Predicting Aromatic Amine Mutagenicity with Confidence: A Case Study Using Conformal Prediction


Abstract
:1. Introduction
2. Results
2.1. Conformal Prediction Model Based on Leadscope Fingerprints
2.2. Conformal Prediction Model Based on Leadscope PAA Features
3. Discussion
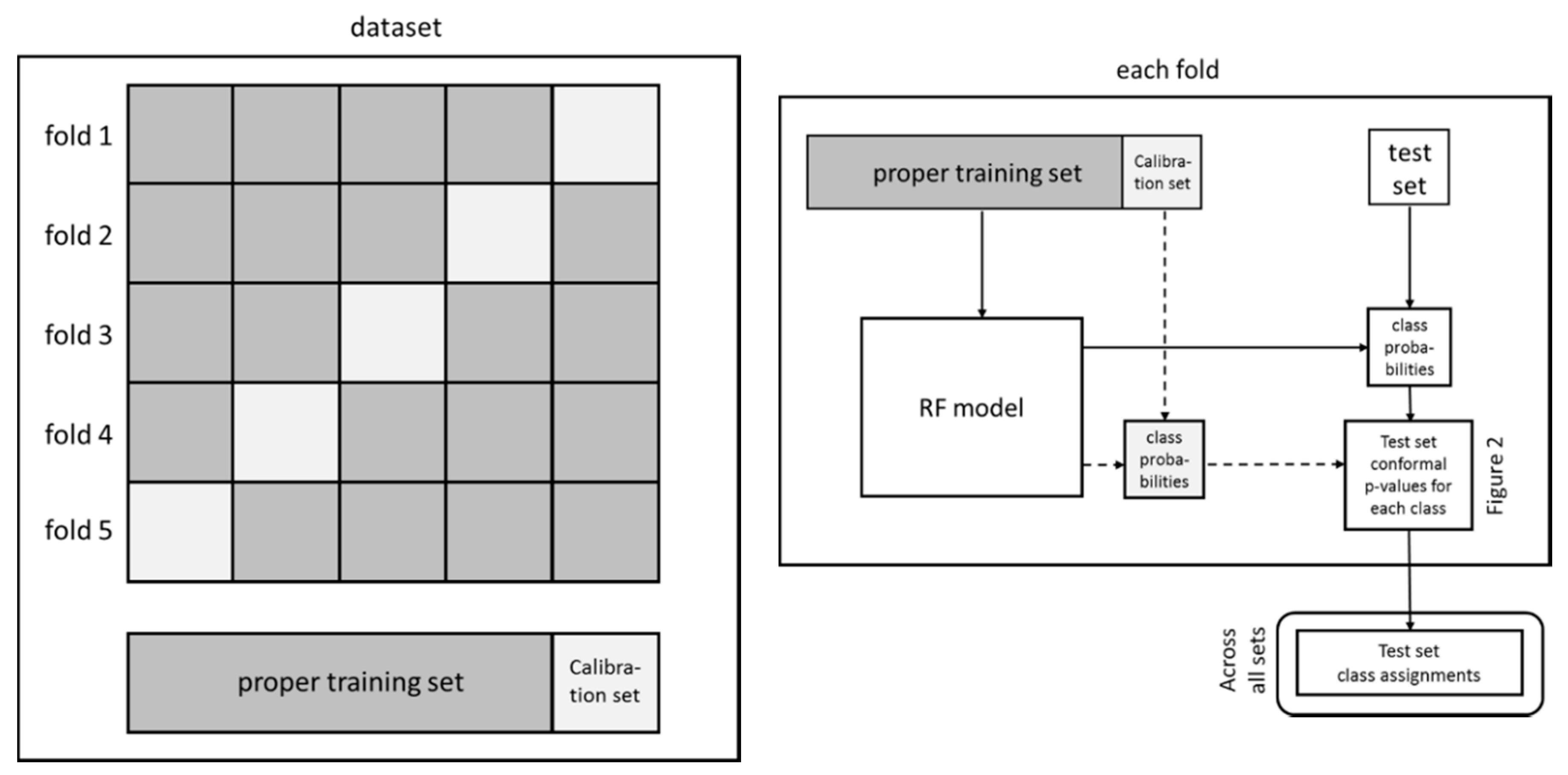
4. Materials and Methods
4.1. Primary Aromatic Amine Dataset
4.2. Leadscope Molecular Descriptors
4.2.1. Leadscope Fingerprints
4.2.2. Leadscope PAA Features
4.3. Conformal Prediction
4.3.1. Model Generation
4.3.2. Model Validation
5. Conclusions
- It is possible to develop mathematically proven valid models using the conformal prediction framework for predicting mutagenicity of primary aromatic amines.
