Mineralocorticoid Receptor and Aldosterone-Related Biomarkers of End-Organ Damage in Cardiometabolic Disease

 ,
, 
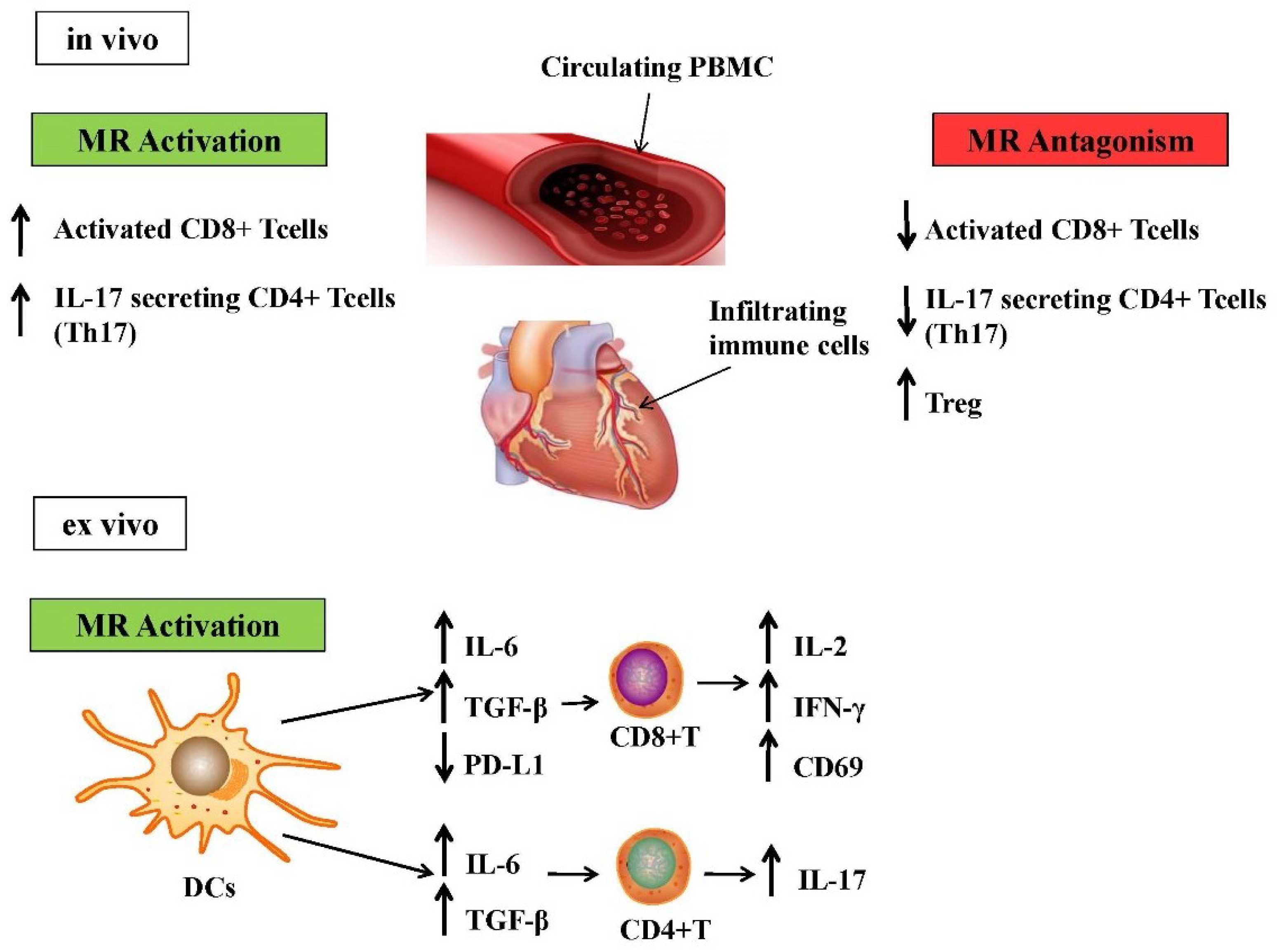{kind=link}
