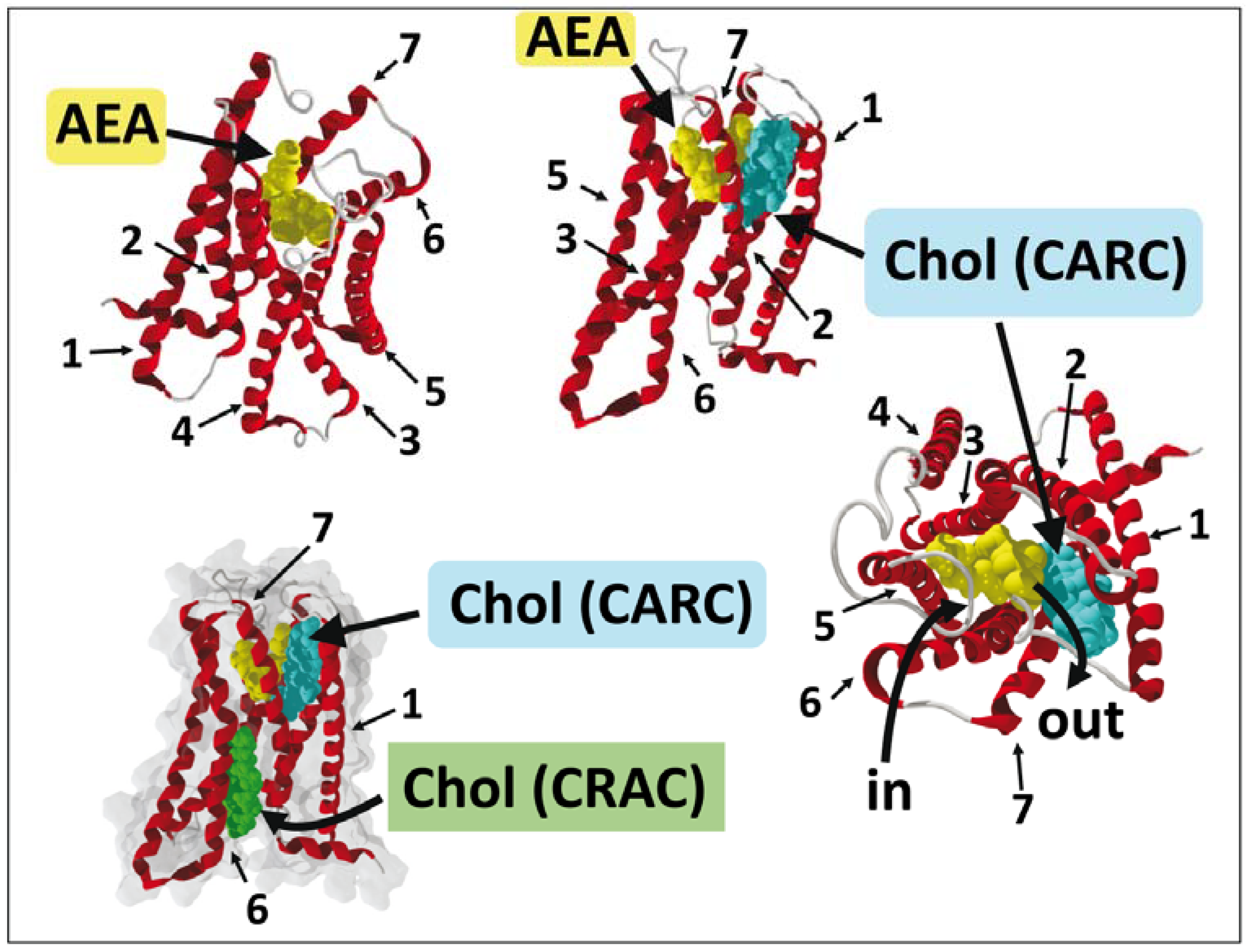
7. Endocannabinoid Binding to CB1 Receptors: In Silico Data
8. G Protein-Coupled Receptors and CB1 Receptors: What Crystallographic Data Have Revealed
9. A Dual Cholesterol–CB1 Receptor Model for Anandamide
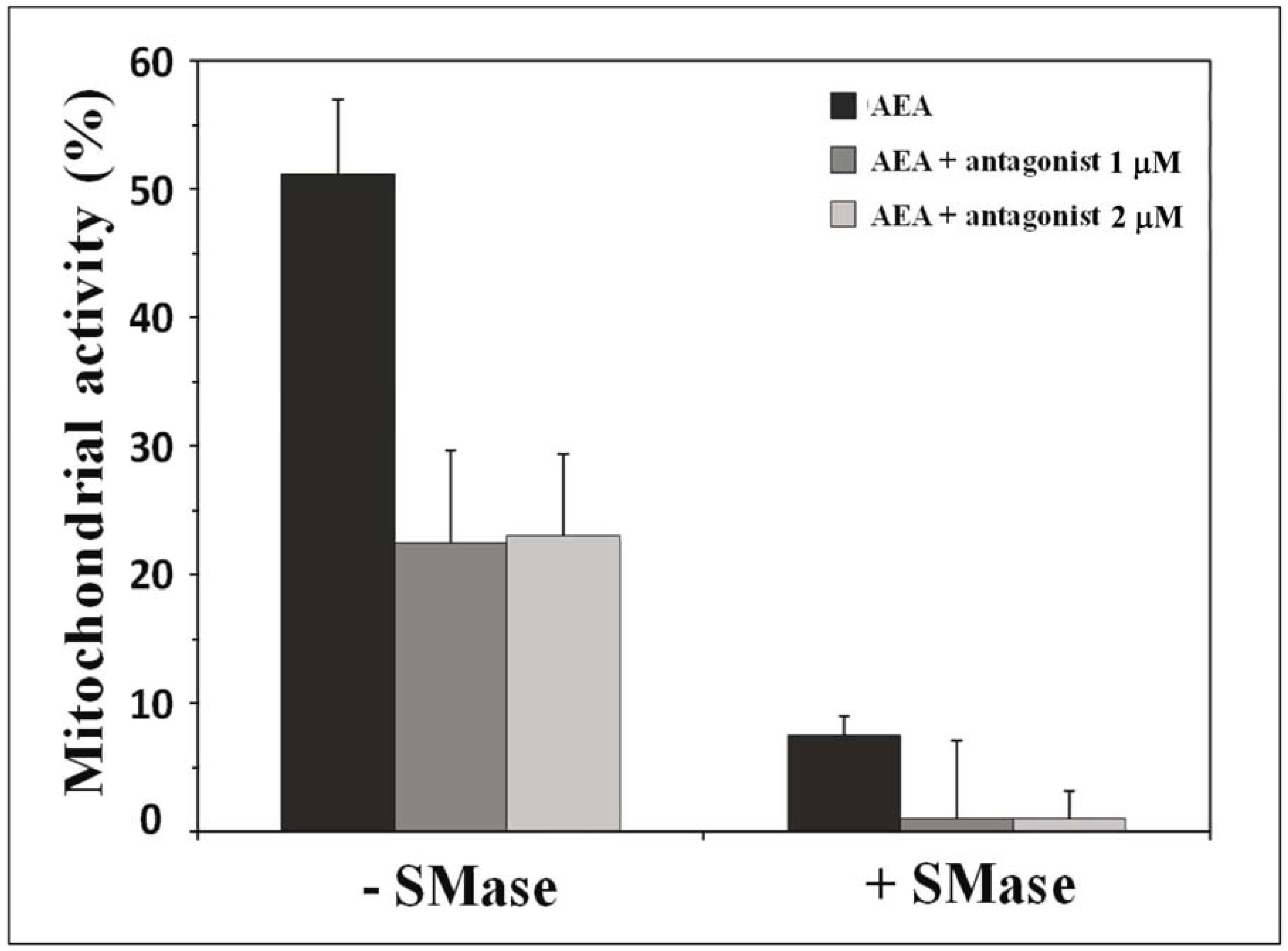
10. Receptor-Independent Signaling of Anandamide
11. How Can We Discriminate between Receptor-Dependent and Receptor-Independent Effects of Anandamide?
Acknowledgments
Conflicts of Interest
References
- Sudhof, T.C. Molecular Neuroscience in the 21(st) Century: A Personal Perspective. Neuron 2017, 96, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Garmy, N.; Mahfoud, R.; Yahi, N. Lipid rafts: Structure, function and role in HIV, Alzheimer’s and prion diseases. Expert Rev. Mol. Med. 2002, 4, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J. Rafts defined: A report on the Keystone Symposium on Lipid Rafts and Cell Function. J. Lipid Res. 2006, 47, 1597–1598. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.M.; Williamson, D.J.; Magenau, A.; Gaus, K. Sub-resolution lipid domains exist in the plasma membrane and regulate protein diffusion and distribution. Nat. Commun. 2012, 3, 1256. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Mukherjee, S.; Maxfield, F.R. Cholesterol depletion induces large scale domain segregation in living cell membranes. Proc. Natl. Acad. Sci. USA 2001, 98, 13072–13077. [Google Scholar] [CrossRef] [PubMed]
- Frisz, J.F.; Lou, K.; Klitzing, H.A.; Hanafin, W.P.; Lizunov, V.; Wilson, R.L.; Carpenter, K.J.; Kim, R.; Hutcheon, I.D.; Zimmerberg, J.; et al. Direct chemical evidence for sphingolipid domains in the plasma membranes of fibroblasts. Proc. Natl. Acad. Sci. USA 2013, 110, E613–E622. [Google Scholar] [CrossRef] [PubMed]
- Frisz, J.F.; Klitzing, H.A.; Lou, K.; Hutcheon, I.D.; Weber, P.K.; Zimmerberg, J.; Kraft, M.L. Sphingolipid domains in the plasma membranes of fibroblasts are not enriched with cholesterol. J. Boil. Chem. 2013, 288, 16855–16861. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Yahi, N. Brain Lipids in Synaptic Function and Neurological Disease. Clues to Innovative Therapeutic Strategies for Brain Disorders, 1st ed.; Elsevier: New York, NY, USA, 2015. [Google Scholar]
- Borroni, M.V.; Valles, A.S.; Barrantes, F.J. The lipid habitats of neurotransmitter receptors in brain. Biochim. Biophys. Acta 2016, 1858, 2662–2670. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Barrantes, F.J. Sphingolipid/cholesterol regulation of neurotransmitter receptor conformation and function. Biochim. Biophys. Acta 2009, 1788, 2345–2361. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.A.; Halverson-Tamboli, R.A.; Rasenick, M.M. Lipid raft microdomains and neurotransmitter signaling. Nat. Rev. Neurosci. 2007, 8, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Bieberich, E. It’s a lipid’s world: Bioactive lipid metabolism and signaling in neural stem cell differentiation. Neurochem. Res. 2012, 37, 1208–1229. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Anderson, H.D. Cannabinoid signaling in health and disease. Can. J. Physiol. Pharmacol. 2017, 95, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Sarne, Y.; Mechoulam, R. Cannabinoids: Between neuroprotection and neurotoxicity, Current drug targets. CNS Neurol. Disord. 2005, 4, 677–684. [Google Scholar] [CrossRef]