- The user of the models can set the acceptable level of errors.
- The existence of uncertain classes of prediction (both and empty) provides the user with information on how to use, further develop or possibly improve future models.
- The use of different sets of fingerprints results in models where the ability to discriminate varies with respect to the set level of acceptable errors.
Author Contributions
Funding
Conflicts of Interest
References
- Ashby, J.; Tennant, R.W. Chemical structure, Salmonella mutagenicity and extent of carcinogenicity as indicators of genotoxic carcinogenesis among 222 chemicals tested in rodents by the U.S. NCI/NTP. Mutat. Res. 1988, 204, 17–115. [Google Scholar] [CrossRef]
- Bailey, A.B.; Chanderbhan, R.; Collazo-Braier, N.; Cheeseman, M.; Twaroski, M.L. The use of structure–activity relationship analysis in the food contact notification program. Regul. Toxicol. Pharmacol. 2005, 42, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Benigni, R.; Bossa, C.; Jeliazkova, N.G.; Netzeva, T.I.; Worth, A.P. The Benigni/Bossa Rulebase for Mutagenicity and Carcinogenicity—A Module of Toxtree. Technical Report EUR 23241 EN. European Commission—Joint Research Centre. 2008. Available online: https://eurl-ecvam.jrc.ec.europa.eu/laboratories-research/predictive_toxicology/doc/EUR_23241_EN.pdf (accessed on 11 May 2018).
- Benigni, R.; Bossa, C. Mechanisms of chemical carcinogenicity and mutagenicity: A review with implications for predictive toxicology. Chem. Rev. 2011, 111, 2507–2536. [Google Scholar] [CrossRef] [PubMed]
- Enoch, S.J.; Cronin, M.T.D. A review of the electrophilic reaction chemistry involved in covalent DNA binding. Crit. Rev. Toxicol. 2010, 40, 728–748. [Google Scholar] [CrossRef] [PubMed]
- Enoch, S.; Cronin, M. Development of new structural alerts suitable for chemical category formation for assigning covalent and non-covalent mechanisms relevant to DNA binding. Mutat. Res. 2012, 743, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Galloway, S.M.; Reddy, M.V.; Mcgettigan, K.; Gealy, R.; Bercu, J. Potentially mutagenic impurities: Analysis of structural classes and carcinogenic potencies of chemical intermediates in pharmaceutical syntheses supports alternative methods to the default TTC for calculating safe levels of impurities. Regul. Toxicol. Pharmacol. 2013, 66, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Shamovsky, I.; Ripa, L.; Börjesson, L.; Mee, C.; Norden, B.; Hansen, P.; Hasselgren, C.; O’Donovan, M.; Sjö, P. Explanation for main features of structure genotoxicity relationships of aromatic amines by theoretical studies of their activation pathways in CYP1A2. J. Am. Chem. Soc. 2011, 133, 16168–16185. [Google Scholar] [CrossRef] [PubMed]
- Ridings, J.E.; Barratt, M.D.; Cary, R.; Earnshaw, C.G.; Eggington, C.E.; Ellis, M.K.; Judson, P.N.; Langowski, J.J.; Marchant, C.A.; Payne, M.P.; et al. Computer prediction of possible toxic action from chemical structure: An update on the DEREK system. Toxicology 1996, 106, 267–279. [Google Scholar] [CrossRef]
- Mostrag-Szlichtyng, A.; Zaldívar Comenges, J.-M.; Worth, A.P. Computational toxicology at the European Commission’s Joint Research Centre. Expert Opin. Drug Metab. Toxicol. 2010, 6, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Myatt, G.J.; Beilke, L.D.; Cross, K.P. In Silico Tools and their Application. Ref. Module Chem. Mol. Sci. Chem. Eng. 2017, 156–176. [Google Scholar] [CrossRef]
- Klopman, G. MULTICASE 1. A Hierarchical Computer Automated Structure Evaluation Program. Quant. Struct.-Activ. Relat. 1992, 11, 176–184. [Google Scholar] [CrossRef]
- CAESAR; 1.1.4; Istituto di Ricerche Farmacologiche Mario Negri: Milan, Italy, 2011; Available online: http://www.caesar-project.eu/ (accessed on 11 May 2018).
