Molecular Modeling Applied to Nucleic Acid-Based Molecule Development


Abstract
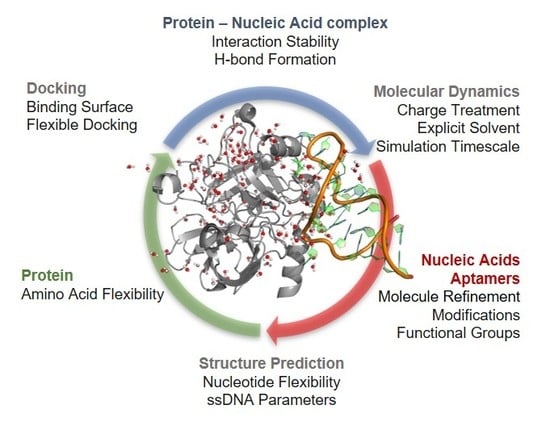
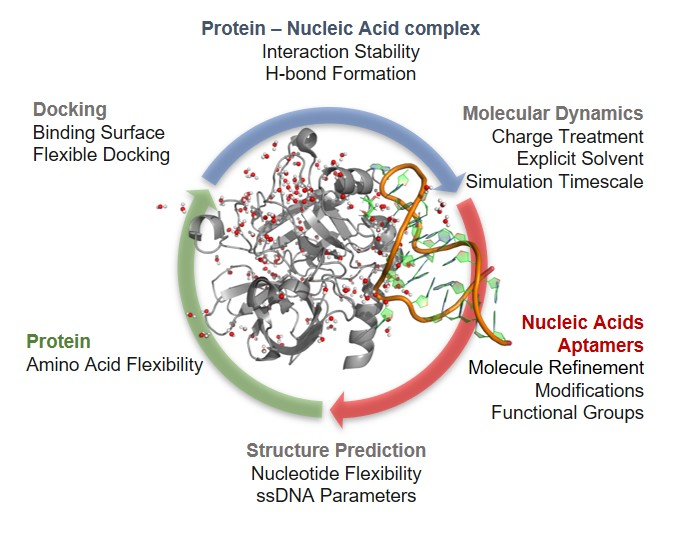
:
1. Introduction—Nucleic Acids as a Class of Drugs and Biomarkers
2. In Silico Approaches for Structure Prediction and Docking
2.1. Systematic Evolution of Ligands by Exponential Enrichment—Alternative Algorithms
2.2. Aptamer Secondary and Tertiary Structure Prediction
3. Historical Overview of Docking Algorithm Development
4. Benchmarking and Quality Tests
5. Evaluating the Quality of Docking
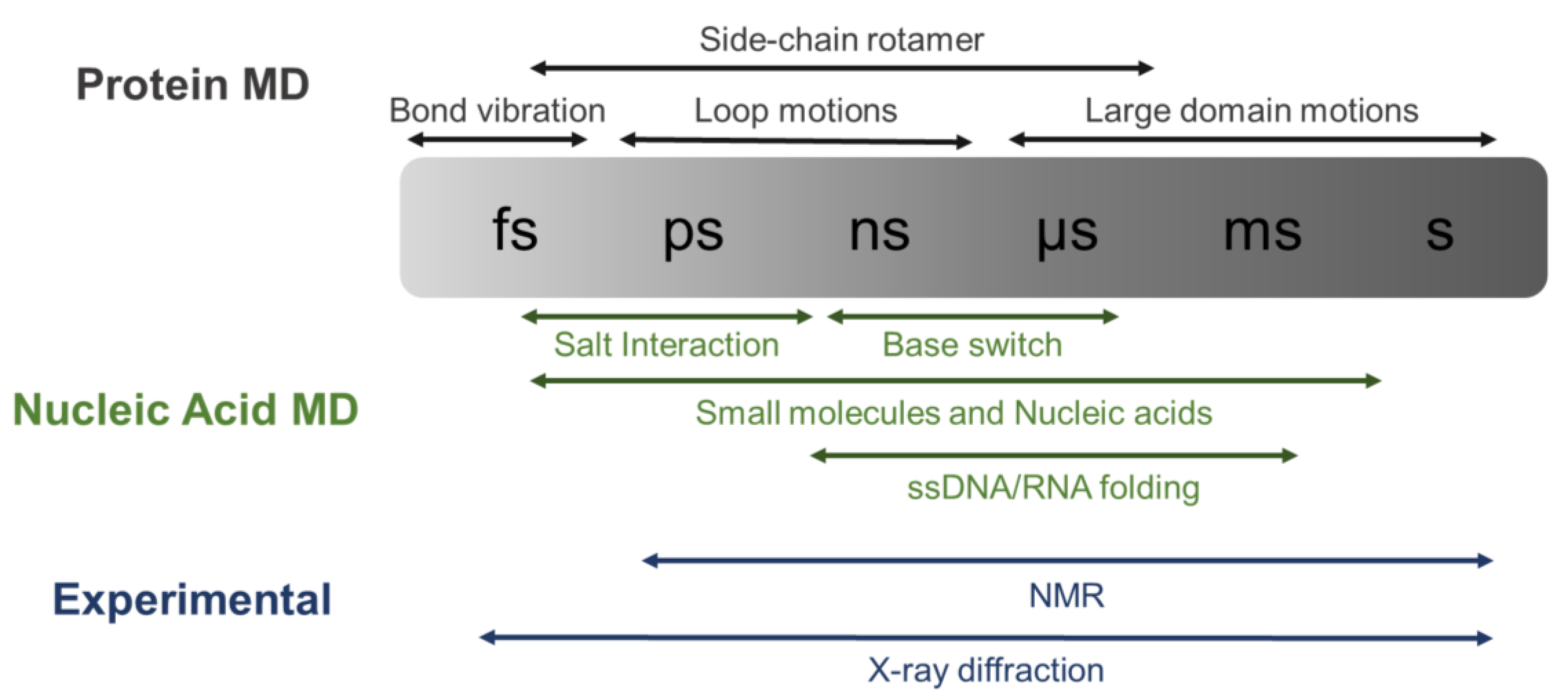
6. Molecular Dynamics Simulations Applied to Nucleic Acids (NA) and Protein–NA Interactions
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Hess, G.P.; Ulrich, H.; Breitinger, H.-G.; Niu, L.; Gameiro, A.M.; Grewer, C.; Srivastava, S.; Ippolito, J.E.; Lee, S.M.; Jayaraman, V.; et al. Mechanism-based discovery of ligands that counteract inhibition of the nicotinic acetylcholine receptor by cocaine and MK-801. Proc. Natl. Acad. Sci. USA 2000, 97, 13895–13900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faria, M.; Ulrich, H. The use of synthetic oligonucleotides as protein inhibitors and anticode drugs in cancer therapy: Accomplishments and limitations. Curr. Cancer Drug Targets 2002, 2, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.; Wrenger, C. Disease-specific biomarker discovery by aptamers. Cytom. Part A 2009, 75A, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Sefah, K.; Shangguan, D.; Xiong, X.; O’Donoghue, M.B.; Tan, W. Development of DNA aptamers using Cell-SELEX. Nat. Protoc. 2010, 5, 1169–1185. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.N.; Jensen, K.B.; Julin, C.M.; Weil, M.; Gold, L. High affinity ligands from in vitro selection: Complex targets. Proc. Natl. Acad. Sci. USA 1998, 95, 2902–2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelinas, A.D.; Davies, D.R.; Janjic, N. Embracing proteins: Structural themes in aptamer-protein complexes. Curr. Opin. Struct. Biol. 2016, 36, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Pabo, C.O.; Sauer, R.T. Protein-DNA recognition. Annu. Rev. Biochem. 1984, 53, 293–321. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.-A.; Williams, L.D.; Koudelka, G.B. Specific minor groove solvation is a crucial determinant of DNA binding site recognition. Nucleic Acids Res. 2014, 42, 14053–14059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendieta, J.; Pérez-Lago, L.; Salas, M.; Camacho, A. Functional specificity of a protein-DNA complex mediated by two arginines bound to the minor groove. J. Bacteriol. 2012, 194, 4727–4735. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.; Martins, A.H.B.; Pesquero, J.B. RNA and DNA aptamers in cytomics analysis. Cytom. Part A 2004, 59, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, D.R.; Gelinas, A.D.; Zhang, C.; Rohloff, J.C.; Carter, J.D.; O’Connell, D.; Waugh, S.M.; Wolk, S.K.; Mayfield, W.S.; Burgin, A.B.; et al. Unique motifs and hydrophobic interactions shape the binding of modified DNA ligands to protein targets. Proc. Natl. Acad. Sci. USA 2012, 109, 19971–19976. [Google Scholar] [CrossRef] [PubMed]
