Plasma Membrane Lipid Domains as Platforms for Vesicle Biogenesis and Shedding?


Abstract
:1. Introduction
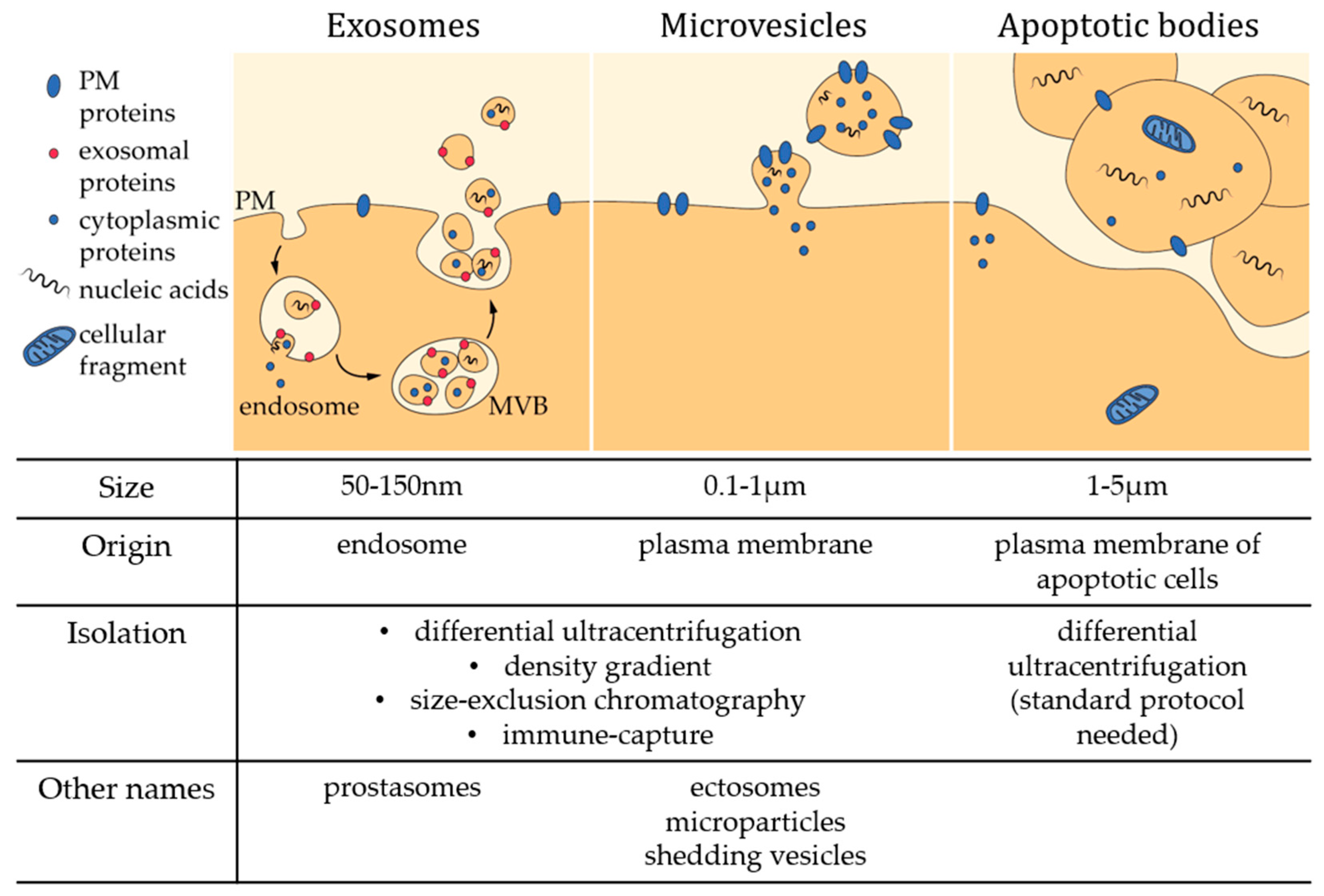
2. Microvesicle Isolation and Characterization
3. Microvesicle Molecular Properties
3.1. Protein Content
3.2. Nucleic Acid Content
3.3. Lipid Content
4. Microvesicle Biogenesis and Shedding—General Mechanisms
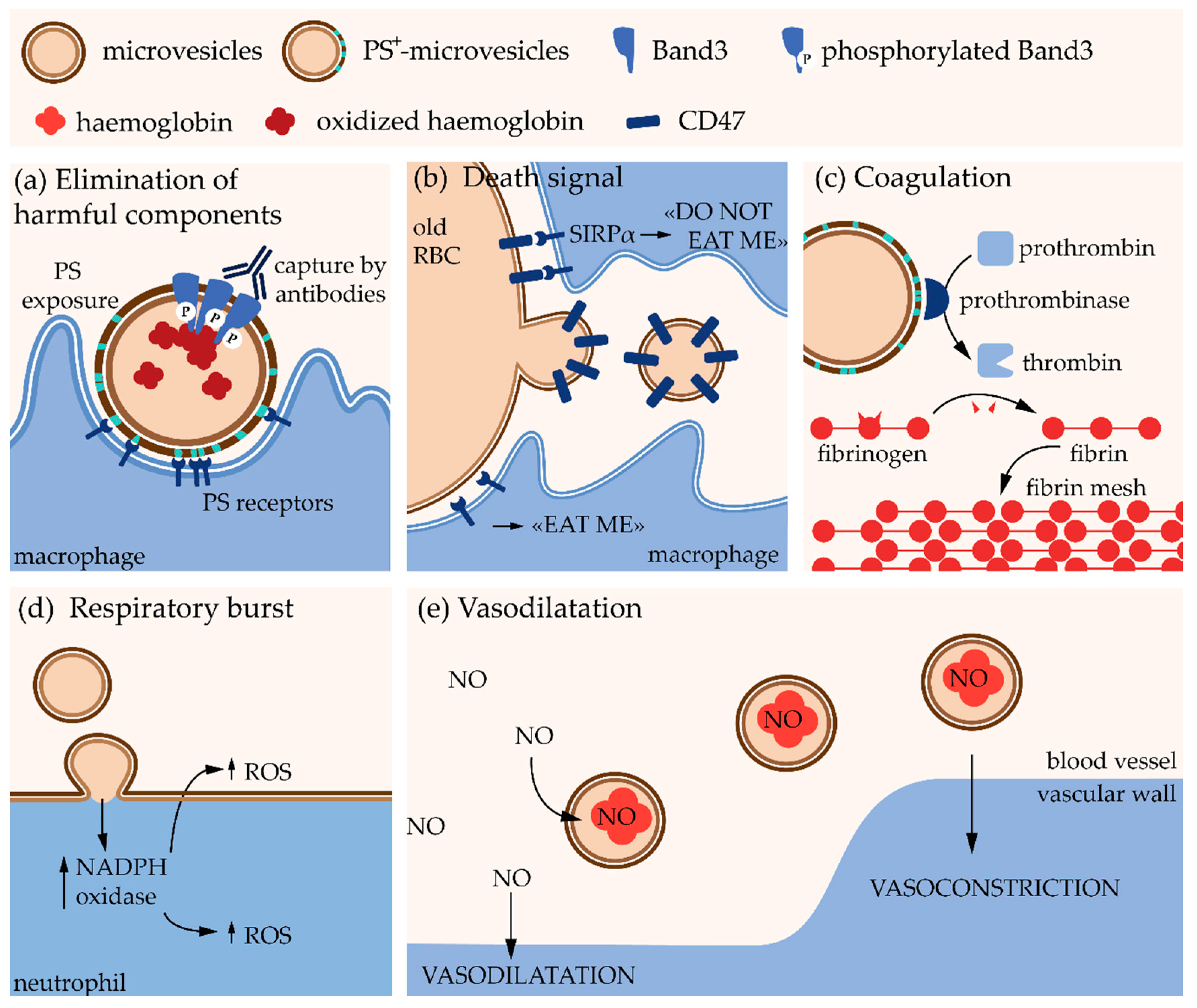
4.1. Red Blood Cells
4.1.1. Main Determinants of Red Blood Cell Integrity Maintenance
4.1.2. Microvesicles upon Red Blood Cell Senescence, Blood Storage and Intracellular Calcium Boost
4.1.3. Microvesicles in Red Blood Cell Hemoglobinopathies
4.1.4. Microvesicles in Red Blood Cell Membrane Fragility Diseases
4.2. Nucleated Cells
5. Microvesicle Biogenesis and Shedding—Role of Plasma Membrane Composition and Biophysical Properties
5.1. Plasma Membrane Lipid Composition
5.1.1. Sphingolipids and Sphingomyelinases
5.1.2. Cholesterol
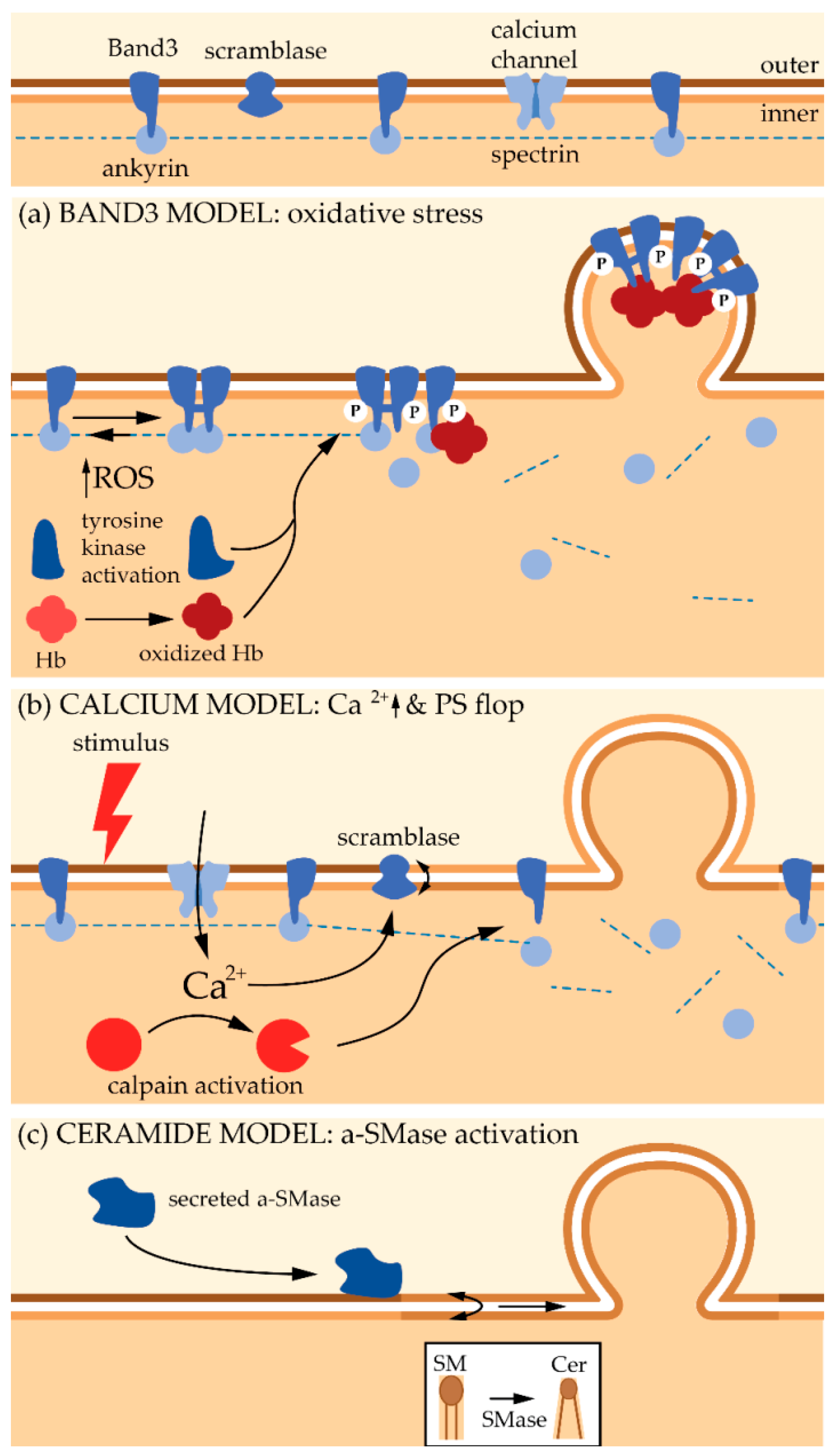
5.2. Plasma Membrane Transversal Asymmetry
5.3. Plasma Membrane Lateral Heterogeneity
5.3.1. General Features
5.3.2. Red Blood Cells
5.3.3. Platelets
5.3.4. Immune Cells
5.3.5. Nervous Cells
5.3.6. Cancer Cells
6. Future Challenges
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Deatherage, B.L.; Cookson, B.T. Membrane vesicle release in bacteria, eukaryotes, and archaea: A conserved yet underappreciated aspect of microbial life. Infect. Immun. 2012, 80, 1948–1957. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.G.; Ding, Y.; Jiang, L. Unconventional protein secretion in plants: A critical assessment. Protoplasma 2016, 253, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.J.; Raposo, G. As we wait: Coping with an imperfect nomenclature for extracellular vesicles. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyorgy, B.; Szabo, T.G.; Pasztoi, M.; Pal, Z.; Misjak, P.; Aradi, B.; Laszlo, V.; Pallinger, E.; Pap, E.; Kittel, A.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, A.E.; D’Souza-Schorey, C. The biology of extracellular microvesicles. Traffic 2018, 19, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Turturici, G.; Tinnirello, R.; Sconzo, G.; Geraci, F. Extracellular membrane vesicles as a mechanism of cell-to-cell communication: Advantages and disadvantages. Am. J. Physiol. Cell Physiol. 2014, 306, C621–C633. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, A.M.; Fang, Y.; Fallon, J.K.; Yang, J.M.; Hildreth, J.E.; Gould, S.J. Exosomes and HIV Gag bud from endosome-like domains of the T cell plasma membrane. J. Cell Biol. 2006, 172, 923–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzesco, A.M.; Janich, P.; Wilsch-Brauninger, M.; Dubreuil, V.; Langenfeld, K.; Corbeil, D.; Huttner, W.B. Release of extracellular membrane particles carrying the stem cell marker prominin-1 (CD133) from neural progenitors and other epithelial cells. J. Cell Sci. 2005, 118, 2849–2858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabhan, J.F.; Hu, R.; Oh, R.S.; Cohen, S.N.; Lu, Q. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4146–4151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciana, A.; Achilli, C.; Gaur, A.; Minetti, G. Membrane Remodelling and Vesicle Formation During Ageing of Human Red Blood Cells. Cell. Physiol. Biochem. 2017, 42, 1127–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuana, Y.; Sturk, A.; Nieuwland, R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 2013, 27, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Pulliam, L. Exosomes as mediators of neuroinflammation. J. Neuroinflamm. 2014, 11, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, B.; Fang, Y.; Wu, N.; Gould, S.J. Biogenesis of the posterior pole is mediated by the exosome/microvesicle protein-sorting pathway. J. Biol. Chem. 2011, 286, 44162–44176. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan-Chari, V.; Clancy, J.W.; Sedgwick, A.; D’Souza-Schorey, C. Microvesicles: Mediators of extracellular communication during cancer progression. J. Cell Sci. 2010, 123, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Barteneva, N.S.; Maltsev, N.; Vorobjev, I.A. Microvesicles and intercellular communication in the context of parasitism. Front. Cell. Infect. Microbiol. 2013, 3, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Xing, F.; Wu, S.Y.; Watabe, K. Extracellular vesicles as emerging targets in cancer: Recent development from bench to bedside. Biochim. Biophys. Acta 2017, 1868, 538–563. [Google Scholar] [CrossRef] [PubMed]
- Lutz, H.U.; Bogdanova, A. Mechanisms tagging senescent red blood cells for clearance in healthy humans. Front. Physiol. 2013, 4, 387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonelou, M.H.; Kriebardis, A.G.; Papassideri, I.S. Aging and death signalling in mature red cells: From basic science to transfusion practice. Blood Transfus. 2010, 8 (Suppl. 3), S39–S47. [Google Scholar]
- Leal, J.K.F.; Adjobo-Hermans, M.J.W.; Bosman, G. Red Blood Cell Homeostasis: Mechanisms and Effects of Microvesicle Generation in Health and Disease. Front. Physiol. 2018, 9, 703. [Google Scholar] [CrossRef] [PubMed]
- Antonelou, M.H.; Seghatchian, J. Update on extracellular vesicles inside red blood cell storage units: Adjust the sails closer to the new wind. Transfus. Apher. Sci. 2016, 55, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Willekens, F.L.; Werre, J.M.; Groenen-Dopp, Y.A.; Roerdinkholder-Stoelwinder, B.; de Pauw, B.; Bosman, G.J. Erythrocyte vesiculation: A self-protective mechanism? Br. J. Haematol. 2008, 141, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.; Urbaniak, S.; Turner, M.; Bessos, H. The application of a new quantitative assay for the monitoring of integrin-associated protein CD47 on red blood cells during storage and comparison with the expression of CD47 and phosphatidylserine with flow cytometry. Transfusion 2005, 45, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Bocci, V.; Pessina, G.P.; Paulesu, L. Studies of factors regulating the ageing of human erythrocytes—III. Metabolism and fate of erythrocytic vesicles. Int. J. Biochem. 1980, 11, 139–142. [Google Scholar] [CrossRef]
- Jank, H.; Salzer, U. Vesicles generated during storage of red blood cells enhance the generation of radical oxygen species in activated neutrophils. Sci. World J. 2011, 11, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Donadee, C.; Raat, N.J.; Kanias, T.; Tejero, J.; Lee, J.S.; Kelley, E.E.; Zhao, X.; Liu, C.; Reynolds, H.; Azarov, I.; et al. Nitric oxide scavenging by red blood cell microparticles and cell-free hemoglobin as a mechanism for the red cell storage lesion. Circulation 2011, 124, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Kriebardis, A.; Antonelou, M.; Stamoulis, K.; Papassideri, I. Cell-derived microparticles in stored blood products: Innocent-bystanders or effective mediators of post-transfusion reactions? Blood Transfus. 2012, 10 (Suppl. 2), S25–S38. [Google Scholar]
- Burnouf, T.; Chou, M.L.; Goubran, H.; Cognasse, F.; Garraud, O.; Seghatchian, J. An overview of the role of microparticles/microvesicles in blood components: Are they clinically beneficial or harmful? Transfus. Apher. Sci. 2015, 53, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Arraud, N.; Linares, R.; Tan, S.; Gounou, C.; Pasquet, J.M.; Mornet, S.; Brisson, A.R. Extracellular vesicles from blood plasma: Determination of their morphology, size, phenotype and concentration. J. Thromb. Haemost. 2014, 12, 614–627. [Google Scholar] [CrossRef] [PubMed]
- Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Scott syndrome, a bleeding disorder caused by defective scrambling of membrane phospholipids. Biochim. Biophys. Acta 2004, 1636, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.C.; Guo, S.C.; Zhang, C.Q. Platelet-derived Extracellular Vesicles: An Emerging Therapeutic Approach. Int. J. Biol. Sci. 2017, 13, 828–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vajen, T.; Benedikter, B.J.; Heinzmann, A.C.A.; Vasina, E.M.; Henskens, Y.; Parsons, M.; Maguire, P.B.; Stassen, F.R.; Heemskerk, J.W.M.; Schurgers, L.J.; et al. Platelet extracellular vesicles induce a pro-inflammatory smooth muscle cell phenotype. J. Extracell. Vesicles 2017, 6, 1322454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprague, D.L.; Elzey, B.D.; Crist, S.A.; Waldschmidt, T.J.; Jensen, R.J.; Ratliff, T.L. Platelet-mediated modulation of adaptive immunity: Unique delivery of CD154 signal by platelet-derived membrane vesicles. Blood 2008, 111, 5028–5036. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, L.C. The role of platelet microvesicles in intercellular communication. Platelets 2017, 28, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.L., III; Kuethe, J.W.; Caldwell, C.C. Neutrophil derived microvesicles: Emerging role of a key mediator to the immune response. Endocr. Metab. Immune Disord. Drug Targets 2014, 14, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Halim, A.T.; Ariffin, N.A.; Azlan, M. Review: The Multiple Roles of Monocytic Microparticles. Inflammation 2016, 39, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Angelillo-Scherrer, A. Leukocyte-derived microparticles in vascular homeostasis. Circ. Res. 2012, 110, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Del Conde, I.; Shrimpton, C.N.; Thiagarajan, P.; Lopez, J.A. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005, 106, 1604–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budnik, V.; Ruiz-Canada, C.; Wendler, F. Extracellular vesicles round off communication in the nervous system. Nat. Rev. Neurosci. 2016, 17, 160–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolicelli, R.C.; Bergamini, G.; Rajendran, L. Cell-to-cell Communication by Extracellular Vesicles: Focus on Microglia. Neuroscience 2018. [Google Scholar] [CrossRef] [PubMed]