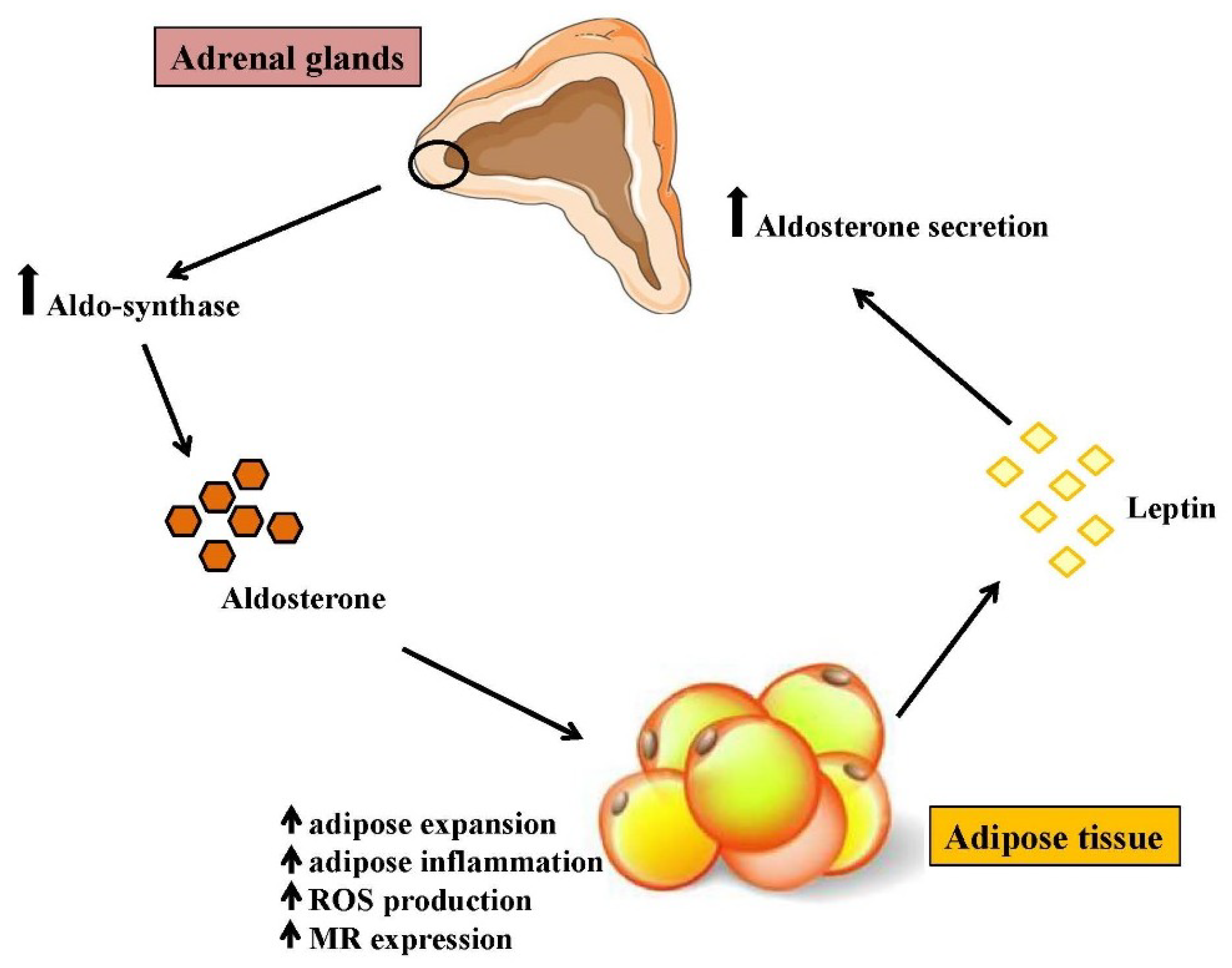{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cardio-Metabolic Effects of Altered Mineralocorticoid Receptor Activation