- Vaught, J.D.; Bock, C.; Carter, J.; Fitzwater, T.; Otis, M.; Schneider, D.; Rolando, J.; Waugh, S.; Wilcox, S.K.; Eaton, B.E. Expanding the chemistry of DNA for in vitro selection. J. Am. Chem. Soc. 2010, 132, 4141–4151. [Google Scholar] [CrossRef] [PubMed]
- Rohloff, J.C.; Gelinas, A.D.; Jarvis, T.C.; Ochsner, U.A.; Schneider, D.J.; Gold, L.; Janjic, N. Nucleic acid ligands with protein-like side chains: Modified aptamers and their use as diagnostic and therapeutic agents. Mol. Ther. Nucleic Acids 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, V.B.; Taylor, A.I.; Cozens, C.; Abramov, M.; Renders, M.; Zhang, S.; Chaput, J.C.; Wengel, J.; Peak-Chew, S.-Y.; McLaughlin, S.H.; et al. Synthetic genetic polymers capable of heredity and evolution. Science 2012, 336, 341–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meek, K.N.; Rangel, A.E.; Heemstra, J.M. Enhancing aptamer function and stability via in vitro selection using modified nucleic acids. Methods 2016, 106, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masaki, Y.; Miyasaka, R.; Ohkubo, A.; Seio, K.; Sekine, M. Linear relationship between deformability and thermal stability of 2′-O-Modified RNA hetero duplexes. J. Phys. Chem. B 2010, 114, 2517–2524. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Rossi, J.J. Future strategies for the discovery of therapeutic aptamers. Expert Opin. Drug Discov. 2017, 12, 317–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolle, F.; Brändle, G.M.; Matzner, D.; Mayer, G. A versatile approach towards nucleobase-modified aptamers. Angew. Chem. Int. Ed. 2015, 54, 10971–10974. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Hongdilokkul, N.; Liu, Z.; Adhikary, R.; Tsuen, S.S.; Romesberg, F.E. Evolution of thermophilic DNA polymerases for the recognition and amplification of C2’-modified DNA. Nat. Chem. 2016, 8, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Team:Heidelberg/Software/Maws-2015.igem.org. Available online: http://2015.igem.org/Team:Heidelberg/software/maws (accessed on 23 May 2018).
- Hu, W.-P.; Lin, H.-T.; Tsai, J.J.P.; Chen, W.-Y. Investigating interactions between proteins and nucleic acids by computational approaches. In Computational Methods with Applications in Bioinformatics Analysis; Advanced Series in Electrical and Computer Engineering; World Scientific: Singapore, 2017; Volume 20, pp. 98–117. ISBN 978-981-320-797-4. [Google Scholar]
- Gong, S.; Wang, Y.; Wang, Z.; Zhang, W. Computational methods for modeling aptamers and designing riboswitches. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; McKeague, M.; Pitre, S.; Dumontier, M.; Green, J.; Golshani, A.; Derosa, M.C.; Dehne, F. Computational approaches toward the design of pools for the in vitro selection of complex aptamers. RNA 2010, 16, 2252–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chushak, Y.; Stone, M.O. In silico selection of RNA aptamers. Nucleic Acids Res. 2009, 37. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.-P.; Kumar, J.V.; Huang, C.-J.; Chen, W.-Y. Computational selection of RNA aptamer against angiopoietin-2 and experimental evaluation. BioMed Res. Int. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ahirwar, R.; Nahar, S.; Aggarwal, S.; Ramachandran, S.; Maiti, S.; Nahar, P. In silico selection of an aptamer to estrogen receptor alpha using computational docking employing estrogen response elements as aptamer-alike molecules. Sci. Rep. 2016, 6, 21285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cataldo, R.; Ciriaco, F.; Alfinito, E. A validation strategy for in silico generated aptamers. arXiv, 2017; arXiv:1711.07397. [Google Scholar]