- Bianco, F.; Pravettoni, E.; Colombo, A.; Schenk, U.; Moller, T.; Matteoli, M.; Verderio, C. Astrocyte-derived ATP induces vesicle shedding and IL-1 beta release from microglia. J. Immunol. 2005, 174, 7268–7277. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Turola, E.; Ruiz, A.; Bergami, A.; Libera, D.D.; Benussi, L.; Giussani, P.; Magnani, G.; Comi, G.; Legname, G.; et al. Microglia convert aggregated amyloid-beta into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2014, 21, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Carandini, T.; Colombo, F.; Finardi, A.; Casella, G.; Garzetti, L.; Verderio, C.; Furlan, R. Microvesicles: What is the Role in Multiple Sclerosis? Front. Neurol. 2015, 6, 111. [Google Scholar] [CrossRef] [PubMed]
- Minagar, A.; Alexander, J.S. Blood-brain barrier disruption in multiple sclerosis. Mult. Scler. 2003, 9, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, S.; Corrado, C.; Raimondi, L.; De Leo, G.; Alessandro, R. Role of Extracellular Vesicles in Hematological Malignancies. Biomed. Res. Int. 2015, 2015, 821613. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.K.; Greening, D.W.; Rai, A.; Chen, M.; Xu, R.; Shafiq, A.; Mathias, R.A.; Zhu, H.J.; Simpson, R.J. Extracellular vesicles: Their role in cancer biology and epithelial-mesenchymal transition. Biochem. J. 2017, 474, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Coumans, F.A.W.; Brisson, A.R.; Buzas, E.I.; Dignat-George, F.; Drees, E.E.E.; El-Andaloussi, S.; Emanueli, C.; Gasecka, A.; Hendrix, A.; Hill, A.F.; et al. Methodological Guidelines to Study Extracellular Vesicles. Circ. Res. 2017, 120, 1632–1648. [Google Scholar] [CrossRef] [PubMed]
- Menck, K.; Bleckmann, A.; Schulz, M.; Ries, L.; Binder, C. Isolation and Characterization of Microvesicles from Peripheral Blood. J. Vis. Exp. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konoshenko, M.Y.; Lekchnov, E.A.; Vlassov, A.V.; Laktionov, P.P. Isolation of Extracellular Vesicles: General Methodologies and Latest Trends. Biomed. Res. Int. 2018, 2018, 8545347. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.I.; Amorim, M.G.; Gadelha, C.; Milic, I.; Welsh, J.A.; Freitas, V.M.; Nawaz, M.; Akbar, N.; Couch, Y.; Makin, L.; et al. Technical challenges of working with extracellular vesicles. Nanoscale 2018, 10, 881–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateescu, B.; Kowal, E.J.; van Balkom, B.W.; Bartel, S.; Bhattacharyya, S.N.; Buzas, E.I.; Buck, A.H.; de Candia, P.; Chow, F.W.; Das, S.; et al. Obstacles and opportunities in the functional analysis of extracellular vesicle RNA—An ISEV position paper. J. Extracell. Vesicles 2017, 6, 1286095. [Google Scholar] [CrossRef] [PubMed]
- Szatanek, R.; Baj-Krzyworzeka, M.; Zimoch, J.; Lekka, M.; Siedlar, M.; Baran, J. The Methods of Choice for Extracellular Vesicles (EVs) Characterization. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Aharon, A.; Brenner, B. Microparticles and pregnancy complications. Thromb. Res. 2011, 127 (Suppl. 3), S67–S71. [Google Scholar] [CrossRef]
- Wilhelm, E.N.; Mourot, L.; Rakobowchuk, M. Exercise-Derived Microvesicles: A Review of the Literature. Sports Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Strohacker, K.; Breslin, W.L.; Carpenter, K.C.; Davidson, T.R.; Agha, N.H.; McFarlin, B.K. Moderate-intensity, premeal cycling blunts postprandial increases in monocyte cell surface CD18 and CD11a and endothelial microparticles following a high-fat meal in young adults. Appl. Physiol. Nutr. Metab. 2012, 37, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Alijotas-Reig, J.; Palacio-Garcia, C.; Llurba, E.; Vilardell-Tarres, M. Cell-derived microparticles and vascular pregnancy complications: A systematic and comprehensive review. Fertil. Steril. 2013, 99, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, C.M.; Shepherd, A.J.; Miller, V.M.; Jayachandran, M. Age- and sex-specific differences in blood-borne microvesicles from apparently healthy humans. Biol. Sex Differ. 2015, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Bernimoulin, M.; Waters, E.K.; Foy, M.; Steele, B.M.; Sullivan, M.; Falet, H.; Walsh, M.T.; Barteneva, N.; Geng, J.G.; Hartwig, J.H.; et al. Differential stimulation of monocytic cells results in distinct populations of microparticles. J. Thromb. Haemost. 2009, 7, 1019–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalra, H.; Simpson, R.J.; Ji, H.; Aikawa, E.; Altevogt, P.; Askenase, P.; Bond, V.C.; Borras, F.E.; Breakefield, X.; Budnik, V.; et al. Vesiclepedia: A compendium for extracellular vesicles with continuous community annotation. PLoS Biol. 2012, 10, e1001450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.K.; Kang, B.; Kim, O.Y.; Choi, D.S.; Lee, J.; Kim, S.R.; Go, G.; Yoon, Y.J.; Kim, J.H.; Jang, S.C.; et al. EVpedia: An integrated database of high-throughput data for systemic analyses of extracellular vesicles. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan-Chari, V.; Clancy, J.; Plou, C.; Romao, M.; Chavrier, P.; Raposo, G.; D’Souza-Schorey, C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr. Biol. 2009, 19, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Keerthikumar, S.; Gangoda, L.; Liem, M.; Fonseka, P.; Atukorala, I.; Ozcitti, C.; Mechler, A.; Adda, C.G.; Ang, C.S.; Mathivanan, S. Proteogenomic analysis reveals exosomes are more oncogenic than ectosomes. Oncotarget 2015, 6, 15375–15396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasser, O.; Hess, C.; Miot, S.; Deon, C.; Sanchez, J.C.; Schifferli, J.A. Characterisation and properties of ectosomes released by human polymorphonuclear neutrophils. Exp. Cell Res. 2003, 285, 243–257. [Google Scholar] [CrossRef]
- Kriebardis, A.G.; Antonelou, M.H.; Stamoulis, K.E.; Economou-Petersen, E.; Margaritis, L.H.; Papassideri, I.S. RBC-derived vesicles during storage: Ultrastructure, protein composition, oxidation, and signaling components. Transfusion 2008, 48, 1943–1953. [Google Scholar] [CrossRef] [PubMed]
- Salzer, U.; Zhu, R.; Luten, M.; Isobe, H.; Pastushenko, V.; Perkmann, T.; Hinterdorfer, P.; Bosman, G.J. Vesicles generated during storage of red cells are rich in the lipid raft marker stomatin. Transfusion 2008, 48, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Salzer, U.; Hinterdorfer, P.; Hunger, U.; Borken, C.; Prohaska, R. Ca++-dependent vesicle release from erythrocytes involves stomatin-specific lipid rafts, synexin (annexin VII), and sorcin. Blood 2002, 99, 2569–2577. [Google Scholar] [CrossRef] [PubMed]
- Di Vizio, D.; Morello, M.; Dudley, A.C.; Schow, P.W.; Adam, R.M.; Morley, S.; Mulholland, D.; Rotinen, M.; Hager, M.H.; Insabato, L.; et al. Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. Am. J. Pathol. 2012, 181, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Millimaggi, D.; Festuccia, C.; Angelucci, A.; D’Ascenzo, S.; Rucci, N.; Flati, S.; Bologna, M.; Teti, A.; Pavan, A.; Dolo, V. Osteoblast-conditioned media stimulate membrane vesicle shedding in prostate cancer cells. Int. J. Oncol. 2006, 28, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Taraboletti, G.; D’Ascenzo, S.; Borsotti, P.; Giavazzi, R.; Pavan, A.; Dolo, V. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am. J. Pathol. 2002, 160, 673–680. [Google Scholar] [CrossRef]
- Shimoda, M.; Khokha, R. Metalloproteinases in extracellular vesicles. Biochim. Biophys. Acta 2017, 1864, 1989–2000. [Google Scholar] [CrossRef] [PubMed]
- Dolo, V.; Ginestra, A.; Cassara, D.; Violini, S.; Lucania, G.; Torrisi, M.R.; Nagase, H.; Canevari, S.; Pavan, A.; Vittorelli, M.L. Selective localization of matrix metalloproteinase 9, beta1 integrins, and human lymphocyte antigen class I molecules on membrane vesicles shed by 8701-BC breast carcinoma cells. Cancer Res. 1998, 58, 4468–4474. [Google Scholar] [PubMed]
- Heijnen, H.F.; Schiel, A.E.; Fijnheer, R.; Geuze, H.J.; Sixma, J.J. Activated platelets release two types of membrane vesicles: Microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood 1999, 94, 3791–3799. [Google Scholar] [PubMed]