3. Contribution of Different Immune Cells Subsets to Aldosterone-Induced Inflammation
4. Aldosterone as a Novel Marker of Obesity
5. Mineralocorticoid Receptor Downstream Molecules: Novel Biomarkers of Cardiometabolic Diseases?
5.1. Neutrophil Gelatinase-Associated Lipocalin Protein
5.2. Galectin-3
5.3. Lipocalin-Like Prostaglandin D2 Synthase
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rossier, B.C. Hormonal regulation of the epithelial sodium channel ENaC: N or Po? J. Gen. Physiol. 2002, 120, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Rossier, B.C. Epithelial sodium channel (ENaC) and the control of blood pressure. Curr. Opin. Pharmacol. 2014, 15, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Rossier, B.C.; Staub, O.; Hummler, E. Genetic dissection of sodium and potassium transport along the aldosterone-sensitive distal nephron: Importance in the control of blood pressure and hypertension. FEBS Lett. 2013, 587, 1929–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arriza, J.L.; Weinberger, C.; Cerelli, G.; Glaser, T.M.; Handelin, B.L.; Housman, D.E.; Evans, R.M. Cloning of human mineralocorticoid receptor complementary DNA: Structural and functional kinship with the glucocorticoid receptor. Science 1987, 237, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Marzolla, V.; Armani, A.; Feraco, A.; De Martino, M.; Fabbri, A.; Rosano, G.; Caprio, M. Mineralocorticoid receptor in adipocytes and macrophages: A promising target to fight metabolic syndrome. Steroids 2014, 91, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Lombes, M.; Oblin, M.E.; Gasc, J.M.; Baulieu, E.E.; Farman, N.; Bonvalet, J.P. Immunohistochemical and biochemical evidence for a cardiovascular mineralocorticoid receptor. Circ. Res. 1992, 71, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Lombes, M.; Alfaidy, N.; Eugene, E.; Lessana, A.; Farman, N.; Bonvalet, J.P. Prerequisite for cardiac aldosterone action. Mineralocorticoid receptor and 11 beta-hydroxysteroid dehydrogenase in the human heart. Circulation 1995, 92, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Rondinone, C.M.; Rodbard, D.; Baker, M.E. Aldosterone stimulated differentiation of mouse 3T3-L1 cells into adipocytes. Endocrinology 1993, 132, 2421–2426. [Google Scholar] [CrossRef] [PubMed]
- DuPont, J.J.; Jaffe, I.Z. 30 years of the mineralocorticoid receptor: The role of the mineralocorticoid receptor in the vasculature. J. Endocrinol. 2017, 234, T67–T82. [Google Scholar] [CrossRef] [PubMed]
- Caprio, M.; Newfell, B.G.; La Sala, A.; Baur, W.; Fabbri, A.; Rosano, G.; Mendelsohn, M.E.; Jaffe, I.Z. Functional mineralocorticoid receptors in human vascular endothelial cells regulate intercellular adhesion molecule-1 expression and promote leukocyte adhesion. Circ. Res. 2008, 102, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Armani, A.; Marzolla, V.; Fabbri, A.; Caprio, M. Cellular mechanisms of MR regulation of adipose tissue physiology and pathophysiology. J. Mol. Endocrinol. 2015, 55, R1–R10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funder, J.W. Aldosterone and mineralocorticoid receptors-physiology and pathophysiology. Int. J. Mol. Sci. 2017, 18, 1032. [Google Scholar] [CrossRef] [PubMed]
- Bentley-Lewis, R.; Adler, G.K.; Perlstein, T.; Seely, E.W.; Hopkins, P.N.; Williams, G.H.; Garg, R. Body mass index predicts aldosterone production in normotensive adults on a high-salt diet. J. Clin. Endocrinol. Metab. 2007, 92, 4472–4475. [Google Scholar] [CrossRef] [PubMed]
- Goodfriend, T.L.; Kelley, D.E.; Goodpaster, B.H.; Winters, S.J. Visceral obesity and insulin resistance are associated with plasma aldosterone levels in women. Obes. Res. 1999, 7, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.S.; Williams, G.H. 50th anniversary of aldosterone. J. Clin. Endocrinol. Metab. 2003, 88, 2364–2372. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, I.Z.; Mendelsohn, M.E. Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circ. Res. 2005, 96, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, I.Z.; Tintut, Y.; Newfell, B.G.; Demer, L.L.; Mendelsohn, M.E. Mineralocorticoid receptor activation promotes vascular cell calcification. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Park, J.B.; Schiffrin, E.L. Cardiac and vascular fibrosis and hypertrophy in aldosterone-infused rats: Role of endothelin-1. Am. J. Hypertens. 2002, 15, 164–169. [Google Scholar] [CrossRef]