- Jarvis, T.C.; Davies, D.R.; Hisaminato, A.; Resnicow, D.I.; Gupta, S.; Waugh, S.M.; Nagabukuro, A.; Wadatsu, T.; Hishigaki, H.; Gawande, B.; et al. Non-helical DNA triplex forms a unique aptamer scaffold for high affinity recognition of nerve growth factor. Structure 2015, 23, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Sim, A.Y.L.; Minary, P.; Levitt, M. Modeling nucleic acids. Curr. Opin. Struct. Biol. 2012, 22, 273–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Hiller, M.; Pudimat, R.; Busch, A.; Backofen, R. Using RNA secondary structures to guide sequence motif finding towards single-stranded regions. Nucleic Acids Res. 2006, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frith, M.C.; Saunders, N.F.W.; Kobe, B.; Bailey, T.L. Discovering sequence motifs with arbitrary insertions and deletions. PLoS Comput. Biol. 2008, 4, e1000071. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoinka, J.; Zotenko, E.; Friedman, A.; Sauna, Z.E.; Przytycka, T.M. Identification of sequence-structure RNA binding motifs for SELEX-derived aptamers. Bioinformatics 2012, 28, i215–i223. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; SantaLucia, J.; Burkard, M.E.; Kierzek, R.; Schroeder, S.J.; Jiao, X.; Cox, C.; Turner, D.H. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry 1998, 37, 14719–14735. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.H.; Sabina, J.; Zuker, M.; Turner, D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999, 288, 911–940. [Google Scholar] [CrossRef] [PubMed]
- Parisien, M.; Major, F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature 2008, 452, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Hermann, T.; Westhof, E. Non-Watson-Crick base pairs in RNA-protein recognition. Chem. Biol. 1999, 6, R335–R343. [Google Scholar] [CrossRef] [Green Version]
- van Dijk, M.; Bonvin, A.M.J.J. Pushing the limits of what is achievable in protein-DNA docking: Benchmarking HADDOCK’s performance. Nucleic Acids Res. 2010, 38, 5634–5647. [Google Scholar] [CrossRef] [PubMed]
- Jeddi, I.; Saiz, L. Three-dimensional modeling of single stranded DNA hairpins for aptamer-based biosensors. Sci. Rep. 2017, 7, 1178. [Google Scholar] [CrossRef] [PubMed]
- Popenda, M.; Szachniuk, M.; Antczak, M.; Purzycka, K.J.; Lukasiak, P.; Bartol, N.; Blazewicz, J.; Adamiak, R.W. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012, 40. [Google Scholar] [CrossRef] [PubMed]
- Biesiada, M.; Purzycka, K.J.; Szachniuk, M.; Blazewicz, J.; Adamiak, R.W. Automated RNA 3D Structure Prediction with RNA Composer. Methods Mol. Biol. 2016, 1490, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Frellsen, J.; Moltke, I.; Thiim, M.; Mardia, K.V.; Ferkinghoff-Borg, J.; Hamelryck, T. A probabilistic model of RNA conformational space. PLoS Comput. Biol. 2009, 5, e1000406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.; Baker, D. Automated de novo prediction of native-like RNA tertiary structures. Proc. Natl. Acad. Sci. USA 2007, 104, 14664–14669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinghorn, A.B.; Fraser, L.A.; Lang, S.; Shiu, S.C.-C.; Tanner, J.A. Aptamer Bioinformatics. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- van Dijk, M.; Bonvin, A.M.J.J. A protein-DNA docking benchmark. Nucleic Acids Res. 2008, 36. [Google Scholar] [CrossRef] [PubMed]