- Pluskota, E.; Woody, N.M.; Szpak, D.; Ballantyne, C.M.; Soloviev, D.A.; Simon, D.I.; Plow, E.F. Expression, activation, and function of integrin alphaMbeta2 (Mac-1) on neutrophil-derived microparticles. Blood 2008, 112, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Mezouar, S.; Darbousset, R.; Dignat-George, F.; Panicot-Dubois, L.; Dubois, C. Inhibition of platelet activation prevents the P-selectin and integrin-dependent accumulation of cancer cell microparticles and reduces tumor growth and metastasis in vivo. Int. J. Cancer 2015, 136, 462–475. [Google Scholar] [CrossRef] [PubMed]
- Deregibus, M.C.; Cantaluppi, V.; Calogero, R.; Lo Iacono, M.; Tetta, C.; Biancone, L.; Bruno, S.; Bussolati, B.; Camussi, G. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood 2007, 110, 2440–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Abdelmohsen, K.; Mustapic, M.; Kapogiannis, D.; Gorospe, M. RNA in extracellular vesicles. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Baj-Krzyworzeka, M.; Szatanek, R.; Weglarczyk, K.; Baran, J.; Urbanowicz, B.; Branski, P.; Ratajczak, M.Z.; Zembala, M. Tumour-derived microvesicles carry several surface determinants and mRNA of tumour cells and transfer some of these determinants to monocytes. Cancer Immunol. Immunother. 2006, 55, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Fatima, F.; Nawaz, M. Vesiculated Long Non-Coding RNAs: Offshore Packages Deciphering Trans-Regulation between Cells, Cancer Progression and Resistance to Therapies. Non-Coding RNA 2017, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.; Farber, E.L.; Rapoport, A.L.; Tejada, D.; Deniskin, R.; Akhmedov, N.B.; Farber, D.B. Transfer of microRNAs by embryonic stem cell microvesicles. PLoS ONE 2009, 4, e4722. [Google Scholar] [CrossRef] [PubMed]
- Fonsato, V.; Collino, F.; Herrera, M.B.; Cavallari, C.; Deregibus, M.C.; Cisterna, B.; Bruno, S.; Romagnoli, R.; Salizzoni, M.; Tetta, C.; et al. Human liver stem cell-derived microvesicles inhibit hepatoma growth in SCID mice by delivering antitumor microRNAs. Stem Cells 2012, 30, 1985–1998. [Google Scholar] [CrossRef] [PubMed]
- Crescitelli, R.; Lasser, C.; Szabo, T.G.; Kittel, A.; Eldh, M.; Dianzani, I.; Buzas, E.I.; Lotvall, J. Distinct RNA profiles in subpopulations of extracellular vesicles: Apoptotic bodies, microvesicles and exosomes. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, L.; Lin, W.; Qi, P.; Xu, M.D.; Wu, X.; Ni, S.; Huang, D.; Weng, W.W.; Tan, C.; Sheng, W.; et al. Circulating Long RNAs in Serum Extracellular Vesicles: Their Characterization and Potential Application as Biomarkers for Diagnosis of Colorectal Cancer. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, Y.; Yamamoto, Y.; Sato, T.A.; Ochiya, T. Extracellular vesicles as trans-genomic agents: Emerging roles in disease and evolution. Cancer Sci. 2017, 108, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Kanada, M.; Bachmann, M.H.; Hardy, J.W.; Frimannson, D.O.; Bronsart, L.; Wang, A.; Sylvester, M.D.; Schmidt, T.L.; Kaspar, R.L.; Butte, M.J.; et al. Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proc. Natl. Acad. Sci. USA 2015, 112, E1433–E1442. [Google Scholar] [CrossRef] [PubMed]
- Osteikoetxea, X.; Balogh, A.; Szabo-Taylor, K.; Nemeth, A.; Szabo, T.G.; Paloczi, K.; Sodar, B.; Kittel, A.; Gyorgy, B.; Pallinger, E.; et al. Improved characterization of EV preparations based on protein to lipid ratio and lipid properties. PLoS ONE 2015, 10, e0121184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haraszti, R.A.; Didiot, M.C.; Sapp, E.; Leszyk, J.; Shaffer, S.A.; Rockwell, H.E.; Gao, F.; Narain, N.R.; DiFiglia, M.; Kiebish, M.A.; et al. High-resolution proteomic and lipidomic analysis of exosomes and microvesicles from different cell sources. J. Extracell. Vesicles 2016, 5, 32570. [Google Scholar] [CrossRef] [PubMed]
- Baig, S.; Lim, J.Y.; Fernandis, A.Z.; Wenk, M.R.; Kale, A.; Su, L.L.; Biswas, A.; Vasoo, S.; Shui, G.; Choolani, M. Lipidomic analysis of human placental syncytiotrophoblast microvesicles in adverse pregnancy outcomes. Placenta 2013, 34, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Weerheim, A.M.; Kolb, A.M.; Sturk, A.; Nieuwland, R. Phospholipid composition of cell-derived microparticles determined by one-dimensional high-performance thin-layer chromatography. Anal. Biochem. 2002, 302, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Bicalho, B.; Holovati, J.L.; Acker, J.P. Phospholipidomics reveals differences in glycerophosphoserine profiles of hypothermically stored red blood cells and microvesicles. Biochim. Biophys. Acta 2013, 1828, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauren, E.; Tigistu-Sahle, F.; Valkonen, S.; Westberg, M.; Valkeajarvi, A.; Eronen, J.; Siljander, P.; Pettila, V.; Kakela, R.; Laitinen, S.; et al. Phospholipid composition of packed red blood cells and that of extracellular vesicles show a high resemblance and stability during storage. Biochim. Biophys. Acta 2018, 1863, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lutz, H.U.; Liu, S.C.; Palek, J. Release of spectrin-free vesicles from human erythrocytes during ATP depletion. I. Characterization of spectrin-free vesicles. J. Cell Biol. 1977, 73, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Wang, M.; Cho, M.S.; Wang, C.; Nick, A.M.; Thiagarajan, P.; Aung, F.M.; Han, X.; Sood, A.K.; Afshar-Kharghan, V. Lipid profile of platelets and platelet-derived microparticles in ovarian cancer. BBA Clin. 2016, 6, 76–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biro, E.; Akkerman, J.W.; Hoek, F.J.; Gorter, G.; Pronk, L.M.; Sturk, A.; Nieuwland, R. The phospholipid composition and cholesterol content of platelet-derived microparticles: A comparison with platelet membrane fractions. J. Thromb. Haemost. 2005, 3, 2754–2763. [Google Scholar] [CrossRef] [PubMed]
- Connor, D.E.; Exner, T.; Ma, D.D.; Joseph, J.E. The majority of circulating platelet-derived microparticles fail to bind annexin V, lack phospholipid-dependent procoagulant activity and demonstrate greater expression of glycoprotein Ib. Thromb. Haemost. 2010, 103, 1044–1052. [Google Scholar] [PubMed]
- Cluitmans, J.C.; Gevi, F.; Siciliano, A.; Matte, A.; Leal, J.K.; De Franceschi, L.; Zolla, L.; Brock, R.; Adjobo-Hermans, M.J.; Bosman, G.J. Red Blood Cell Homeostasis: Pharmacological Interventions to Explore Biochemical, Morphological and Mechanical Properties. Front. Mol. Biosci. 2016, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Dinkla, S.; Wessels, K.; Verdurmen, W.P.; Tomelleri, C.; Cluitmans, J.C.; Fransen, J.; Fuchs, B.; Schiller, J.; Joosten, I.; Brock, R.; et al. Functional consequences of sphingomyelinase-induced changes in erythrocyte membrane structure. Cell Death Dis. 2012, 3, e410. [Google Scholar] [CrossRef] [PubMed]
- Boilard, E. Extracellular vesicles and their content in bioactive lipid mediators: More than a sack of microRNA. J. Lipid Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, M.; Belleannee, C.; Duchez, A.C.; Cloutier, N.; Levesque, T.; Jacques, F.; Perron, J.; Nigrovic, P.A.; Dieude, M.; Hebert, M.J.; et al. Detection and quantification of microparticles from different cellular lineages using flow cytometry. Evaluation of the impact of secreted phospholipase A2 on microparticle assessment. PLoS ONE 2015, 10, e0116812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchez, A.C.; Boudreau, L.H.; Naika, G.S.; Bollinger, J.; Belleannee, C.; Cloutier, N.; Laffont, B.; Mendoza-Villarroel, R.E.; Levesque, T.; Rollet-Labelle, E.; et al. Platelet microparticles are internalized in neutrophils via the concerted activity of 12-lipoxygenase and secreted phospholipase A2-IIA. Proc. Natl. Acad. Sci. USA 2015, 112, E3564–E3573. [Google Scholar] [CrossRef] [PubMed]
- Lux, S.E.T. Anatomy of the red cell membrane skeleton: Unanswered questions. Blood 2016, 127, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Manno, S.; Takakuwa, Y.; Mohandas, N. Modulation of erythrocyte membrane mechanical function by protein 4.1 phosphorylation. J. Biol. Chem. 2005, 280, 7581–7587. [Google Scholar] [CrossRef] [PubMed]
- Picas, L.; Rico, F.; Deforet, M.; Scheuring, S. Structural and mechanical heterogeneity of the erythrocyte membrane reveals hallmarks of membrane stability. ACS Nano 2013, 7, 1054–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muravyov, A.V.; Tikhomirova, I.A. Role molecular signaling pathways in changes of red blood cell deformability. Clin. Hemorheol. Microcirc. 2013, 53, 45–59. [Google Scholar] [PubMed]
- An, X.; Zhang, X.; Debnath, G.; Baines, A.J.; Mohandas, N. Phosphatidylinositol-4,5-biphosphate (PIP2) differentially regulates the interaction of human erythrocyte protein 4.1 (4.1R) with membrane proteins. Biochemistry 2006, 45, 5725–5732. [Google Scholar] [CrossRef] [PubMed]
- Kapus, A.; Janmey, P. Plasma membrane—Cortical cytoskeleton interactions: A cell biology approach with biophysical considerations. Compr. Physiol. 2013, 3, 1231–1281. [Google Scholar] [PubMed]