- Harvey, A.P.; Montezano, A.C.; Hood, K.Y.; Lopes, R.A.; Rios, F.; Ceravolo, G.; Graham, D.; Touyz, R.M. Vascular dysfunction and fibrosis in stroke-prone spontaneously hypertensive rats: The aldosterone-mineralocorticoid receptor-Nox1 axis. Life Sci. 2017, 179, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, E.; Gomez-Sanchez, C.E. The multifaceted mineralocorticoid receptor. Compr. Physiol. 2014, 4, 965–994. [Google Scholar] [PubMed]
- Shibata, S.; Nagase, M.; Yoshida, S.; Kawarazaki, W.; Kurihara, H.; Tanaka, H.; Miyoshi, J.; Takai, Y.; Fujita, T. Modification of mineralocorticoid receptor function by Rac1 GTPase: Implication in proteinuric kidney disease. Nat. Med. 2008, 14, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Nagase, M.; Fujita, T. Role of Rac1-mineralocorticoid-receptor signalling in renal and cardiac disease. Nat. Rev. Nephrol. 2013, 9, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Ayuzawa, N.; Nagase, M.; Ueda, K.; Nishimoto, M.; Kawarazaki, W.; Marumo, T.; Aiba, A.; Sakurai, T.; Shindo, T.; Fujita, T. Rac1-mediated activation of mineralocorticoid receptor in pressure overload-induced cardiac injury. Hypertension 2016, 67, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Nagase, M.; Ayuzawa, N.; Kawarazaki, W.; Ishizawa, K.; Ueda, K.; Yoshida, S.; Fujita, T. Oxidative stress causes mineralocorticoid receptor activation in rat cardiomyocytes: Role of small GTPase Rac1. Hypertension 2012, 59, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Castillo, A.; Carvajal, C.A.; Campino, C.; Hill, C.; Allende, F.; Vecchiola, A.; Carrasco, C.; Bancalari, R.; Valdivia, C.; Lagos, C.; et al. The expression of Rac1 and mineralocorticoid pathway-dependent genes are associated with different responses to salt intake. Am. J. Hypertens. 2015, 28, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Ruhs, S.; Nolze, A.; Hubschmann, R.; Grossmann, C. 30 years of the mineralocorticoid receptor: Nongenomic effects via the mineralocorticoid receptor. J. Endocrinol. 2017, 234, T107–T124. [Google Scholar] [CrossRef] [PubMed]
- Wehling, M. Rapid actions of aldosterone revisited: Receptors in the limelight. J. Steroid Biochem. Mol. Biol. 2018, 176, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Gekle, M.; Mildenberger, S.; Freudinger, R.; Grossmann, C. Altered collagen homeostasis in human aortic smooth muscle cells (HAoSMCs) induced by aldosterone. Pflug. Arch. 2007, 454, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.L.; Nikolic-Paterson, D.J.; Ma, F.Y.; Tesch, G.H. Aldosterone induces kidney fibroblast proliferation via activation of growth factor receptors and PI3K/MAPK signalling. Nephron Exp. Nephrol. 2012, 120, e115–e122. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.W.; Allenhofer, L.; Monticone, R.; Spinetti, G.; Gekle, M.; Wang, M.; Lakatta, E.G. Elevated mineralocorticoid receptor activity in aged rat vascular smooth muscle cells promotes a proinflammatory phenotype via extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase and epidermal growth factor receptor-dependent pathways. Hypertension 2010, 55, 1476–1483. [Google Scholar] [PubMed]
- Zennaro, M.C.; Caprio, M.; Feve, B. Mineralocorticoid receptors in the metabolic syndrome. Trends Endocrinol. Metab. 2009, 20, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Marney, A.M.; Brown, N.J. Aldosterone and end-organ damage. Clin. Sci. (Lond.) 2007, 113, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Catena, C.; Colussi, G.; Sechi, L.A. Aldosterone, organ damage and dietary salt. Clin. Exp. Pharmacol. Physiol. 2013, 40, 922–928. [Google Scholar] [CrossRef] [PubMed]
- Brilla, C.G.; Weber, K.T. Mineralocorticoid excess, dietary sodium, and myocardial fibrosis. J. Lab. Clin. Med. 1992, 120, 893–901. [Google Scholar] [PubMed]