- Roberts, V.A.; Pique, M.E.; Ten Eyck, L.F.; Li, S. Predicting protein-DNA interactions by full search computational docking. Proteins 2013, 81, 2106–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruvkun, G. Molecular biology. Glimpses of a tiny RNA world. Science 2001, 294, 797–799. [Google Scholar] [CrossRef] [PubMed]
- Kruger, K.; Grabowski, P.J.; Zaug, A.J.; Sands, J.; Gottschling, D.E.; Cech, T.R. Self-splicing RNA: Autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 1982, 31, 147–157. [Google Scholar] [CrossRef]
- Tuszynska, I.; Magnus, M.; Jonak, K.; Dawson, W.; Bujnicki, J.M. NPDock: A web server for protein-nucleic acid docking. Nucleic Acids Res. 2015, 43, W425–W430. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Cui, J.; Cheng, J.; Wu, R. Computational Prediction of RNA-Binding Proteins and Binding Sites. Int. J. Mol. Sci. 2015, 16, 26303–26317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katchalski-Katzir, E.; Shariv, I.; Eisenstein, M.; Friesem, A.A.; Aflalo, C.; Vakser, I.A. Molecular surface recognition: Determination of geometric fit between proteins and their ligands by correlation techniques. Proc. Natl. Acad. Sci. USA 1992, 89, 2195–2199. [Google Scholar] [CrossRef] [PubMed]
- Gabb, H.A.; Jackson, R.M.; Sternberg, M.J. Modelling protein docking using shape complementarity, electrostatics and biochemical information. J. Mol. Biol. 1997, 272, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, M.J.; Aloy, P.; Gabb, H.A.; Jackson, R.M.; Moont, G.; Querol, E.; Aviles, F.X. A computational system for modelling flexible protein-protein and protein-DNA docking. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1998, 6, 183–192. [Google Scholar] [PubMed]
- Carter, P.; Lesk, V.I.; Islam, S.A.; Sternberg, M.J.E. Protein-protein docking using 3D-Dock in rounds 3, 4, and 5 of CAPRI. Proteins 2005, 60, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.W.; Kemp, G.J. Protein docking using spherical polar Fourier correlations. Proteins 2000, 39, 178–194. [Google Scholar] [CrossRef] [Green Version]
- Mandell, J.G.; Roberts, V.A.; Pique, M.E.; Kotlovyi, V.; Mitchell, J.C.; Nelson, E.; Tsigelny, I.; Ten Eyck, L.F. Protein docking using continuum electrostatics and geometric fit. Protein Eng. 2001, 14, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, V.A.; Thompson, E.E.; Pique, M.E.; Perez, M.S.; Ten Eyck, L.F. DOT2: Macromolecular docking with improved biophysical models. J. Comput. Chem. 2013, 34, 1743–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Banitt, I.; Wolfson, H.J. ParaDock: A flexible non-specific DNA—Rigid protein docking algorithm. Nucleic Acids Res. 2011, 39. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.-Y. HDOCK: A web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Huang, S. A New pairwise shape-based scoring function to consider long-range interactions for protein-protein docking. Biophys. J. 2017, 112. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Tuszynska, I.; Bujnicki, J.M. DARS-RNP and QUASI-RNP: New statistical potentials for protein-RNA docking. BMC Bioinform. 2011, 12, 348. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Westhof, E. A Large-Scale Assessment of Nucleic Acids Binding Site Prediction Programs. PLoS Comput. Biol. 2015, 11, e1004639. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brown, S.J. BindN: A web-based tool for efficient prediction of DNA and RNA binding sites in amino acid sequences. Nucleic Acids Res. 2006, 34, W243–W248. [Google Scholar] [CrossRef] [PubMed]
- Torabi, R.; Bagherzadeh, K.; Ghourchian, H.; Amanlou, M. An investigation on the interaction modes of a single-strand DNA aptamer and RBP4 protein: A molecular dynamic simulations approach. Org. Biomol. Chem. 2016, 14, 8141–8153. [Google Scholar] [CrossRef] [PubMed]
- Lensink, M.F.; Wodak, S.J. Docking, scoring, and affinity prediction in CAPRI. Proteins 2013, 81, 2082–2095. [Google Scholar] [CrossRef] [PubMed]
- Lensink, M.F.; Velankar, S.; Wodak, S.J. Modeling protein-protein and protein-peptide complexes: CAPRI 6th edition. Proteins 2017, 85, 359–377. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cano, L.; Jiménez-García, B.; Fernández-Recio, J. A protein-RNA docking benchmark (II): Extended set from experimental and homology modeling data. Proteins 2012, 80, 1872–1882. [Google Scholar] [CrossRef] [PubMed]
- Nithin, C.; Mukherjee, S.; Bahadur, R.P. A non-redundant protein-RNA docking benchmark version 2.0. Proteins 2017, 85, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Zou, X. A nonredundant structure dataset for benchmarking protein-RNA computational docking. J. Comput. Chem. 2013, 34, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Lim, C. Predicting RNA-binding sites from the protein structure based on electrostatics, evolution and geometry. Nucleic Acids Res. 2008, 36. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Shanahan, H.P.; Berman, H.M.; Thornton, J.M. Using electrostatic potentials to predict DNA-binding sites on DNA-binding proteins. Nucleic Acids Res. 2003, 31, 7189–7198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandel-Gutfreund, Y.; Schueler, O.; Margalit, H. Comprehensive analysis of hydrogen bonds in regulatory protein DNA-complexes: In search of common principles. J. Mol. Biol. 1995, 253, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Lin, S.L.; Nussinov, R. Protein binding versus protein folding: The role of hydrophilic bridges in protein associations. J. Mol. Biol. 1997, 265, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Theobald, D.L.; Schultz, S.C. Nucleotide shuffling and ssDNA recognition in Oxytricha nova telomere end-binding protein complexes. EMBO J. 2003, 22, 4314–4324. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S. Principles of Nucleic Acid Structure; Elsevier: New York, NY, USA, 2010; ISBN 978-0-08-055352-8. [Google Scholar]
- Steiner, T. The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements; Elsevier: New York, NY, USA, 2012; ISBN 978-0-08-050109-3. [Google Scholar]