- Bogdanova, A.; Makhro, A.; Wang, J.; Lipp, P.; Kaestner, L. Calcium in red blood cells-a perilous balance. Int. J. Mol. Sci. 2013, 14, 9848–9872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huisjes, R.; Bogdanova, A.; van Solinge, W.W.; Schiffelers, R.M.; Kaestner, L.; van Wijk, R. Squeezing for Life—Properties of Red Blood Cell Deformability. Front. Physiol. 2018, 9, 656. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.; Mohandas, N.; Leung, A. Static and dynamic rigidities of normal and sickle erythrocytes. Major influence of cell hemoglobin concentration. J. Clin. Investig. 1984, 73, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Lew, V.L.; Tiffert, T. On the Mechanism of Human Red Blood Cell Longevity: Roles of Calcium, the Sodium Pump, PIEZO1, and Gardos Channels. Front. Physiol. 2017, 8, 977. [Google Scholar] [CrossRef] [PubMed]
- Betz, T.; Lenz, M.; Joanny, J.F.; Sykes, C. ATP-dependent mechanics of red blood cells. Proc. Natl. Acad. Sci. USA 2009, 106, 15320–15325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.; Best, C.A.; Auth, T.; Gov, N.S.; Safran, S.A.; Popescu, G.; Suresh, S.; Feld, M.S. Metabolic remodeling of the human red blood cell membrane. Proc. Natl. Acad. Sci. USA 2010, 107, 1289–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, Y.Z.; Kotar, J.; Yoon, G.; Cicuta, P. The nonlinear mechanical response of the red blood cell. Phys. Biol. 2008, 5, 036007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rifkind, J.M.; Nagababu, E. Hemoglobin redox reactions and red blood cell aging. Antioxid. Redox Signal. 2013, 18, 2274–2283. [Google Scholar] [CrossRef] [PubMed]
- Rubin, O.; Crettaz, D.; Canellini, G.; Tissot, J.D.; Lion, N. Microparticles in stored red blood cells: An approach using flow cytometry and proteomic tools. Vox Sang. 2008, 95, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Kriebardis, A.G.; Antonelou, M.H.; Stamoulis, K.E.; Economou-Petersen, E.; Margaritis, L.H.; Papassideri, I.S. Storage-dependent remodeling of the red blood cell membrane is associated with increased immunoglobulin G binding, lipid raft rearrangement, and caspase activation. Transfusion 2007, 47, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Bosman, G.J.; Lasonder, E.; Groenen-Dopp, Y.A.; Willekens, F.L.; Werre, J.M.; Novotny, V.M. Comparative proteomics of erythrocyte aging in vivo and in vitro. J. Proteom. 2010, 73, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Salehyar, S.; Cabrales, P.; Asaro, R.J. Prospects for Human Erythrocyte Skeleton-Bilayer Dissociation during Splenic Flow. Biophys. J. 2017, 113, 900–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fricke, K.; Sackmann, E. Variation of frequency spectrum of the erythrocyte flickering caused by aging, osmolarity, temperature and pathological changes. Biochim. Biophys. Acta 1984, 803, 145–152. [Google Scholar] [CrossRef]
- Gov, N.S.; Safran, S.A. Red blood cell membrane fluctuations and shape controlled by ATP-induced cytoskeletal defects. Biophys. J. 2005, 88, 1859–1874. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.L.; Scales, M.T.; Loughlin, C.; Bennett, G.G.; Harris-Peterson, S.; De Castro, L.M.; Whitworth, E.; Abrams, M.; Feliu, M.; Johnson, S.; et al. A brief review of the pathophysiology, associated pain, and psychosocial issues in sickle cell disease. Int. J. Behav. Med. 2005, 12, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Bartosz, G.; Gaczynska, M.; Grzelinska, E.; Soszynski, M.; Michalak, W.; Gondko, R. Aged erythrocytes exhibit decreased anion exchange. Mech. Ageing Dev. 1987, 39, 245–250. [Google Scholar] [CrossRef]
- Bartosz, G.; Tannert, C.; Fried, R.; Leyko, W. Superoxide dismutase activity decreases during erythrocyte aging. Experientia 1978, 34, 1464. [Google Scholar] [CrossRef] [PubMed]
- Fornaini, G.; Magnani, M.; Fazi, A.; Accorsi, A.; Stocchi, V.; Dacha, M. Regulatory properties of human erythrocyte hexokinase during cell ageing. Arch. Biochem. Biophys. 1985, 239, 352–358. [Google Scholar] [CrossRef]
- Azouzi, S.; Romana, M.; Arashiki, N.; Takakuwa, Y.; El Nemer, W.; Peyrard, T.; Colin, Y.; Amireault, P.; Le Van Kim, C. Band 3 phosphorylation induces irreversible alterations of stored red blood cells. Am. J. Hematol. 2018, 93, E110–E112. [Google Scholar] [CrossRef] [PubMed]
- Szigyarto, I.C.; Deak, R.; Mihaly, J.; Rocha, S.; Zsila, F.; Varga, Z.; Beke-Somfai, T. Flow Alignment of Extracellular Vesicles: Structure and Orientation of Membrane-Associated Bio-macromolecules Studied with Polarized Light. ChemBioChem 2018, 19, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Wither, M.; Dzieciatkowska, M.; Nemkov, T.; Strop, P.; D’Alessandro, A.; Hansen, K.C. Hemoglobin oxidation at functional amino acid residues during routine storage of red blood cells. Transfusion 2016, 56, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Arashiki, N.; Kimata, N.; Manno, S.; Mohandas, N.; Takakuwa, Y. Membrane peroxidation and methemoglobin formation are both necessary for band 3 clustering: Mechanistic insights into human erythrocyte senescence. Biochemistry 2013, 52, 5760–5769. [Google Scholar] [CrossRef] [PubMed]
- Lutz, H.U.; Flepp, R.; Stringaro-Wipf, G. Naturally occurring autoantibodies to exoplasmic and cryptic regions of band 3 protein, the major integral membrane protein of human red blood cells. J. Immunol. 1984, 133, 2610–2618. [Google Scholar] [PubMed]
- Stowell, S.R.; Smith, N.H.; Zimring, J.C.; Fu, X.; Palmer, A.F.; Fontes, J.; Banerjee, U.; Yazer, M.H. Addition of ascorbic acid solution to stored murine red blood cells increases posttransfusion recovery and decreases microparticles and alloimmunization. Transfusion 2013, 53, 2248–2257. [Google Scholar] [CrossRef] [PubMed]
- Freikman, I.; Fibach, E. Distribution and shedding of the membrane phosphatidylserine during maturation and aging of erythroid cells. Biochim. Biophys. Acta 2011, 1808, 2773–2780. [Google Scholar] [CrossRef] [PubMed]
- Repsold, L.; Joubert, A.M. Eryptosis: An Erythrocyte’s Suicidal Type of Cell Death. Biomed. Res. Int. 2018, 2018, 9405617. [Google Scholar] [CrossRef] [PubMed]
- Allan, D.; Thomas, P.; Limbrick, A.R. The isolation and characterization of 60 nm vesicles (‘nanovesicles’) produced during ionophore A23187-induced budding of human erythrocytes. Biochem. J. 1980, 188, 881–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butikofer, P.; Kuypers, F.A.; Xu, C.M.; Chiu, D.T.; Lubin, B. Enrichment of two glycosyl-phosphatidylinositol-anchored proteins, acetylcholinesterase and decay accelerating factor, in vesicles released from human red blood cells. Blood 1989, 74, 1481–1485. [Google Scholar] [PubMed]
- Nguyen, D.B.; Ly, T.B.; Wesseling, M.C.; Hittinger, M.; Torge, A.; Devitt, A.; Perrie, Y.; Bernhardt, I. Characterization of Microvesicles Released from Human Red Blood Cells. Cell. Physiol. Biochem. 2016, 38, 1085–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allan, D.; Michell, R.H. Calcium ion-dependent diacylglycerol accumulation in erythrocytes is associated with microvesiculation but not with efflux of potassium ions. Biochem. J. 1977, 166, 495–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.B.; Wagner-Britz, L.; Maia, S.; Steffen, P.; Wagner, C.; Kaestner, L.; Bernhardt, I. Regulation of phosphatidylserine exposure in red blood cells. Cell. Physiol. Biochem. 2011, 28, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Morel, O.; Jesel, L.; Freyssinet, J.M.; Toti, F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Westerman, M.; Porter, J.B. Red blood cell-derived microparticles: An overview. Blood Cells Mol. Dis. 2016, 59, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Rachmilewitz, E.A.; Thorell, B. Hemichromes in single inclusion bodies in red cells of beta thalassemia. Blood 1972, 39, 794–800. [Google Scholar] [PubMed]
- Ferru, E.; Pantaleo, A.; Carta, F.; Mannu, F.; Khadjavi, A.; Gallo, V.; Ronzoni, L.; Graziadei, G.; Cappellini, M.D.; Turrini, F. Thalassemic erythrocytes release microparticles loaded with hemichromes by redox activation of p72Syk kinase. Haematologica 2014, 99, 570–578. [Google Scholar] [CrossRef] [PubMed]
- George, A.; Pushkaran, S.; Konstantinidis, D.G.; Koochaki, S.; Malik, P.; Mohandas, N.; Zheng, Y.; Joiner, C.H.; Kalfa, T.A. Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood 2013, 121, 2099–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camus, S.M.; De Moraes, J.A.; Bonnin, P.; Abbyad, P.; Le Jeune, S.; Lionnet, F.; Loufrani, L.; Grimaud, L.; Lambry, J.C.; Charue, D.; et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 2015, 125, 3805–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eber, S.; Lux, S.E. Hereditary spherocytosis—Defects in proteins that connect the membrane skeleton to the lipid bilayer. Semin. Hematol. 2004, 41, 118–141. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, S.; Gallagher, P.G.; Mohandas, N. Hereditary spherocytosis. Lancet 2008, 372, 1411–1426. [Google Scholar] [CrossRef]
- Diez-Silva, M.; Dao, M.; Han, J.; Lim, C.T.; Suresh, S. Shape and Biomechanical Characteristics of Human Red Blood Cells in Health and Disease. MRS Bull. 2010, 35, 382–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Lykotrafitis, G. Vesiculation of healthy and defective red blood cells. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2015, 92, 012715. [Google Scholar] [CrossRef] [PubMed]