- Derek Nexus; 6.0.0; Lhasa Ltd.: Leeds, UK, 2018; Available online: https://www.lhasalimited.org (accessed on 11 May 2018).
- Helma, C. Lazy structure–activity relationships (lazar) for the prediction of rodent carcinogenicity and Salmonella mutagenicity. Mol. Diver. 2006, 10, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Roberts, G.; Myatt, G.; Johnson, W.; Cross, K.; Blower, P. LeadScope: Software for Exploring Large Sets of Screening Data. J. Chem. Inf. Model. 2000, 40, 1302–1314. [Google Scholar] [CrossRef]
- MultiCASE; 1.5.2.0; MultiCASE Inc.: Cleveland, OH, USA; Available online: http://www.multicase.com/ (accessed on 11 May 2018).
- OECD Toolbox; 4.2; OECD: Paris, France, 2018; Available online: http://www.oecd.org/chemicalsafety/risk-assessment/oecd-qsar-toolbox.htm (accessed on 11 May 2018).
- Joint Meeting of the Chemicals Committee and Working Party on Chemicals, Pesticides and Biotechnology. 2004. Available online: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?doclanguage=en&cote=env/jm/mono(2004)24 (accessed on 24 August 2018).
- Vovk, V.; Gammerman, A.; Shafer, G. Algorithmic Learning in a Random World; Springer: New York, NY, USA, 2005. [Google Scholar]
- Norinder, U.; Carlsson, L.; Boyer, S.; Eklund, M. Introducing Conformal Prediction in Predictive Modeling. A Transparent and Flexible Alternative to Applicability Domain Determination. J. Chem. Inf. Model. 2014, 54, 1596–1603. [Google Scholar] [PubMed]
- Norinder, U.; Boyer, S. Conformal prediction classification of a large data set of environmental chemicals from ToxCast and Tox21 estrogen receptor assays. Chem. Res. Toxicol. 2016, 29, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Norinder, U.; Rybacka, A.; Andersson, P.L. Conformal prediction to define applicability domain—A case study on predicting ER and AR binding. SAR QSAR Environ. Res. 2016, 27, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Norinder, U.; Boyer, S. Binary classification of imbalanced datasets using conformal prediction. J. Mol. Graph. Model. 2017, 72, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Stavitskaya, L.; Minnier, B.L.; Benz, R.D.; Kruhlak, N.L. Development of Improved Salmonella Mutagenicity QSAR Models Using Structural Fingerprints of Known Toxicophores. Poster presented at Society of Toxicology, 52nd Annual Meeting, San Antonio, TX, USA, 10–14 March 2013; Available online: https://www.leadscope.com/media/SOT%202013%20Stavitskaya%20CDER%20poster.pdf (accessed on 1 June 2018).