- Okamoto, Y.; Wang, J.; Morishita, J.; Ueda, N. Biosynthetic pathways of the endocannabinoid anandamide. Chem. Biodivers. 2007, 4, 1842–1857. [Google Scholar]
- Fride, E. Endocannabinoids in the central nervous system—An overview. Prostaglandins Leukot. Essent. Fatty Acids 2002, 66, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Brash, A.R. Arachidonic acid as a bioactive molecule. J. Clin. Investig. 2001, 107, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Klinman, J.P. Nature of rate-limiting steps in the soybean lipoxygenase-1 reaction. Biochemistry 1995, 34, 14077–14092. [Google Scholar] [CrossRef] [PubMed]
- Bild, G.S.; Ramadoss, C.S.; Lim, S.; Axelrod, B. Double dioxygenation of arachidonic acid by soybean lipoxygenase-1. Biochem. Biophys. Res. Commun. 1977, 74, 949–954. [Google Scholar] [CrossRef]
- Reggio, P.H.; Traore, H. Conformational requirements for endocannabinoid interaction with the cannabinoid receptors, the anandamide transporter and fatty acid amidohydrolase. Chem. Phys. Lipids 2000, 108, 15–35. [Google Scholar] [CrossRef]
- Di Scala, C.; Mazzarino, M.; Yahi, N.; Varini, K.; Garmy, N.; Fantini, J.; Chahinian, H. Anandamide-ceramide interactions in a membrane environment: Molecular dynamic simulations data. Data Brief 2017, 14, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Di Scala, C.; Fantini, J. Hybrid In Silico/In Vitro Approaches for the Identification of Functional Cholesterol-Binding Domains in Membrane Proteins. Methods Mol. Boil. 2017, 1583, 7–19. [Google Scholar]
- Alger, B.E. Endocannabinoids: Getting the message across. Proc. Natl. Acad. Sci. USA 2004, 101, 8512–8513. [Google Scholar] [CrossRef] [PubMed]
- Kreitzer, A.C.; Regehr, W.G. Retrograde signaling by endocannabinoids. Curr. Opin. Neurobiol. 2002, 12, 324–330. [Google Scholar] [CrossRef]
- Thakur, G.; Micic, M.; Leblanc, R.M. Surface chemistry of Alzheimer’s disease: A Langmuir monolayer approach. Colloids Surf. B Biointerfaces 2009, 74, 436–456. [Google Scholar] [CrossRef] [PubMed]
- Di Scala, C.; Yahi, N.; Fantini, J.; Chahinian, H. Disruption of anandamide aggregates studied by surface pressure measurements. Unpublished data. 2017. [Google Scholar]
- Castagnet, P.I.; Golovko, M.Y.; Barcelo-Coblijn, G.C.; Nussbaum, R.L.; Murphy, E.J. Fatty acid incorporation is decreased in astrocytes cultured from α-synuclein gene-ablated mice. J. Neurochem. 2005, 94, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Chivet, M.; Hemming, F.; Pernet-Gallay, K.; Fraboulet, S.; Sadoul, R. Emerging role of neuronal exosomes in the central nervous system. Front. Physiol. 2012, 3, 145. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.; Oliva, R.; del Vecchio, P.; Paolantoni, M.; Morresi, A.; Sassi, P. DMSO-induced perturbation of thermotropic properties of cholesterol-containing DPPC liposomes. Biochim. Biophys. Acta 2016, 1858, 3024–3031. [Google Scholar] [CrossRef] [PubMed]
- Gurtovenko, A.A.; Anwar, J. Modulating the structure and properties of cell membranes: The molecular mechanism of action of dimethyl sulfoxide. J. Phys. Chem. B 2007, 111, 10453–10460. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, L. Brain lipid changes after ethanol exposure. Upsala J. Med. Sci. Suppl. 1990, 48, 245–266. [Google Scholar]