- Alaarg, A.; Schiffelers, R.M.; van Solinge, W.W.; van Wijk, R. Red blood cell vesiculation in hereditary hemolytic anemia. Front. Physiol. 2013, 4, 365. [Google Scholar] [CrossRef] [PubMed]
- Reliene, R.; Mariani, M.; Zanella, A.; Reinhart, W.H.; Ribeiro, M.L.; del Giudice, E.M.; Perrotta, S.; Iolascon, A.; Eber, S.; Lutz, H.U. Splenectomy prolongs in vivo survival of erythrocytes differently in spectrin/ankyrin- and band 3-deficient hereditary spherocytosis. Blood 2002, 100, 2208–2215. [Google Scholar] [PubMed]
- Peters, L.L.; Shivdasani, R.A.; Liu, S.C.; Hanspal, M.; John, K.M.; Gonzalez, J.M.; Brugnara, C.; Gwynn, B.; Mohandas, N.; Alper, S.L.; et al. Anion exchanger 1 (band 3) is required to prevent erythrocyte membrane surface loss but not to form the membrane skeleton. Cell 1996, 86, 917–927. [Google Scholar] [CrossRef]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and biogenesis of shed microvesicles. Small GTPases 2017, 8, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, M.; Camussi, G.; Valadi, H.; Nazarenko, I.; Ekstrom, K.; Wang, X.; Principe, S.; Shah, N.; Ashraf, N.M.; Fatima, F.; et al. The emerging role of extracellular vesicles as biomarkers for urogenital cancers. Nat. Rev. Urol. 2014, 11, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Hugel, B.; Martinez, M.C.; Kunzelmann, C.; Freyssinet, J.M. Membrane microparticles: Two sides of the coin. Physiology 2005, 20, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Muhsin-Sharafaldine, M.R.; McLellan, A.D. Tumor-Derived Apoptotic Vesicles: With Death They Do Part. Front. Immunol. 2018, 9, 957. [Google Scholar] [CrossRef] [PubMed]
- McMahon, H.T.; Boucrot, E. Membrane curvature at a glance. J. Cell Sci. 2015, 128, 1065–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanez-Mo, M.; Barreiro, O.; Gordon-Alonso, M.; Sala-Valdes, M.; Sanchez-Madrid, F. Tetraspanin-enriched microdomains: A functional unit in cell plasma membranes. Trends Cell Biol. 2009, 19, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Bari, R.; Guo, Q.; Xia, B.; Zhang, Y.H.; Giesert, E.E.; Levy, S.; Zheng, J.J.; Zhang, X.A. Tetraspanins regulate the protrusive activities of cell membrane. Biochem. Biophys. Res. Commun. 2011, 415, 619–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clancy, J.W.; Sedgwick, A.; Rosse, C.; Muralidharan-Chari, V.; Raposo, G.; Method, M.; Chavrier, P.; D’Souza-Schorey, C. Regulated delivery of molecular cargo to invasive tumour-derived microvesicles. Nat. Commun. 2015, 6, 6919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza-Schorey, C.; Chavrier, P. ARF proteins: Roles in membrane traffic and beyond. Nat. Rev. Mol. Cell Biol. 2006, 7, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Antonyak, M.A.; Zhang, J.; Cerione, R.A. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene 2012, 31, 4740–4749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Gilkes, D.M.; Takano, N.; Xiang, L.; Luo, W.; Bishop, C.J.; Chaturvedi, P.; Green, J.J.; Semenza, G.L. Hypoxia-inducible factors and RAB22A mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. USA 2014, 111, E3234–E3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verderio, C.; Gabrielli, M.; Giussani, P. Role of sphingolipids in the biogenesis and biological activity of extracellular vesicles. J. Lipid Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Pfrieger, F.W.; Vitale, N. Cholesterol and the journey of extracellular vesicles. J. Lipid Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Morad, S.A.; Cabot, M.C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 2013, 13, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Veldman, R.J.; Maestre, N.; Aduib, O.M.; Medin, J.A.; Salvayre, R.; Levade, T. A neutral sphingomyelinase resides in sphingolipid-enriched microdomains and is inhibited by the caveolin-scaffolding domain: Potential implications in tumour necrosis factor signalling. Biochem. J. 2001, 355, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Menck, K.; Sonmezer, C.; Worst, T.S.; Schulz, M.; Dihazi, G.H.; Streit, F.; Erdmann, G.; Kling, S.; Boutros, M.; Binder, C.; et al. Neutral sphingomyelinases control extracellular vesicles budding from the plasma membrane. J. Extracell. Vesicles 2017, 6, 1378056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianco, F.; Perrotta, C.; Novellino, L.; Francolini, M.; Riganti, L.; Menna, E.; Saglietti, L.; Schuchman, E.H.; Furlan, R.; Clementi, E.; et al. Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO J. 2009, 28, 1043–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, B.; Ziobro, R.; Becker, K.A.; Kolesnick, R.; Gulbins, E. Acid sphingomyelinase. Handb. Exp. Pharmacol. 2013, 77–88. [Google Scholar] [CrossRef]
- Xu, M.; Xia, M.; Li, X.X.; Han, W.Q.; Boini, K.M.; Zhang, F.; Zhang, Y.; Ritter, J.K.; Li, P.L. Requirement of translocated lysosomal V1 H+-ATPase for activation of membrane acid sphingomyelinase and raft clustering in coronary endothelial cells. Mol. Biol. Cell 2012, 23, 1546–1557. [Google Scholar] [CrossRef] [PubMed]
- Awojoodu, A.O.; Keegan, P.M.; Lane, A.R.; Zhang, Y.; Lynch, K.R.; Platt, M.O.; Botchwey, E.A. Acid sphingomyelinase is activated in sickle cell erythrocytes and contributes to inflammatory microparticle generation in SCD. Blood 2014, 124, 1941–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoehn, R.S.; Jernigan, P.L.; Japtok, L.; Chang, A.L.; Midura, E.F.; Caldwell, C.C.; Kleuser, B.; Lentsch, A.B.; Edwards, M.J.; Gulbins, E.; et al. Acid Sphingomyelinase Inhibition in Stored Erythrocytes Reduces Transfusion-Associated Lung Inflammation. Ann. Surg. 2017, 265, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pendurthi, U.R.; Rao, L.V.M. Sphingomyelin encrypts tissue factor: ATP-induced activation of A-SMase leads to tissue factor decryption and microvesicle shedding. Blood Adv. 2017, 1, 849–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Montero, I.; Rodriguez, N.; Cribier, S.; Pohl, A.; Velez, M.; Devaux, P.F. Rapid transbilayer movement of ceramides in phospholipid vesicles and in human erythrocytes. J. Biol. Chem. 2005, 280, 25811–25819. [Google Scholar] [CrossRef] [PubMed]
- Slotte, J.P.; Hedstrom, G.; Rannstrom, S.; Ekman, S. Effects of sphingomyelin degradation on cell cholesterol oxidizability and steady-state distribution between the cell surface and the cell interior. Biochim. Biophys. Acta 1989, 985, 90–96. [Google Scholar] [CrossRef]
- Fanani, M.L.; De Tullio, L.; Hartel, S.; Jara, J.; Maggio, B. Sphingomyelinase-induced domain shape relaxation driven by out-of-equilibrium changes of composition. Biophys. J. 2009, 96, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Castro, B.M.; Prieto, M.; Silva, L.C. Ceramide: A simple sphingolipid with unique biophysical properties. Prog. Lipid Res. 2014, 54, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Goni, F.M.; Alonso, A. Effects of ceramide and other simple sphingolipids on membrane lateral structure. Biochim. Biophys. Acta 2009, 1788, 169–177. [Google Scholar] [CrossRef] [PubMed]
- White, A.B.; Givogri, M.I.; Lopez-Rosas, A.; Cao, H.; van Breemen, R.; Thinakaran, G.; Bongarzone, E.R. Psychosine accumulates in membrane microdomains in the brain of krabbe patients, disrupting the raft architecture. J. Neurosci. 2009, 29, 6068–6077. [Google Scholar] [CrossRef] [PubMed]
- D’Auria, L.; Reiter, C.; Ward, E.; Moyano, A.L.; Marshall, M.S.; Nguyen, D.; Scesa, G.; Hauck, Z.; van Breemen, R.; Givogri, M.I.; et al. Psychosine enhances the shedding of membrane microvesicles: Implications in demyelination in Krabbe’s disease. PLoS ONE 2017, 12, e0178103. [Google Scholar] [CrossRef] [PubMed]
- Vind-Kezunovic, D.; Nielsen, C.H.; Wojewodzka, U.; Gniadecki, R. Line tension at lipid phase boundaries regulates formation of membrane vesicles in living cells. Biochim. Biophys. Acta 2008, 1778, 2480–2486. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.L.; Reilly, M.P.; Casasanto, P.; McKenzie, S.E.; Williams, K.J. Cholesterol enrichment of human monocyte/macrophages induces surface exposure of phosphatidylserine and the release of biologically-active tissue factor-positive microvesicles. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Carquin, M.; D’Auria, L.; Pollet, H.; Bongarzone, E.R.; Tyteca, D. Recent progress on lipid lateral heterogeneity in plasma membranes: From rafts to submicrometric domains. Prog. Lipid Res. 2016, 62, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipowsky, R. Budding of membranes induced by intramembrane domains. J. Phys. II EDP Sci. 1992, 2, 1825–1840. [Google Scholar] [CrossRef]
- Baumgart, T.; Hess, S.T.; Webb, W.W. Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature 2003, 425, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-T.; Kiessling, V.; Tamm, L.K. Line tension at lipid phase boundaries as driving force for HIV fusion peptide-mediated fusion. Nat. Commun. 2016, 7, 11401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bretscher, M.S. Phosphatidyl-ethanolamine: Differential labelling in intact cells and cell ghosts of human erythrocytes by a membrane-impermeable reagent. J. Mol. Biol. 1972, 71, 523–528. [Google Scholar] [CrossRef]