- Stavitskaya, L.; Minnier, B.L.; Benz, R.D.; Kruhlak, N.L. Development of Improved QSAR Models for Predicting A-T Base Pair Mutations. In Proceedings of the Genetic Toxicity Association (GTA) Meeting, Newark, DE, USA, 16–17 October 2013; Available online: http://www.leadscope.com/media/GTA_LStavitskaya.pdf (accessed on 1 June 2018).
- Leadscope SAR Genetox Database 2015 User Guide. Available online: https://www.leadscope.com/toxicity_databases/ (accessed on 1 June 2018).
- Matthews, E.J.; Kruhlak, N.L.; Cimino, M.C.; Benz, R.D.; Contrera, J.F. An analysis of genetic toxicity, reproductive and developmental toxicity, and carcinogenicity data: I. Identification of carcinogens using surrogate endpoints. Regul. Toxicol. Pharmacol. 2006, 44, 83–96. [Google Scholar] [CrossRef] [PubMed]
- FDA CFSAN. Food and Drug Administration CFSAN EAFUS, U.S. FDA Center for Food Safety and Applied Nutrition. Everything Added to Food in the United States. 2007. Available online: http://www.fda.gov/food/ingredientspackaginglabeling/ucm115326.htm (accessed on 1 June 2018).
- FDA CDER. Drugs@FDA: FDA Approved Drug Products. 2015. Available online: http://www.accessdata.fda.gov/scripts/cder/drugsatfda (accessed on 1 June 2018).
- CCRIS. 2011. Available online: http://toxnet.nlm.nih.gov/cgi-bin/sis/htmlgen?CCRIS (accessed on 1 June 2018).
- Tennant, R.W. The genetic toxicity database of the National Toxicology Program: Evaluation of the relationships between genetic toxicity and carcinogenicity. Environ. Health Perspect. 1991, 96, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Tokyo-Eiken. Tokyo Metropolitan Institute of Public Health Providing Primary Mutagenicity of Food Additives for about 300 Chemicals. Available online: http://www.tokyo-eiken.go.jp/henigen/ (accessed on 1 June 2018).
- Ahlberg, E.; Amberg, A.; Beilke, L.D.; Bower, D.; Cross, K.P.; Custer, L.; Ford, K.A.; Gompel, J.V.; Harvey, J.; Honma, M.; et al. Extending (Q)SARs to incorporate proprietary knowledge for regulatory purposes: A case study using aromatic amine mutagenicity. Regul. Toxicol. Pharmacol. 2016, 77, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Nonconformist Package; 1.2.5; Henrik Linusson: Borås, Sweden, 2015; Available online: https://github.com/donlnz/nonconformist (accessed on 9 June 2015).
- Sun, J.; Carlsson, L.; Ahlberg, E.; Norinder, U.; Engkvist, O.; Chen, H. Applying Mondrian cross-conformal prediction to estimate prediction confidence on large imbalanced bioactivity data sets. J. Chem. Inf. Model. 2017, 57, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, D.; Grisoni, F.; Todeschini, R. Multivariate comparison of classification performance measures. Chemom. Intell. Lab. Syst. 2018, 174, 33–44. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Descriptors a | Set b | No Of Compounds Per Set | Significance Level c | Validity Mutagenic Class | Validity Nonmutagenic Class | Efficiency d | BA e | Kappa f | MCC g | Sensitivity h | Specificity h | Percentage Class Both i | Percentage Class Empty j |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LS fingerprints | internal | 656 | 0.15 | 0.851 | 0.849 | 0.695 | 0.537 | 0.543 | 0.784 | 0.786 | 0.782 | 30.5 | 0 |