- Di Pasquale, E.; Chahinian, H.; Sanchez, P.; Fantini, J. The insertion and transport of anandamide in synthetic lipid membranes are both cholesterol-dependent. PLoS ONE 2009, 4, e4989. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C.; Abood, M.E. CB1 and CB2 Receptor Pharmacology. Adv. Pharmacol. 2017, 80, 169–206. [Google Scholar] [PubMed]
- Barnett-Norris, J.; Hurst, D.P.; Lynch, D.L.; Guarnieri, F.; Makriyannis, A.; Reggio, P.H. Conformational memories and the endocannabinoid binding site at the cannabinoid CB1 receptor. J. Med. Chem. 2002, 45, 3649–3659. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Guo, J.; Yao, F.; Yang, D.P.; Makriyannis, A. The conformation, location, and dynamic properties of the endocannabinoid ligand anandamide in a membrane bilayer. J. Boil. Chem. 2005, 280, 29788–29795. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C.; Reggio, P.H.; Childers, S.R.; Hampson, R.E.; Ulloa, N.M.; Deutsch, D.G. Endocannabinoid tone versus constitutive activity of cannabinoid receptors. Br. J. Pharmacol. 2011, 163, 1329–1343. [Google Scholar] [CrossRef] [PubMed]
- Fowler, C.J. Transport of endocannabinoids across the plasma membrane and within the cell. FEBS J. 2013, 280, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Fegley, D.; Kathuria, S.; Mercier, R.; Li, C.; Goutopoulos, A.; Makriyannis, A.; Piomelli, D. Anandamide transport is independent of fatty-acid amide hydrolase activity and is blocked by the hydrolysis-resistant inhibitor AM1172. Proc. Natl. Acad. Sci. USA 2004, 101, 8756–8761. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M. Metabolism of the Endocannabinoid Anandamide: Open Questions after 25 Years. Front. Mol. Neurosci. 2017, 10, 166. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.T.; Kaczocha, M.; Deutsch, D.G. Anandamide transport: A critical review. Life Sci. 2005, 77, 1584–1604. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Kaczocha, M.; Deutsch, D.G. 2-Arachidonoylglycerol (2-AG) membrane transport: History and outlook. AAPS J. 2006, 8, E409–E412. [Google Scholar] [CrossRef] [PubMed]
- Aureli, M.; Grassi, S.; Prioni, S.; Sonnino, S.; Prinetti, A. Lipid membrane domains in the brain. Biochim. Biophys. Acta 2015, 1851, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, D.; Kucerka, N.; Wassall, S.R.; Harroun, T.A.; Katsaras, J. Cholesterol’s location in lipid bilayers. Chem. Phys. Lipids 2016, 199, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.L.; Davis, J.H. Liquid Disordered-Liquid Ordered Phase Coexistence in Lipid/Cholesterol Mixtures: A Deuterium 2D NMR Exchange Study. Langmuir 2017, 33, 1881–1890. [Google Scholar] [CrossRef] [PubMed]
- Thakur, G.; Pao, C.; Micic, M.; Johnson, S.; Leblanc, R.M. Surface chemistry of lipid raft and amyloid Aβ (1–40) Langmuir monolayer. Colloids Surf. B Biointerfaces 2011, 87, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Hammache, D.; Pieroni, G.; Maresca, M.; Ivaldi, S.; Yahi, N.; Fantini, J. Reconstitution of sphingolipid-cholesterol plasma membrane microdomains for studies of virus-glycolipid interactions. Methods Enzymol. 2000, 312, 495–506. [Google Scholar] [PubMed]
- Di Scala, C.; Chahinian, H.; Yahi, N.; Garmy, N.; Fantini, J. Interaction of Alzheimer’s β-amyloid peptides with cholesterol: Mechanistic insights into amyloid pore formation. Biochemistry 2014, 53, 4489–4502. [Google Scholar] [CrossRef] [PubMed]