- Yeung, T.; Grinstein, S. Lipid signaling and the modulation of surface charge during phagocytosis. Immunol. Rev. 2007, 219, 17–36. [Google Scholar] [CrossRef] [PubMed]
- Bevers, E.M.; Comfurius, P.; van Rijn, J.L.; Hemker, H.C.; Zwaal, R.F. Generation of prothrombin-converting activity and the exposure of phosphatidylserine at the outer surface of platelets. Eur. J. Biochem. FEBS 1982, 122, 429–436. [Google Scholar] [CrossRef]
- Lentz, B.R. Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog. Lipid Res. 2003, 42, 423–438. [Google Scholar] [CrossRef]
- Larson, M.C.; Karafin, M.S.; Hillery, C.A.; Hogg, N. Phosphatidylethanolamine is progressively exposed in RBCs during storage. Transfus. Med. 2017, 27, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Suzuki, J.; Segawa, K.; Fujii, T. Exposure of phosphatidylserine on the cell surface. Cell Death Differ. 2016, 23, 952–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratton, D.; Moore, C.; Zheng, L.; Lange, S.; Inal, J. Prostate cancer cells stimulated by calcium-mediated activation of protein kinase C undergo a refractory period before re-releasing calcium-bearing microvesicles. Biochem. Biophys. Res. Commun. 2015, 460, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Tepper, A.D.; Ruurs, P.; Wiedmer, T.; Sims, P.J.; Borst, J.; van Blitterswijk, W.J. Sphingomyelin hydrolysis to ceramide during the execution phase of apoptosis results from phospholipid scrambling and alters cell-surface morphology. J. Cell Biol. 2000, 150, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Parton, R.G.; del Pozo, M.A. Caveolae as plasma membrane sensors, protectors and organizers. Nat. Rev. Mol. Cell Biol. 2013, 14, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J. Rafts defined: A report on the Keystone Symposium on Lipid Rafts and Cell Function. J. Lipid Res. 2006, 47, 1597–1598. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, T.; Hammond, A.T.; Sengupta, P.; Hess, S.T.; Holowka, D.A.; Baird, B.A.; Webb, W.W. Large-scale fluid/fluid phase separation of proteins and lipids in giant plasma membrane vesicles. Proc. Natl. Acad. Sci. USA 2007, 104, 3165–3170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardino de la Serna, J.; Perez-Gil, J.; Simonsen, A.C.; Bagatolli, L.A. Cholesterol rules: Direct observation of the coexistence of two fluid phases in native pulmonary surfactant membranes at physiological temperatures. J. Biol. Chem. 2004, 279, 40715–40722. [Google Scholar] [CrossRef] [PubMed]
- Kahya, N.; Scherfeld, D.; Bacia, K.; Poolman, B.; Schwille, P. Probing lipid mobility of raft-exhibiting model membranes by fluorescence correlation spectroscopy. J. Biol. Chem. 2003, 278, 28109–28115. [Google Scholar] [CrossRef] [PubMed]
- Plasencia, I.; Norlen, L.; Bagatolli, L.A. Direct visualization of lipid domains in human skin stratum corneum’s lipid membranes: Effect of pH and temperature. Biophys. J. 2007, 93, 3142–3155. [Google Scholar] [CrossRef] [PubMed]
- Carquin, M.; Pollet, H.; Veiga-da-Cunha, M.; Cominelli, A.; Van Der Smissen, P.; N’Kuli, F.; Emonard, H.; Henriet, P.; Mizuno, H.; Courtoy, P.J.; et al. Endogenous sphingomyelin segregates into submicrometric domains in the living erythrocyte membrane. J. Lipid Res. 2014, 55, 1331–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Auria, L.; Fenaux, M.; Aleksandrowicz, P.; Van Der Smissen, P.; Chantrain, C.; Vermylen, C.; Vikkula, M.; Courtoy, P.J.; Tyteca, D. Micrometric segregation of fluorescent membrane lipids: Relevance for endogenous lipids and biogenesis in erythrocytes. J. Lipid Res. 2013, 54, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, S.A.; Tricerri, M.A.; Gratton, E. Laurdan generalized polarization fluctuations measures membrane packing micro-heterogeneity in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 7314–7319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carquin, M.; Conrard, L.; Pollet, H.; Van Der Smissen, P.; Cominelli, A.; Veiga-da-Cunha, M.; Courtoy, P.J.; Tyteca, D. Cholesterol segregates into submicrometric domains at the living erythrocyte membrane: Evidence and regulation. Cell. Mol. Life Sci. 2015, 72, 4633–4651. [Google Scholar] [CrossRef] [PubMed]
- Tyteca, D.; D’Auria, L.; Van Der Smissen, P.; Medts, T.; Carpentier, S.; Monbaliu, J.C.; de Diesbach, P.; Courtoy, P.J. Three unrelated sphingomyelin analogs spontaneously cluster into plasma membrane micrometric domains. Biochim. Biophys. Acta 2010, 1798, 909–927. [Google Scholar] [CrossRef] [PubMed]
- Bach, J.N.; Bramkamp, M. Flotillins functionally organize the bacterial membrane. Mol. Microbiol. 2013, 88, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, G.; Opekarova, M.; Malinsky, J.; Weig-Meckl, I.; Tanner, W. Membrane potential governs lateral segregation of plasma membrane proteins and lipids in yeast. EMBO J. 2007, 26, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Leonard, C.; Alsteens, D.; Dumitru, A.; Mingeot-Leclercq, M.; Tyteca, D. Lipid domains and membrane (re)shaping: From biophysics to biology. In The Role of the Physical Properties of Membranes in Influencing Biological Phenomena; Ruysschaert, J., Epand, R., Eds.; Springer Series in Biophysics: Basel, Switzerland, 2017; Volume 19, pp. 121–175. [Google Scholar]
- Grassme, H.; Jendrossek, V.; Riehle, A.; von Kurthy, G.; Berger, J.; Schwarz, H.; Weller, M.; Kolesnick, R.; Gulbins, E. Host defense against Pseudomonas aeruginosa requires ceramide-rich membrane rafts. Nat. Med. 2003, 9, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Stancevic, B.; Kolesnick, R. Ceramide-rich platforms in transmembrane signaling. FEBS Lett. 2010, 584, 1728–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, C.; Bagatolli, L.A.; Volovyk, Z.N.; Thompson, N.L.; Levi, M.; Jacobson, K.; Gratton, E. Lipid Rafts Reconstituted in Model Membranes. Biophys. J. 2001, 80, 1417–1428. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, K.; Mouritsen, O.G.; Anderson, R.G.W. Lipid rafts: At a crossroad between cell biology and physics. Nat. Cell Biol. 2007, 9, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Sezgin, E.; Gutmann, T.; Buhl, T.; Dirkx, R.; Grzybek, M.; Coskun, Ü.; Solimena, M.; Simons, K.; Levental, I.; Schwille, P. Adaptive lipid packing and bioactivity in membrane domains. PLoS ONE 2015, 10, e0123930. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.B.; Shelby, S.A.; Núñez, M.F.; Wisser, K.; Veatch, S.L. Protein sorting by lipid phase-like domains supports emergent signaling function in B lymphocyte plasma membranes. eLife 2017, 6, e19891. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.M.; Oddos, S.; Kumar, S.; Davis, D.M.; Neil, M.A.A.; French, P.M.W.; Dustin, M.L.; Magee, A.I.; Cebecauer, M. High plasma membrane lipid order imaged at the immunological synapse periphery in live T cells. Mol. Membr. Biol. 2010, 27, 178–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rentero, C.; Zech, T.; Quinn, C.M.; Engelhardt, K.; Williamson, D.; Grewal, T.; Jessup, W.; Harder, T.; Gaus, K. Functional Implications of Plasma Membrane Condensation for T Cell Activation. PLoS ONE 2008, 3, e2262. [Google Scholar] [CrossRef] [PubMed]
- Gaus, K.; Chklovskaia, E.; Fazekas de St Groth, B.; Jessup, W.; Harder, T. Condensation of the plasma membrane at the site of T lymphocyte activation. J. Cell Biol. 2005, 171, 121–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, C.; Pollet, H.; Vermylen, C.; Gov, N.S.; Tyteca, D.; Mingeot-Leclercq, M.-P. Tuning of differential lipid order between submicrometric domains and surrounding membrane upon erythrocyte reshaping. Cell. Physiol. Biochem. 2018, 48, 2563–2582. [Google Scholar] [CrossRef] [PubMed]
- Ursell, T.S.; Klug, W.S.; Phillips, R. Morphology and interaction between lipid domains. Proc. Natl. Acad. Sci. USA 2009, 106, 13301–13306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skocaj, M.; Yu, Y.; Grundner, M.; Resnik, N.; Bedina Zavec, A.; Leonardi, A.; Krizaj, I.; Guella, G.; Macek, P.; Kreft, M.E.; et al. Characterisation of plasmalemmal shedding of vesicles induced by the cholesterol/sphingomyelin binding protein, ostreolysin A-mCherry. Biochim. Biophys. Acta 2016, 1858, 2882–2893. [Google Scholar] [CrossRef] [PubMed]
- D’Auria, L.; Van der Smissen, P.; Bruyneel, F.; Courtoy, P.J.; Tyteca, D. Segregation of fluorescent membrane lipids into distinct micrometric domains: Evidence for phase compartmentation of natural lipids? PLoS ONE 2011, 6, e17021. [Google Scholar] [CrossRef] [PubMed]
- Leonard, C.; Conrard, L.; Guthmann, M.; Pollet, H.; Carquin, M.; Vermylen, C.; Gailly, P.; Van Der Smissen, P.; Mingeot-Leclercq, M.P.; Tyteca, D. Contribution of plasma membrane lipid domains to red blood cell (re)shaping. Sci. Rep. 2017, 7, 4264. [Google Scholar] [CrossRef] [PubMed]
- Conrard, L.; Stommen, A.; Steinkhühler, J.; Dimova, R.; Pollet, H.; Tyteca, D. Spatial relationship and functional relevance of three lipid domain populations at the erythrocyte surface. Submitted.