- Dougherty, R.C. Temperature and pressure dependence of hydrogen bond strength: A perturbation molecular orbital approach. J. Chem. Phys. 1998, 109, 7372–7378. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Dai, D.-Q.; Yan, H. PRL-Dock: Protein-ligand docking based on hydrogen bond matching and probabilistic relaxation labeling. Proteins 2012, 80, 2137–2153. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Wilson, P.; Schomburg, D. Hydrogen bonding and molecular surface shape complementarity as a basis for protein docking. J. Mol. Biol. 1996, 264, 199–210. [Google Scholar] [CrossRef] [PubMed]
- In Silico Aptamer Docking Studies: From a Retrospective Validation to a Prospective Case Study’TIM3 Aptamers Binding. Mol. Ther. Nucleic Acids 2016. Available online: https://www.cell.com/molecular-therapy-family/nucleic-acids/fulltext/S2162-2531(17)30103-8 (accessed on 24 July 2018). [CrossRef]
- Charifson, P.S.; Corkery, J.J.; Murcko, M.A.; Walters, W.P. Consensus Scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J. Med. Chem. 1999, 42, 5100–5109. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Salsbury, F.R. Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr. Opin. Pharmacol. 2010, 10, 738–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dynamics of DNA Oligomers. J. Biomol. Struct. Dyn. 1983. Available online: https://www.tandfonline.com/doi/abs/10.1080/07391102.1983.10507437 (accessed on 27 April 2018). [CrossRef]
- Levitt, M. Computer simulation of DNA double-helix dynamics. Cold Spring Harb. Symp. Quant. Biol. 1983, 47 Pt 1, 251–262. [Google Scholar] [CrossRef]
- Mackerell, A.D.; Nilsson, L. Molecular dynamics simulations of nucleic acid-protein complexes. Curr. Opin. Struct. Biol. 2008, 18, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Cheatham, T.E.I.; Miller, J.L.; Fox, T.; Darden, T.A.; Kollman, P.A. Molecular dynamics simulations on solvated biomolecular systems: the particle mesh Ewald method leads to stable trajectories of DNA, RNA, and proteins. J. Am. Chem. Soc. 1995, 117, 4193–4194. [Google Scholar] [CrossRef]
- Weiner, P.K.; Kollman, P.A. AMBER: Assisted model building with energy refinement. A general program for modeling molecules and their interactions. J. Comput. Chem. 1981, 2, 287–303. [Google Scholar] [CrossRef]
- Galindo-Murillo, R.; Robertson, J.C.; Zgarbová, M.; Šponer, J.; Otyepka, M.; Jurečka, P.; Cheatham, T.E. Assessing the current state of amber force field modifications for DNA. J. Chem. Theory Comput. 2016, 12, 4114–4127. [Google Scholar] [CrossRef] [PubMed]
- Drew, H.R.; Wing, R.M.; Takano, T.; Broka, C.; Tanaka, S.; Itakura, K.; Dickerson, R.E. Structure of a B-DNA dodecamer: Conformation and dynamics. Proc. Natl. Acad. Sci. USA 1981, 78, 2179–2183. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svozil, D.; Šponer, J.E.; Marchan, I.; Pérez, A.; Cheatham, T.E., III; Forti, F.; Luque, F.J.; Orozco, M.; Šponer, J. Geometrical and Electronic Structure Variability of the Sugar−Phosphate Backbone in Nucleic Acids. Available online: https://pubs.acs.org/doi/abs/10.1021/jp801245h (accessed on 10 August 2018).
- Zgarbová, M.; Šponer, J.; Otyepka, M.; Cheatham, T.E., III; Galindo-Murillo, R.; Jurečka, P. Refinement of the Sugar–Phosphate Backbone Torsion Beta for AMBER Force Fields Improves the Description of Z- and B-DNA. Available online: https://pubs.acs.org/doi/abs/10.1021/acs.jctc.5b00716 (accessed on 10 August 2018).