- Di Scala, C.; Mazzarino, M.; Yahi, N.; Varini, K.; Garmy, N.; Fantini, J.; Chahinian, H. Ceramide binding to anandamide increases its half-life and potentiates its cytotoxicity in human neuroblastoma cells. Chem. Phys. Lipids 2017, 205, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Lonnfors, M.; Langvik, O.; Bjorkbom, A.; Slotte, J.P. Cholesteryl phosphocholine—A study on its interactions with ceramides and other membrane lipids. Langmuir 2013, 29, 2319–2329. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, R.; Pandit, S.A. Mixing properties of sphingomyelin ceramide bilayers: A simulation study. J. Phys. Chem. B. 2012, 116, 4500–4509. [Google Scholar] [CrossRef] [PubMed]
- Bari, M.; Battista, N.; Fezza, F.; Finazzi-Agro, A.; Maccarrone, M. Lipid rafts control signaling of type-1 cannabinoid receptors in neuronal cells. Implications for anandamide-induced apoptosis. J. Boil. Chem. 2005, 280, 12212–12220. [Google Scholar] [CrossRef] [PubMed]
- Sarnataro, D.; Grimaldi, C.; Pisanti, S.; Gazzerro, P.; Laezza, C.; Zurzolo, C.; Bifulco, M. Plasma membrane and lysosomal localization of CB1 cannabinoid receptor are dependent on lipid rafts and regulated by anandamide in human breast cancer cells. FEBS Lett. 2005, 579, 6343–6349. [Google Scholar] [CrossRef] [PubMed]
- Rukmini, R.; Rawat, S.S.; Biswas, S.C.; Chattopadhyay, A. Cholesterol organization in membranes at low concentrations: Effects of curvature stress and membrane thickness. Biophys. J. 2001, 81, 2122–2134. [Google Scholar] [CrossRef]
- Mukherjee, S.; Chattopadhyay, A. Membrane organization at low cholesterol concentrations: A study using 7-nitrobenz-2-oxa-1,3-diazol-4-yl-labeled cholesterol. Biochemistry 1996, 35, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- Bennett, W.F.; Tieleman, D.P. Molecular simulation of rapid translocation of cholesterol, diacylglycerol, and ceramide in model raft and nonraft membranes. J. Lipid Res. 2012, 53, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; di Scala, C.; Evans, L.S.; Williamson, P.T.; Barrantes, F.J. A mirror code for protein-cholesterol interactions in the two leaflets of biological membranes. Sci. Rep. 2016, 6, 21907. [Google Scholar] [CrossRef] [PubMed]
- Kaczocha, M.; Glaser, S.T.; Deutsch, D.G. Identification of intracellular carriers for the endocannabinoid anandamide. Proc. Natl. Acad. Sci. USA 2009, 106, 6375–6380. [Google Scholar] [CrossRef] [PubMed]
- Fezza, F.; de Simone, C.; Amadio, D.; Maccarrone, M. Fatty acid amide hydrolase: A gate-keeper of the endocannabinoid system. Sub-Cell. Biochem. 2008, 49, 101–132. [Google Scholar]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Reggio, P.H. Endocannabinoid binding to the cannabinoid receptors: What is known and what remains unknown. Curr. Med. Chem. 2010, 17, 1468–1486. [Google Scholar] [CrossRef] [PubMed]
- Picone, R.P.; Khanolkar, A.D.; Xu, W.; Ayotte, L.A.; Thakur, G.A.; Hurst, D.P.; Abood, M.E.; Reggio, P.H.; Fournier, D.J.; Makriyannis, A. (−)-7′-Isothiocyanato-11-hydroxy-1′,1′-dimethylheptylhexahydrocannabinol (AM841), a high-affinity electrophilic ligand, interacts covalently with a cysteine in helix six and activates the CB1 cannabinoid receptor. Mol. Pharmacol. 2005, 68, 1623–1635. [Google Scholar] [PubMed]