- Mikhalyov, I.; Samsonov, A. Lipid raft detecting in membranes of live erythrocytes. Biochim. Biophys. Acta 2011, 1808, 1930–1939. [Google Scholar] [CrossRef] [PubMed]
- Murate, M.; Abe, M.; Kasahara, K.; Iwabuchi, K.; Umeda, M.; Kobayashi, T. Transbilayer distribution of lipids at nano scale. J. Cell Sci. 2015, 128, 1627–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montes, L.R.; Lopez, D.J.; Sot, J.; Bagatolli, L.A.; Stonehouse, M.J.; Vasil, M.L.; Wu, B.X.; Hannun, Y.A.; Goni, F.M.; Alonso, A. Ceramide-enriched membrane domains in red blood cells and the mechanism of sphingomyelinase-induced hot-cold hemolysis. Biochemistry 2008, 47, 11222–11230. [Google Scholar] [CrossRef] [PubMed]
- Gousset, K.; Wolkers, W.F.; Tsvetkova, N.M.; Oliver, A.E.; Field, C.L.; Walker, N.J.; Crowe, J.H.; Tablin, F. Evidence for a physiological role for membrane rafts in human platelets. J. Cell Physiol. 2002, 190, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Gaus, K.; Gratton, E.; Kable, E.P.; Jones, A.S.; Gelissen, I.; Kritharides, L.; Jessup, W. Visualizing lipid structure and raft domains in living cells with two-photon microscopy. Proc. Natl. Acad. Sci. USA 2003, 100, 15554–15559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Mouton, C.; Abad, J.L.; Mira, E.; Lacalle, R.A.; Gallardo, E.; Jimenez-Baranda, S.; Illa, I.; Bernad, A.; Manes, S.; Martinez, A.C. Segregation of leading-edge and uropod components into specific lipid rafts during T cell polarization. Proc. Natl. Acad. Sci. USA 2001, 98, 9642–9647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekyalongo, R.C.; Nakayama, H.; Kina, K.; Kaga, N.; Iwabuchi, K. Organization and functions of glycolipid-enriched microdomains in phagocytes. Biochim. Biophys. Acta 2015, 1851, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, H.; Abe, M.; Dedecker, P.; Makino, A.; Rocha, S.; Ohno-Iwashita, Y.; Hofkens, J.; Kobayashi, T.; Miyawaki, A. Fluorescent probes for superresolution imaging of lipid domains on the plasma membrane. Chem. Sci. 2011, 2, 1548–1553. [Google Scholar] [CrossRef]
- Rungaldier, S.; Oberwagner, W.; Salzer, U.; Csaszar, E.; Prohaska, R. Stomatin interacts with GLUT1/SLC2A1, band 3/SLC4A1, and aquaporin-1 in human erythrocyte membrane domains. Biochim. Biophys. Acta 2013, 1828, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Salzer, U.; Prohaska, R. Stomatin, flotillin-1, and flotillin-2 are major integral proteins of erythrocyte lipid rafts. Blood 2001, 97, 1141–1143. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.C.; Samuel, B.U.; Harrison, T.; Speicher, K.D.; Speicher, D.W.; Reid, M.E.; Prohaska, R.; Low, P.S.; Tanner, M.J.; Mohandas, N.; et al. Erythrocyte detergent-resistant membrane proteins: Their characterization and selective uptake during malarial infection. Blood 2004, 103, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Zhao, W.; Shang, X.; Jiang, J.; Ji, H.; Tang, Z.; Wang, H. Direct evidence of lipid rafts by in situ atomic force microscopy. Small 2012, 8, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Dumitru, A.C.; Poncin, M.A.; Conrard, L.; Dufrêne, Y.; Tyteca, D.; Alsteens, D. Nanoscale membrane architecture of healthy and pathological red blood cells. Nanoscale Horiz. 2018, 3, 293–304. [Google Scholar] [CrossRef]
- Lutz, H.U.; Barber, R.; McGuire, R.F. Glycoprotein-enriched vesicles from sheep erythrocyte ghosts obtained by spontaneous vesiculation. J. Biol. Chem. 1976, 251, 3500–3510. [Google Scholar] [PubMed]
- Salzer, U.H.; Hunger, U.; Prohaska, R. Chapter three: Insights in the organization and dynamics of erythrocyte lipid rafts. Adv. Planar Lipid Bilayers Liposomes 2008, 6, 49–80. [Google Scholar]
- Civenni, G.; Test, S.T.; Brodbeck, U.; Butikofer, P. In vitro incorporation of GPI-anchored proteins into human erythrocytes and their fate in the membrane. Blood 1998, 91, 1784–1792. [Google Scholar] [PubMed]
- Whitlow, M.; Iida, K.; Marshall, P.; Silber, R.; Nussenzweig, V. Cells lacking glycan phosphatidylinositol-linked proteins have impaired ability to vesiculate. Blood 1993, 81, 510–516. [Google Scholar] [PubMed]
- Wilkinson, D.K.; Turner, E.J.; Parkin, E.T.; Garner, A.E.; Harrison, P.J.; Crawford, M.; Stewart, G.W.; Hooper, N.M. Membrane raft actin deficiency and altered Ca2+-induced vesiculation in stomatin-deficient overhydrated hereditary stomatocytosis. Biochim. Biophys. Acta 2008, 1778, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Santos, N.C.; Martins-Silva, J.; Saldanha, C. Gramicidin D and dithiothreitol effects on erythrocyte exovesiculation. Cell Biochem. Biophys. 2005, 43, 419–430. [Google Scholar] [CrossRef]
- Kunzelmann-Marche, C.; Freyssinet, J.M.; Martinez, M.C. Loss of plasma membrane phospholipid asymmetry requires raft integrity. Role of transient receptor potential channels and ERK pathway. J. Biol. Chem. 2002, 277, 19876–19881. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.J.; Gibbons, E.; Bailey, R.W.; Fairbourn, J.; Nguyen, T.; Smith, S.K.; Best, K.B.; Nelson, J.; Judd, A.M.; Bell, J.D. The influence of membrane physical properties on microvesicle release in human erythrocytes. PMC Biophys. 2009, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Dorahy, D.J.; Lincz, L.F.; Meldrum, C.J.; Burns, G.F. Biochemical isolation of a membrane microdomain from resting platelets highly enriched in the plasma membrane glycoprotein CD36. Biochem. J. 1996, 319, 67–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bali, R.; Savino, L.; Ramirez, D.A.; Tsvetkova, N.M.; Bagatolli, L.; Tablin, F.; Crowe, J.H.; Leidy, C. Macroscopic domain formation during cooling in the platelet plasma membrane: An issue of low cholesterol content. Biochim. Biophys. Acta 2009, 1788, 1229–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodin, S.; Tronchere, H.; Payrastre, B. Lipid rafts are critical membrane domains in blood platelet activation processes. Biochim. Biophys. Acta 2003, 1610, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Bodin, S.; Giuriato, S.; Ragab, J.; Humbel, B.M.; Viala, C.; Vieu, C.; Chap, H.; Payrastre, B. Production of phosphatidylinositol 3,4,5-trisphosphate and phosphatidic acid in platelet rafts: Evidence for a critical role of cholesterol-enriched domains in human platelet activation. Biochemistry 2001, 40, 15290–15299. [Google Scholar] [CrossRef] [PubMed]
- Caroni, P. New EMBO members’ review: Actin cytoskeleton regulation through modulation of PI(4,5)P(2) rafts. EMBO J. 2001, 20, 4332–4336. [Google Scholar] [CrossRef] [PubMed]
- Flaumenhaft, R. Formation and fate of platelet microparticles. Blood Cells Mol. Dis. 2006, 36, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Mairhofer, M.; Steiner, M.; Mosgoeller, W.; Prohaska, R.; Salzer, U. Stomatin is a major lipid-raft component of platelet alpha granules. Blood 2002, 100, 897–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Malcor, J.M.; Harper, M.T. Lipid rafts are essential for release of phosphatidylserine-exposing extracellular vesicles from platelets. Sci. Rep. 2018, 8, 9987. [Google Scholar] [CrossRef] [PubMed]
- Larive, R.M.; Baisamy, L.; Urbach, S.; Coopman, P.; Bettache, N. Cell membrane extensions, generated by mechanical constraint, are associated with a sustained lipid raft patching and an increased cell signaling. Biochim. Biophys. Acta 2010, 1798, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Heijnen, H.F.; Van Lier, M.; Waaijenborg, S.; Ohno-Iwashita, Y.; Waheed, A.A.; Inomata, M.; Gorter, G.; Mobius, W.; Akkerman, J.W.; Slot, J.W. Concentration of rafts in platelet filopodia correlates with recruitment of c-Src and CD63 to these domains. J. Thromb. Haemost. 2003, 1, 1161–1173. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, D.J.; Rozenvayn, N.; Flaumenhaft, R. Phosphatidylinositol 4,5-bisphosphate regulates activation-induced platelet microparticle formation. Biochemistry 2005, 44, 6361–6370. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Mouton, C.; Lacalle, R.A.; Mira, E.; Jimenez-Baranda, S.; Barber, D.F.; Carrera, A.C.; Martinez, A.C.; Manes, S. Dynamic redistribution of raft domains as an organizing platform for signaling during cell chemotaxis. J. Cell Biol. 2004, 164, 759–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seveau, S.; Eddy, R.J.; Maxfield, F.R.; Pierini, L.M. Cytoskeleton-dependent membrane domain segregation during neutrophil polarization. Mol. Biol. Cell 2001, 12, 3550–3562. [Google Scholar] [CrossRef] [PubMed]
- Millan, J.; Montoya, M.C.; Sancho, D.; Sanchez-Madrid, F.; Alonso, M.A. Lipid rafts mediate biosynthetic transport to the T lymphocyte uropod subdomain and are necessary for uropod integrity and function. Blood 2002, 99, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Pierini, L.M.; Eddy, R.J.; Fuortes, M.; Seveau, S.; Casulo, C.; Maxfield, F.R. Membrane lipid organization is critical for human neutrophil polarization. J. Biol. Chem. 2003, 278, 10831–10841. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Yoshizaki, F.; Prinetti, A.; Sonnino, S.; Mauri, L.; Takamori, K.; Ogawa, H.; Iwabuchi, K. Lyn-coupled LacCer-enriched lipid rafts are required for CD11b/CD18-mediated neutrophil phagocytosis of nonopsonized microorganisms. J. Leukoc. Biol. 2008, 83, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.K.; Dallo, S.F.; Detmar, A.L.; Osmulski, P.; Gaczynska, M.; Huang, T.H.; Ramasubramanian, A.K. Cellular cholesterol regulates monocyte deformation. J. Biomech. 2017, 52, 83–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothmeier, A.S.; Marchese, P.; Petrich, B.G.; Furlan-Freguia, C.; Ginsberg, M.H.; Ruggeri, Z.M.; Ruf, W. Caspase-1-mediated pathway promotes generation of thromboinflammatory microparticles. J. Clin. Investig. 2015, 125, 1471–1484. [Google Scholar] [CrossRef] [PubMed]