- Richard Sinden DNA Structure and Function. DNA Structure and Function; Academic Press: San Diego, CA, USA, 2012; pp. 1–57. ISBN 978-0-08-057173-7. [Google Scholar]
- Ahmad, S.; Keskin, O.; Sarai, A.; Nussinov, R. Protein–DNA interactions: Structural, thermodynamic and clustering patterns of conserved residues in DNA-binding proteins. Nucleic Acids Res. 2008, 36, 5922–5932. [Google Scholar] [CrossRef] [PubMed]
- Etheve, L.; Martin, J.; Lavery, R. Dynamics and recognition within a protein–DNA complex: A molecular dynamics study of the SKN-1/DNA interaction. Nucleic Acids Res. 2016, 44. [Google Scholar] [CrossRef] [PubMed]
- Etheve, L.; Martin, J.; Lavery, R. Protein–DNA interfaces: A molecular dynamics analysis of time-dependent recognition processes for three transcription factors. Nucleic Acids Res. 2016, 44, 9990–10002. [Google Scholar] [CrossRef] [PubMed]
- Zandarashvili, L.; Esadze, A.; Iwahara, J. NMR studies on the dynamics of hydrogen bonds and ion pairs involving lysine side chains of proteins. Adv. Protein Chem. Struct. Biol. 2013, 93, 37–80. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Esadze, A.; Zandarashvili, L.; Nguyen, D.; Pettitt, B.M.; Iwahara, J. Dynamic equilibria of short-range electrostatic interactions at molecular interfaces of protein–DNA complexes. J. Phys. Chem. Lett. 2015, 6, 2733–2737. [Google Scholar] [CrossRef] [PubMed]
- Caruso, I.P.; Panwalkar, V.; Coronado, M.A.; Dingley, A.J.; Cornélio, M.L.; Willbold, D.; Arni, R.K.; Eberle, R.J. Structure and interaction of Corynebacterium pseudotuberculosis cold shock protein A with Y-box single-stranded DNA fragment. FEBS J. 2017, 285, 372–390. [Google Scholar] [CrossRef] [PubMed]
- La Penna, G.; Chelli, R. Structural insights into the osteopontin-aptamer complex by molecular dynamics simulations. Front. Chem. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.-H.; Tsai, C.-W.; Wu, J.W.; Ruaan, R.-C.; Chen, W.-Y. Molecular dynamics simulation of the induced-fit binding process of DNA aptamer and L-argininamide. Biotechnol. J. 2012, 7, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Henzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Murillo, R.; Roe, D.R.; Cheatham, T.E. Convergence and reproducibility in molecular dynamics simulations of the DNA duplex d(GCACGAACGAACGAACGC). Biochim. Biophys. Acta BBA Gen. Subj. 2015, 1850, 1041–1058. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Murillo, R.; Roe, D.R.; Iii, T.E.C. On the absence of intrahelical DNA dynamics on the μs to ms timescale. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012. Available online: https://pubs.acs.org/doi/abs/10.1021/ct200909j (accessed on 27 April 2018). [CrossRef]
- Matsumoto, A.; Olson, W.K. Sequence-Dependent Motions of DNA: A normal mode analysis at the base-pair level. Biophys. J. 2002, 83, 22–41. [Google Scholar] [CrossRef]
- Alexandrov, V.; Lehnert, U.; Echols, N.; Milburn, D.; Engelman, D.; Gerstein, M. Normal modes for predicting protein motions: A comprehensive database assessment and associated Web tool. Protein Sci. 2005, 14, 633–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da, L.-T.; Sheong, F.K.; Silva, D.-A.; Huang, X. Application of Markov state models to simulate long timescale dynamics of biological macromolecules. In Protein Conformational Dynamics; Han, K., Zhang, X., Yang, M., Eds.; Springer International Publishing: Cham, Switzerland, 2014; pp. 29–66. ISBN 978-3-319-02970-2. [Google Scholar]
- Wako, H.; Endo, S. Normal mode analysis as a method to derive protein dynamics information from the Protein Data Bank. Biophys. Rev. 2017, 9, 877–893. [Google Scholar] [CrossRef]
- Xiao, J.; Salsbury, F.R. Molecular dynamics simulations of aptamer-binding reveal generalized allostery in thrombin. J. Biomol. Struct. Dyn. 2017, 35, 3354–3369. [Google Scholar] [CrossRef] [PubMed]
- Nimjee, S.M.; Oney, S.; Volovyk, Z.; Bompiani, K.M.; Long, S.B.; Hoffman, M.; Sullenger, B.A. Synergistic effect of aptamers that inhibit exosites 1 and 2 on thrombin. RNA 2009, 15, 2105–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, X.; Zhang, L.; Xiao, X.; Jiang, Y.; Guo, Y.; Yu, X.; Pu, X.; Li, M. Unfolding mechanism of thrombin-binding aptamer revealed by molecular dynamics simulation and Markov State Model. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-C.; Hyeon, C.; Thirumalai, D. Sequence-dependent folding landscapes of adenine riboswitch aptamers. Phys. Chem. Chem. Phys. 2014, 16, 6376–6382. [Google Scholar] [CrossRef] [PubMed]