- Gimpl, G. Interaction of G protein coupled receptors and cholesterol. Chem. Phys. Lipids 2016, 199, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Paila, Y.D.; Tiwari, S.; Chattopadhyay, A. Are specific nonannular cholesterol binding sites present in G-protein coupled receptors? Biochim. Biophys. Acta 2009, 1788, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Gimpl, G.; Burger, K.; Fahrenholz, F. Cholesterol as modulator of receptor function. Biochemistry 1997, 36, 10959–10974. [Google Scholar] [CrossRef] [PubMed]
- Paila, Y.D.; Chattopadhyay, A. Membrane cholesterol in the function and organization of G-protein coupled receptors. Sub-Cell. Biochem. 2010, 51, 439–466. [Google Scholar]
- Ma, P.; Weichert, D.; Aleksandrov, L.A.; Jensen, T.J.; Riordan, J.R.; Liu, X.; Kobilka, B.K.; Caffrey, M. The cubicon method for concentrating membrane proteins in the cubic mesophase. Nat. Protoc. 2017, 12, 1745–1762. [Google Scholar] [CrossRef] [PubMed]
- Grisshammer, R. New approaches towards the understanding of integral membrane proteins: A structural perspective on G protein-coupled receptors. Protein Sci. 2017, 26, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, M. Membrane protein crystallization. J. Struct. Boil. 2003, 142, 108–132. [Google Scholar] [CrossRef]
- Yin, X.; Xu, H.; Hanson, M.; Liu, W. GPCR crystallization using lipidic cubic phase technique. Curr. Pharm. Biotechnol. 2014, 15, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.A.; Cherezov, V.; Griffith, M.T.; Roth, C.B.; Jaakola, V.P.; Chien, E.Y.; Velasquez, J.; Kuhn, P.; Stevens, R.C. A specific cholesterol binding site is established by the 2.8 Å structure of the human β2-adrenergic receptor. Structure 2008, 16, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Di Scala, C.; Baier, C.J.; Evans, L.S.; Williamson, P.T.F.; Fantini, J.; Barrantes, F.J. Relevance of CARC and CRAC Cholesterol-Recognition Motifs in the Nicotinic Acetylcholine Receptor and Other Membrane-Bound Receptors. Curr. Top. Membr. 2017, 80, 3–23. [Google Scholar] [PubMed]
- Jamin, N.; Neumann, J.M.; Ostuni, M.A.; Vu, T.K.; Yao, Z.X.; Murail, S.; Robert, J.C.; Giatzakis, C.; Papadopoulos, V.; Lacapere, J.J. Characterization of the cholesterol recognition amino acid consensus sequence of the peripheral-type benzodiazepine receptor. Mol. Endocrinol. 2005, 19, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Baier, C.J.; Fantini, J.; Barrantes, F.J. Disclosure of cholesterol recognition motifs in transmembrane domains of the human nicotinic acetylcholine receptor. Sci. Rep. 2011, 1, 69. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; di Scala, C.; Baier, C.J.; Barrantes, F.J. Molecular mechanisms of protein-cholesterol interactions in plasma membranes: Functional distinction between topological (tilted) and consensus (CARC/CRAC) domains. Chem. Phys. Lipids 2016, 199, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Vemuri, K.; Nikas, S.P.; Laprairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.H.; et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 2017, 547, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Gater, D.L.; Saurel, O.; Iordanov, I.; Liu, W.; Cherezov, V.; Milon, A. Two classes of cholesterol binding sites for the β2AR revealed by thermostability and NMR. Biophys. J. 2014, 107, 2305–2312. [Google Scholar] [CrossRef] [PubMed]