| LS fingerprints | external | 280 | 0.15 | 0.857 | 0.871 | 0.718 | 0.581 | 0.589 | 0.809 | 0.804 | 0.814 | 28.2 | 0 |
| LS fingerprints | internal | 656 | 0.20 | 0.803 | 0.797 | 0.824 | 0.485 | 0.493 | 0.759 | 0.764 | 0.753 | 17.5 | 0.2 |
| LS fingerprints | external | 280 | 0.20 | 0.794 | 0.818 | 0.861 | 0.508 | 0.518 | 0.773 | 0.763 | 0.782 | 13.9 | 0 |
| LS fingerprints | internal | 656 | 0.25 | 0.753 | 0.746 | 0.914 | 0.452 | 0.460 | 0.742 | 0.746 | 0.738 | 7.1 | 1.4 |
| LS fingerprints | external | 280 | 0.25 | 0.748 | 0.766 | 0.952 | 0.467 | 0.476 | 0.751 | 0.742 | 0.759 | 4.2 | 0.6 |
| LS fingerprints | internal | 656 | 0.30 | 0.707 | 0.697 | 0.927 | 0.449 | 0.457 | 0.741 | 0.742 | 0.740 | 1.6 | 5.7 |
| LS fingerprints | external | 280 | 0.30 | 0.696 | 0.717 | 0.931 | 0.478 | 0.490 | 0.759 | 0.741 | 0.776 | 0.2 | 6.7 |
| LS PAA features | internal | 656 | 0.15 | 0.853 | 0.851 | 0.793 | 0.596 | 0.601 | 0.813 | 0.815 | 0.811 | 20.7 | 0 |
| LS PAA features | external | 280 | 0.15 | 0.857 | 0.855 | 0.826 | 0.625 | 0.630 | 0.826 | 0.826 | 0.826 | 17.4 | 0.0 |
| LS PAA features | internal | 656 | 0.20 | 0.802 | 0.795 | 0.908 | 0.541 | 0.548 | 0.786 | 0.790 | 0.782 | 8.5 | 0.7 |
| LS PAA features | external | 280 | 0.20 | 0.810 | 0.802 | 0.937 | 0.567 | 0.573 | 0.798 | 0.802 | 0.794 | 5.9 | 0.4 |
| LS PAA features | internal | 656 | 0.25 | 0.753 | 0.746 | 0.930 | 0.532 | 0.539 | 0.782 | 0.783 | 0.781 | 2.3 | 4.7 |
| LS PAA features | external | 280 | 0.25 | 0.758 | 0.748 | 0.945 | 0.559 | 0.565 | 0.794 | 0.798 | 0.791 | 0.3 | 5.2 |
| LS PAA features | internal | 656 | 0.30 | 0.703 | 0.697 | 0.879 | 0.556 | 0.563 | 0.794 | 0.795 | 0.794 | 0.3 | 11.9 |
| LS PAA features | external | 280 | 0.30 | 0.706 | 0.705 | 0.862 | 0.608 | 0.613 | 0.818 | 0.820 | 0.816 | 0.0 | 13.8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Norinder, U.; Myatt, G.; Ahlberg, E. Predicting Aromatic Amine Mutagenicity with Confidence: A Case Study Using Conformal Prediction. Biomolecules 2018, 8, 85. https://doi.org/10.3390/biom8030085
Norinder U, Myatt G, Ahlberg E. Predicting Aromatic Amine Mutagenicity with Confidence: A Case Study Using Conformal Prediction. Biomolecules. 2018; 8(3):85. https://doi.org/10.3390/biom8030085
Chicago/Turabian StyleNorinder, Ulf, Glenn Myatt, and Ernst Ahlberg. 2018. "Predicting Aromatic Amine Mutagenicity with Confidence: A Case Study Using Conformal Prediction" Biomolecules 8, no. 3: 85. https://doi.org/10.3390/biom8030085
APA StyleNorinder, U., Myatt, G., & Ahlberg, E. (2018). Predicting Aromatic Amine Mutagenicity with Confidence: A Case Study Using Conformal Prediction. Biomolecules, 8(3), 85. https://doi.org/10.3390/biom8030085