- Mechanical Unfolding of RNA: From hairpins to structures with internal multiloops. Biophys. J. 2007. Available online: https://www.cell.com/biophysj/abstract/S0006-3495(07)70884-6 (accessed on 2 May 2018). [CrossRef]
- Boniecki, M.J.; Lach, G.; Dawson, W.K.; Tomala, K.; Lukasz, P.; Soltysinski, T.; Rother, K.M.; Bujnicki, J.M. SimRNA: A coarse-grained method for RNA folding simulations and 3D structure prediction. Nucleic Acids Res. 2016, 44. [Google Scholar] [CrossRef] [PubMed]
- Dufour, D.; Marti-Renom, M.A. Software for predicting the 3D structure of RNA molecules. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 5, 56–61. [Google Scholar] [CrossRef]
- Liu, K.; Watanabe, E.; Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations. J. Comput. Aided Mol. Des. 2017, 31, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations: A cross-docking study. J. Chem. Inf. Model. 2017, 57, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Šponer, J.; Banáš, P.; Jurečka, P.; Zgarbová, M.; Kührová, P.; Havrila, M.; Krepl, M.; Stadlbauer, P.; Otyepka, M. Molecular dynamics simulations of nucleic acids. From tetranucleotides to the ribosome. J. Phys. Chem. Lett. 2014, 5, 1771–1782. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A. Molecular dynamics and NMR spin relaxation in proteins. Acc. Chem. Res. 2002, 35, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.A.; Draper, D.E.; Pappu, R.V. Molecular simulation studies of monovalent counterion-mediated interactions in a model RNA kissing loop. J. Mol. Biol. 2009, 390, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Cheatham, T.E.; Case, D.A. Twenty-five years of nucleic acid simulations. Biopolymers 2013, 99, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Lemkul, J.A.; Alexander, D.; MacKerell, J. Polarizable Force Field for DNA Based on the Classical Drude Oscillator: II. Microsecond Molecular Dynamics Simulations of Duplex DNA. Available online: https://pubs.acs.org/doi/abs/10.1021/acs.jctc.7b00068 (accessed on 10 August 2018).


{kind=link}
{kind=link}
| Algorithm | Milestone | Reference |
|---|---|---|
| GRAMM | Rigid docking, six-dimensional shape complementarity; fast Fourier transformation | [55] |
| FTDock | Implementation of electrostatics and biochemical information | [56,57] |
| 3D-Dock | Additionally, energy calculations, side chain optimization, and backbone refinement | [58] |
| Hex | Spherical polar Fourier correlation method | [59] |
| Dot/Dot2 | Implementation of Poisson–Boltzmann methods | [60,61] |
| HADDOCK | Flexibility of amino acid side chains | [62] |
| PatchDock | Local feature matching instead of six-dimensional transformation fitting | [63] |
| ParaDock | Shape complementarity but flexible NA structure prediction | [64] |
| NPDock | Rigid body docking while considering the specific features of NA | [53] |
| HDOCK | Docking between two big molecules; template-based and template-free rigid docking mode | [65,66] |
| Gold | Full flexibility or rotamer-based search for both ligand and selected amino acids residues; docking in a determined binding pocket. Presents a range of different scoring functions, from machine-learning-based to physicochemical-based ones | [67] |
| Autodock Autodock Vina | Full flexibility or rotamer-based search for both ligand and selected amino acids residues; docking in a determined binding pocket. Energy-based scoring function and ability to handle surface pockets | [68] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krüger, A.; Zimbres, F.M.; Kronenberger, T.; Wrenger, C. Molecular Modeling Applied to Nucleic Acid-Based Molecule Development. Biomolecules 2018, 8, 83. https://doi.org/10.3390/biom8030083
Krüger A, Zimbres FM, Kronenberger T, Wrenger C. Molecular Modeling Applied to Nucleic Acid-Based Molecule Development. Biomolecules. 2018; 8(3):83. https://doi.org/10.3390/biom8030083
Chicago/Turabian StyleKrüger, Arne, Flávia M. Zimbres, Thales Kronenberger, and Carsten Wrenger. 2018. "Molecular Modeling Applied to Nucleic Acid-Based Molecule Development" Biomolecules 8, no. 3: 83. https://doi.org/10.3390/biom8030083
APA StyleKrüger, A., Zimbres, F. M., Kronenberger, T., & Wrenger, C. (2018). Molecular Modeling Applied to Nucleic Acid-Based Molecule Development. Biomolecules, 8(3), 83. https://doi.org/10.3390/biom8030083