- Manna, M.; Niemela, M.; Tynkkynen, J.; Javanainen, M. Mechanism of allosteric regulation of β2-adrenergic receptor by cholesterol. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167, 750–762.e714. [Google Scholar] [CrossRef] [PubMed]
- Sabatucci, A.; Tortolani, D.; Dainese, E. In silico mapping of allosteric ligand binding sites in type-1 cannabinoid receptor. Biotechnol. Appl. Biochem. 2018, 65, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Scheerer, P.; Hofmann, K.P.; Choe, H.W.; Ernst, O.P. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 2008, 454, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, P.W.; Scheerer, P.; Park, J.H.; Choe, H.W.; Piechnick, R.; Ernst, O.P.; Hofmann, K.P.; Heck, M. A ligand channel through the G protein coupled receptor opsin. PLoS ONE 2009, 4, e4382. [Google Scholar] [CrossRef] [PubMed]
- Ledeen, R.W.; Wu, G. Nuclear sphingolipids: Metabolism and signaling. J. Lipid Res. 2008, 49, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Parolaro, D.; Massi, P.; Rubino, T.; Monti, E. Endocannabinoids in the immune system and cancer. Prostaglandins Leukot. Essent. Fatty Acids 2002, 66, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Biswas, K.K.; Sarker, K.P.; Abeyama, K.; Kawahara, K.; Iino, S.; Otsubo, Y.; Saigo, K.; Izumi, H.; Hashiguchi, T.; Yamakuchi, M.; et al. Membrane cholesterol but not putative receptors mediates anandamide-induced hepatocyte apoptosis. Hepatology 2003, 38, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Dainese, E.; Oddi, S.; Bari, M.; Maccarrone, M. Modulation of the endocannabinoid system by lipid rafts. Curr. Med. Chem. 2007, 14, 2702–2715. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Liu, H.Y.; Zhang, Y.W.; Wu, W.J.; Tang, W.X. Anandamide induces cell death through lipid rafts in hepatic stellate cells. J. Gastroenterol. Hepatol. 2010, 25, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- McFarland, M.J.; Barker, E.L. Lipid rafts: A nexus for endocannabinoid signaling? Life Sci. 2005, 77, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- Sarker, K.P.; Maruyama, I. Anandamide induces cell death independently of cannabinoid receptors or vanilloid receptor 1: Possible involvement of lipid rafts. Cell. Mol. Life Sci. 2003, 60, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Kaczocha, M.; Lin, Q.; Nelson, L.D.; McKinney, M.K.; Cravatt, B.F.; London, E.; Deutsch, D.G. Anandamide externally added to lipid vesicles containing trapped fatty acid amide hydrolase (FAAH) is readily hydrolyzed in a sterol-modulated fashion. ACS Chem. Neurosci. 2012, 3, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Dainese, E.; de Fabritiis, G.; Sabatucci, A.; Oddi, S.; Angelucci, C.B.; di Pancrazio, C.; Giorgino, T.; Stanley, N.; del Carlo, M.; Cravatt, B.F.; et al. Membrane lipids are key modulators of the endocannabinoid-hydrolase FAAH. Biochem. J. 2014, 457, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Sanson, B.; Wang, T.; Sun, J.; Wang, L.; Kaczocha, M.; Ojima, I.; Deutsch, D.; Li, H. Crystallographic study of FABP5 as an intracellular endocannabinoid transporter. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Luberto, C. Ceramide in the eukaryotic stress response. Trends Cell Boil. 2000, 10, 73–80. [Google Scholar] [CrossRef]