- Roduit, C.; van der Goot, F.G.; De Los Rios, P.; Yersin, A.; Steiner, P.; Dietler, G.; Catsicas, S.; Lafont, F.; Kasas, S. Elastic membrane heterogeneity of living cells revealed by stiff nanoscale membrane domains. Biophys. J. 2008, 94, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- Boggs, J.M.; Wang, H. Co-clustering of galactosylceramide and membrane proteins in oligodendrocyte membranes on interaction with polyvalent carbohydrate and prevention by an intact cytoskeleton. J. Neurosci. Res. 2004, 76, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Boggs, J.M.; Gao, W.; Zhao, J.; Park, H.J.; Liu, Y.; Basu, A. Participation of galactosylceramide and sulfatide in glycosynapses between oligodendrocyte or myelin membranes. FEBS Lett. 2010, 584, 1771–1778. [Google Scholar] [CrossRef] [PubMed]
- Gonnord, P.; Delarasse, C.; Auger, R.; Benihoud, K.; Prigent, M.; Cuif, M.H.; Lamaze, C.; Kanellopoulos, J.M. Palmitoylation of the P2X7 receptor, an ATP-gated channel, controls its expression and association with lipid rafts. FASEB J. 2009, 23, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Ohmi, Y.; Ohkawa, Y.; Yamauchi, Y.; Tajima, O.; Furukawa, K.; Furukawa, K. Essential roles of gangliosides in the formation and maintenance of membrane microdomains in brain tissues. Neurochem. Res. 2012, 37, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.; Eder, C. Importance of lipid rafts for lysophosphatidylcholine-induced caspase-1 activation and reactive oxygen species generation. Cell. Immunol. 2010, 265, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.A.; Halverson-Tamboli, R.A.; Rasenick, M.M. Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 2007, 8, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Decker, L.; ffrench-Constant, C. Lipid rafts and integrin activation regulate oligodendrocyte survival. J. Neurosci. 2004, 24, 3816–3825. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Fabelo, N.; Santpere, G.; Puig, B.; Marin, R.; Ferrer, I.; Diaz, M. Lipid alterations in lipid rafts from Alzheimer’s disease human brain cortex. J. Alzheimers Dis. 2010, 19, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Yuyama, K.; Mitsutake, S.; Igarashi, Y. Pathological roles of ceramide and its metabolites in metabolic syndrome and Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1841, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Hein, L.K.; Rozaklis, T.; Adams, M.K.; Hopwood, J.J.; Karageorgos, L. Lipid composition of microdomains is altered in neuronopathic Gaucher disease sheep brain and spleen. Mol. Genet. Metab. 2017, 121, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Kerbel, R.S.; Allison, A.C.; Rak, J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc. Natl. Acad. Sci. USA 2009, 106, 3794–3799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orsini, F.; Cremona, A.; Arosio, P.; Corsetto, P.A.; Montorfano, G.; Lascialfari, A.; Rizzo, A.M. Atomic force microscopy imaging of lipid rafts of human breast cancer cells. Biochim. Biophys. Acta 2012, 1818, 2943–2949. [Google Scholar] [CrossRef] [PubMed]
- Kiyokawa, E.; Baba, T.; Otsuka, N.; Makino, A.; Ohno, S.; Kobayashi, T. Spatial and functional heterogeneity of sphingolipid-rich membrane domains. J. Biol. Chem. 2005, 280, 24072–24084. [Google Scholar] [CrossRef] [PubMed]
- Kokkonen, N.; Khosrowabadi, E.; Hassinen, A.; Harrus, D.; Glumoff, T.; Kietzmann, T.; Kellokumpu, S. Abnormal Golgi pH Homeostasis in Cancer Cells Impairs Apical Targeting of Carcinoembryonic Antigen by Inhibiting Its Glycosyl-Phosphatidylinositol Anchor-Mediated Association with Lipid Rafts. Antioxid. Redox Signal. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hirpara, J.L.; Loh, T.; Ng, S.B.; Chng, W.J.; Pervaiz, S. Aberrant localization of apoptosis protease activating factor-1 in lipid raft sub-domains of diffuse large B cell lymphomas. Oncotarget 2016, 7, 83964–83975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavie, Y.; Fiucci, G.; Czarny, M.; Liscovitch, M. Changes in membrane microdomains and caveolae constituents in multidrug-resistant cancer cells. Lipids 1999, 34, S57–S63. [Google Scholar] [CrossRef] [PubMed]
- Ausili, A.; Martinez-Valera, P.; Torrecillas, A.; Gomez-Murcia, V.; de Godos, A.M.; Corbalan-Garcia, S.; Teruel, J.A.; Gomez Fernandez, J.C. Anticancer Agent Edelfosine Exhibits a High Affinity for Cholesterol and Disorganizes Liquid-Ordered Membrane Structures. Langmuir 2018, 34, 8333–8346. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; Gajate, C. Lipid rafts as major platforms for signaling regulation in cancer. Adv. Biol. Regul. 2015, 57, 130–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorent, J.H.; Quetin-Leclercq, J.; Mingeot-Leclercq, M.P. The amphiphilic nature of saponins and their effects on artificial and biological membranes and potential consequences for red blood and cancer cells. Org. Biomol. Chem. 2014, 12, 8803–8822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.K.; Lee, E.J.; Lee, S.H.; Koo, K.H.; Sung, J.Y.; Hwang, E.H.; Park, J.H.; Kim, C.W.; Jeong, K.C.; Park, B.K.; et al. Induction of apoptosis by the ginsenoside Rh2 by internalization of lipid rafts and caveolae and inactivation of Akt. Br. J. Pharmacol. 2010, 160, 1212–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, J.S.; Choo, H.J.; Cho, B.R.; Kim, H.M.; Kim, Y.N.; Ham, Y.M.; Ko, Y.G. Ginsenoside Rh2 induces ligand-independent Fas activation via lipid raft disruption. Biochem. Biophys. Res. Commun. 2009, 385, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, S.L.; Albert, M.; Paquot, A.; Muccioli, G.G.; Tyteca, D.; Mingeot-Leclercq, M.P. Membrane cholesterol delays cellular apoptosis induced by ginsenoside Rh2, a steroid saponin. Toxicol. Appl. Pharmacol. 2018, 352, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Van Blitterswijk, W.J.; Emmelot, P.; Hilkmann, H.A.; Hilgers, J.; Feltkamp, C.A. Rigid plasma-membrane-derived vesicles, enriched in tumour-associated surface antigens (MLr), occurring in the ascites fluid of a murine leukaemia (GRSL). Int. J. Cancer 1979, 23, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, C.; Gehrmann, M.; Madl, J.; Romer, W.; Multhoff, G.; Ebner, A. Molecular AFM imaging of Hsp70-1A association with dipalmitoyl phosphatidylserine reveals membrane blebbing in the presence of cholesterol. Cell Stress Chaperones 2018, 23, 673–683. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type/Body Fluid | Lipid Analysis | MV Size (nm) | % of Total Lipid Content (MV/Cell Ratio) | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|
| SM | Cer | PC | PS | PE | Chol/PLPs | ||||
| U87 glioblastoma cells | MS | 50–600 | 15% (2) | 0.7% (2.4) | 25% (0.9) | 17% (1) | 9.6% (0.9) | - | [88] |
| Huh7 hepatocellular carcinoma cells | MS | 50–600 | 15% (3.6) | 0.3% (1.5) | 27% (1.3) | 7.4% (0.6) | 6.8% (0.7) | - | [88] |
| Bone marrow derived stem cells | MS | 50–600 | 8.6% (1.6) | 0.5% (1.9) | 25% (1.2) | 8% (0.7) | 3.7% (0.5) | - | [88] |
| Placenta | MS | - | 37% | 1% | 15% | 17% | 2% | - | [89] |
| Plasma EVs (ratio on platelets) | TLC | - | 21% (1) | - | 59% (1.8) | 3.6% (0.3) | 9.4% (0.3) | - | [90] |
| Stored RBCs (53 ± 4 days) | MS | - | (1) | - | (1) | (7) | (0.8) | - | [91] |
| Stored RBCs (35–42 days) | MS | 100–300 | 33% (1) | - | 26% (1) | 10% (1) | 30% (1) | - | [92] |
| ATP-depleted RBCs | Enzymatic | ~200 | 26% (0.9) | - | 27% (0.9) | - | 25% (0.8) | (0.9) | [93] |
| Platelets | MS | - | 14% (1.3) | 0.7% (1.3) | 27% (0.7) | 17% (1) | 37% (1) | - | [94] |
| Platelets (ratio on PM platelets) | TLC | - | 25% (1.1) | - | 31% (1.1) | 14% (0.8) | 30% (1) | (1.5) | [95] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pollet, H.; Conrard, L.; Cloos, A.-S.; Tyteca, D. Plasma Membrane Lipid Domains as Platforms for Vesicle Biogenesis and Shedding? Biomolecules 2018, 8, 94. https://doi.org/10.3390/biom8030094
Pollet H, Conrard L, Cloos A-S, Tyteca D. Plasma Membrane Lipid Domains as Platforms for Vesicle Biogenesis and Shedding? Biomolecules. 2018; 8(3):94. https://doi.org/10.3390/biom8030094
Chicago/Turabian StylePollet, Hélène, Louise Conrard, Anne-Sophie Cloos, and Donatienne Tyteca. 2018. "Plasma Membrane Lipid Domains as Platforms for Vesicle Biogenesis and Shedding?" Biomolecules 8, no. 3: 94. https://doi.org/10.3390/biom8030094
APA StylePollet, H., Conrard, L., Cloos, A. -S., & Tyteca, D. (2018). Plasma Membrane Lipid Domains as Platforms for Vesicle Biogenesis and Shedding? Biomolecules, 8(3), 94. https://doi.org/10.3390/biom8030094