- Sarda, L.; Desnuelle, P. Actions of pancreatic lipase on esters in emulsions. Biochim. Biophys. Acta 1958, 30, 513–521. [Google Scholar] [CrossRef]
- Chahinian, H.; Fantini, J.; Garmy, N.; Manco, G.; Sarda, L. Non-lipolytic and lipolytic sequence-related carboxylesterases: A comparative study of the structure-function relationships of rabbit liver esterase 1 and bovine pancreatic bile-salt-activated lipase. Biochim. Biophys. Acta 2010, 1801, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Airola, M.V.; Allen, W.J.; Pulkoski-Gross, M.J.; Obeid, L.M.; Rizzo, R.C.; Hannun, Y.A. Structural Basis for Ceramide Recognition and Hydrolysis by Human Neutral Ceramidase. Structure 2015, 23, 1482–1491. [Google Scholar] [CrossRef] [PubMed]
- Ira; Johnston, L.J. Sphingomyelinase generation of ceramide promotes clustering of nanoscale domains in supported bilayer membranes. Biochim. Biophys. Acta 2008, 1778, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Somerharju, P.; Virtanen, J.A.; Cheng, K.H.; Hermansson, M. The superlattice model of lateral organization of membranes and its implications on membrane lipid homeostasis. Biochim. Biophys. Acta 2009, 1788, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.W.; Goldberg, E.M.; Zidovetzki, R. Ceramide induces structural defects into phosphatidylcholine bilayers and activates phospholipase A2. Biochem. Biophys. Res. Commun. 1996, 220, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Zha, X.; Pierini, L.M.; Leopold, P.L.; Skiba, P.J.; Tabas, I.; Maxfield, F.R. Sphingomyelinase treatment induces ATP-independent endocytosis. J. Cell Boil. 1998, 140, 39–47. [Google Scholar] [CrossRef]
- Movsesyan, V.A.; Stoica, B.A.; Yakovlev, A.G.; Knoblach, S.M.; Lea, P.M.T.; Cernak, I.; Vink, R.; Faden, A.I. Anandamide-induced cell death in primary neuronal cultures: Role of calpain and caspase pathways. Cell Death Differ. 2004, 11, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, A.; Beltramo, M.; Piomelli, D. Mechanisms of endocannabinoid inactivation: Biochemistry and pharmacology. J. Pharmacol. Exp. Ther. 2001, 298, 7–14. [Google Scholar] [PubMed]
- Di Scala, C.; Yahi, N.; Fantini, J.; Chahinian, H. Effect of sphingomyelinase on the mitochondrial toxicity of anandamide in cultured neural cells. Unpublished data. 2017. [Google Scholar]
- Smart, D.; Gunthorpe, M.J.; Jerman, J.C.; Nasir, S.; Gray, J.; Muir, A.I.; Chambers, J.K.; Randall, A.D.; Davis, J.B. The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1). Br. J. Pharmacol. 2000, 129, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Ryberg, E.; Larsson, N.; Sjogren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
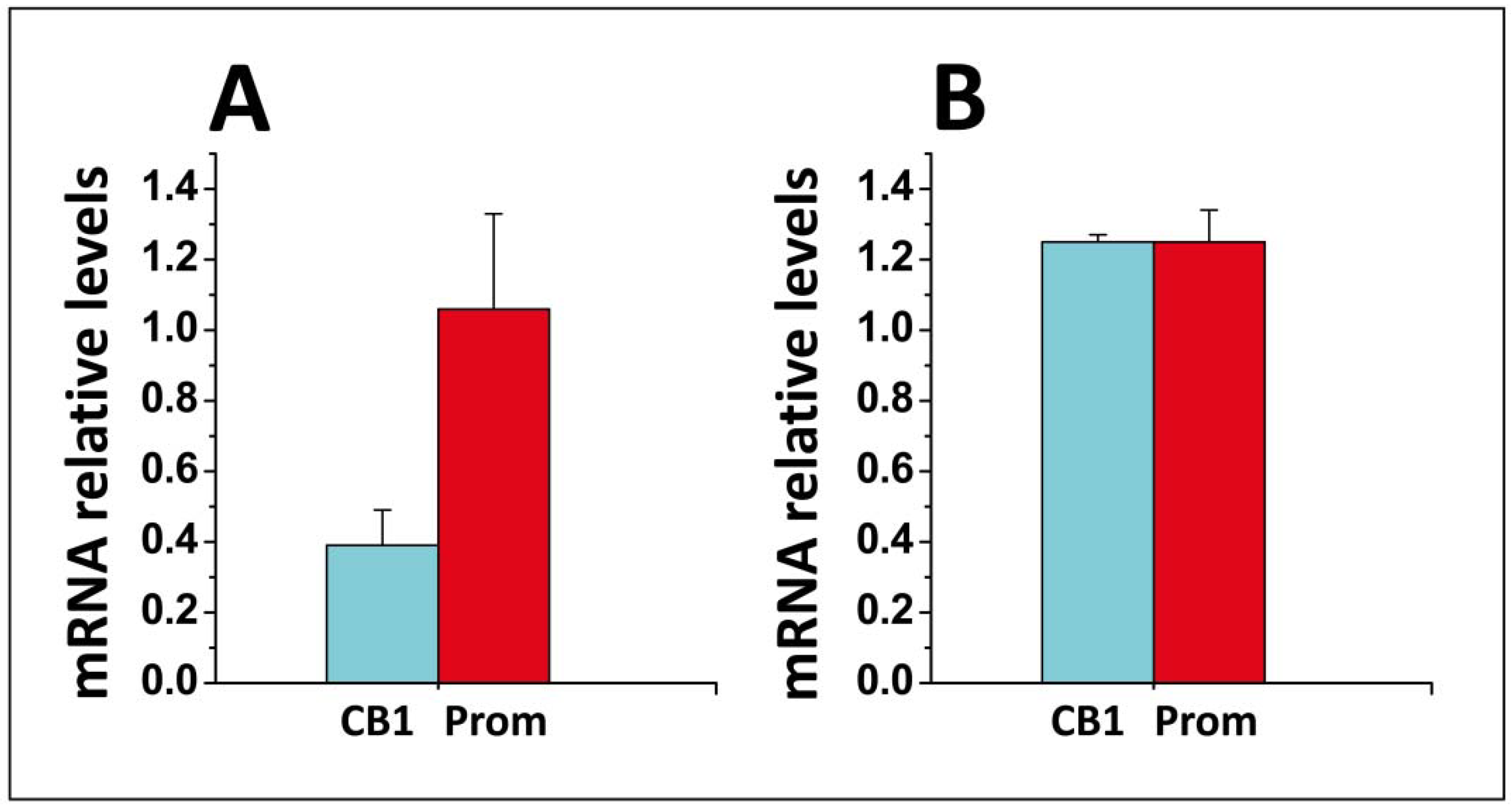
- Di Scala, C.; Yahi, N.; Fantini, J.; Chahinian, H. Real-time PCR determinations of CB1 and prominin mRNAs expression profile in anandamide-treated cells. Unpublished data. 2017. [Google Scholar]
- De, L.; Petrocellis, M.; Nabissi, G.; Santoni, A. Ligresti, Actions and Regulation of Ionotropic Cannabinoid Receptors. Adv. Pharmacol. 2017, 80, 249–289. [Google Scholar]
- Shimasue, K.; Urushidani, T.; Hagiwara, M.; Nagao, T. Effects of anandamide and arachidonic acid on specific binding of (+)-PN200-110, diltiazem and (−)-desmethoxyverapamil to L-type Ca2+ channel. Eur. J. Pharmacol. 1996, 296, 347–350. [Google Scholar] [CrossRef]
- Barann, M.; Molderings, G.; Bruss, M.; Bonisch, H.; Urban, B.W.; Gothert, M. Direct inhibition by cannabinoids of human 5-HT3A receptors: Probable involvement of an allosteric modulatory site. Br. J. Pharmacol. 2002, 137, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Huang, W.; Wu, D.; Priestley, J.V. TRPV1, but not P2X, requires cholesterol for its function and membrane expression in rat nociceptors. Eur. J. Neurosci. 2006, 24, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Nothdurfter, C.; Tanasic, S.; di Benedetto, B.; Rammes, G.; Wagner, E.M.; Kirmeier, T.; Ganal, V.; Kessler, J.S.; Rein, T.; Holsboer, F.; et al. Impact of lipid raft integrity on 5-HT3 receptor function and its modulation by antidepressants. Neuropsychopharmacology 2010, 35, 1510–1519. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, H.; Song, Y.; Watanabe, M.; Masumiya, H.; Gupte, S.A.; Ochi, R.; Okada, T. Cholesterol depletion modulates basal L-type Ca2+ current and abolishes its -adrenergic enhancement in ventricular myocytes, American journal of physiology. Heart Circ. Physiol. 2008, 294, H285–H292. [Google Scholar] [CrossRef] [PubMed]
- Chik, C.L.; Li, B.; Karpinski, E.; Ho, A.K. Ceramide inhibits L-type calcium channel currents in GH3 cells. Mol. Cell. Endocrinol. 2004, 218, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Galve-Roperh, I.; Sanchez, C.; Blazquez, C.; Haro, A.; Guzman, M. Cannabinoids and ceramide: Two lipids acting hand-by-hand. Life Sci. 2005, 77, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Truman, J.P.; Garcia-Barros, M.; Obeid, L.M.; Hannun, Y.A. Evolving concepts in cancer therapy through targeting sphingolipid metabolism. Biochim. Biophys. Acta 2014, 1841, 1174–1188. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, K.; Sander, B.; Bielawski, J.; Hannun, Y.A.; Flygare, J. Potentiation of cannabinoid-induced cytotoxicity in mantle cell lymphoma through modulation of ceramide metabolism. Mol. Cancer Res. 2009, 7, 1086–1098. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scala, C.D.; Fantini, J.; Yahi, N.; Barrantes, F.J.; Chahinian, H. Anandamide Revisited: How Cholesterol and Ceramides Control Receptor-Dependent and Receptor-Independent Signal Transmission Pathways of a Lipid Neurotransmitter. Biomolecules 2018, 8, 31. https://doi.org/10.3390/biom8020031
Scala CD, Fantini J, Yahi N, Barrantes FJ, Chahinian H. Anandamide Revisited: How Cholesterol and Ceramides Control Receptor-Dependent and Receptor-Independent Signal Transmission Pathways of a Lipid Neurotransmitter. Biomolecules. 2018; 8(2):31. https://doi.org/10.3390/biom8020031
Chicago/Turabian StyleScala, Coralie Di, Jacques Fantini, Nouara Yahi, Francisco J. Barrantes, and Henri Chahinian. 2018. "Anandamide Revisited: How Cholesterol and Ceramides Control Receptor-Dependent and Receptor-Independent Signal Transmission Pathways of a Lipid Neurotransmitter" Biomolecules 8, no. 2: 31. https://doi.org/10.3390/biom8020031
APA StyleScala, C. D., Fantini, J., Yahi, N., Barrantes, F. J., & Chahinian, H. (2018). Anandamide Revisited: How Cholesterol and Ceramides Control Receptor-Dependent and Receptor-Independent Signal Transmission Pathways of a Lipid Neurotransmitter. Biomolecules, 8(2), 31. https://doi.org/10.3390/biom8020031