- Rossi, G.P. Primary aldosteronism: A needle in a haystack or a yellow cab on fifth avenue? Curr. Hypertens. Rep. 2004, 6, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Milliez, P.; Girerd, X.; Plouin, P.F.; Blacher, J.; Safar, M.E.; Mourad, J.J. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J. Am. Coll. Cardiol. 2005, 45, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Conn, J.W. Hypertension, the potassium ion and impaired carbohydrate tolerance. N. Engl. J. Med. 1965, 273, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Kidambi, S.; Kotchen, J.M.; Grim, C.E.; Raff, H.; Mao, J.; Singh, R.J.; Kotchen, T.A. Association of adrenal steroids with hypertension and the metabolic syndrome in blacks. Hypertension 2007, 49, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Fallo, F.; Veglio, F.; Bertello, C.; Sonino, N.; Della Mea, P.; Ermani, M.; Rabbia, F.; Federspil, G.; Mulatero, P. Prevalence and characteristics of the metabolic syndrome in primary aldosteronism. J. Clin. Endocrinol. Metab. 2006, 91, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.M.; Zhou, D.C.; Gu, H.F.; Qiao, Q.Y.; Fu, S.K.; Liu, X.L.; Pan, Y. Antioxidant N-acetylcysteine protects pancreatic β-cells against aldosterone-induced oxidative stress and apoptosis in female db/db mice and insulin-producing MIN6 cells. Endocrinology 2013, 154, 4068–4077. [Google Scholar] [CrossRef] [PubMed]
- Luther, J.M. Effects of aldosterone on insulin sensitivity and secretion. Steroids 2014, 91, 54–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacchetti, G.; Ronconi, V.; Turchi, F.; Agostinelli, L.; Mantero, F.; Rilli, S.; Boscaro, M. Aldosterone as a key mediator of the cardiometabolic syndrome in primary aldosteronism: An observational study. J. Hypertens. 2007, 25, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Sindelka, G.; Widimsky, J.; Haas, T.; Prazny, M.; Hilgertova, J.; Skrha, J. Insulin action in primary hyperaldosteronism before and after surgical or pharmacological treatment. Exp. Clin. Endocrinol. Diabetes 2000, 108, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Widimsky, J., Jr.; Sindelka, G.; Haas, T.; Prazny, M.; Hilgertova, J.; Skrha, J. Impaired insulin action in primary hyperaldosteronism. Physiol. Res. 2000, 49, 241–244. [Google Scholar] [PubMed]
- Catena, C.; Lapenna, R.; Baroselli, S.; Nadalini, E.; Colussi, G.; Novello, M.; Favret, G.; Melis, A.; Cavarape, A.; Sechi, L.A. Insulin sensitivity in patients with primary aldosteronism: A follow-up study. J. Clin. Endocrinol. Metab. 2006, 91, 3457–3463. [Google Scholar] [CrossRef] [PubMed]
- Ingelsson, E.; Pencina, M.J.; Tofler, G.H.; Benjamin, E.J.; Lanier, K.J.; Jacques, P.F.; Fox, C.S.; Meigs, J.B.; Levy, D.; Larson, M.G.; et al. Multimarker approach to evaluate the incidence of the metabolic syndrome and longitudinal changes in metabolic risk factors: The framingham offspring study. Circulation 2007, 116, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Freel, E.M.; Mark, P.B.; Weir, R.A.; McQuarrie, E.P.; Allan, K.; Dargie, H.J.; McClure, J.D.; Jardine, A.G.; Davies, E.; Connell, J.M. Demonstration of blood pressure-independent noninfarct myocardial fibrosis in primary aldosteronism: A cardiac magnetic resonance imaging study. Circ. Cardiovasc. Imaging 2012, 5, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Zennaro, M.C.; Boulkroun, S.; Fernandes-Rosa, F. An update on novel mechanisms of primary aldosteronism. J. Endocrinol. 2015, 224, R63–R77. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.P.; Sacchetto, A.; Pavan, E.; Palatini, P.; Graniero, G.R.; Canali, C.; Pessina, A.C. Remodeling of the left ventricle in primary aldosteronism due to conn’s adenoma. Circulation 1997, 95, 1471–1478. [Google Scholar] [CrossRef] [PubMed]
- Hillaert, M.A.; Lentjes, E.G.; Beygui, F.; Kemperman, H.; Asselbergs, F.W.; Nathoe, H.M.; Agostoni, P.; Voskuil, M.; Ivanes, F.; Jude, B.; et al. Measuring and targeting aldosterone and renin in atherosclerosis—A review of clinical data. Am. Heart J. 2011, 162, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Reil, J.C.; Hohl, M.; Selejan, S.; Lipp, P.; Drautz, F.; Kazakow, A.; Munz, B.M.; Muller, P.; Steendijk, P.; Reil, G.H.; et al. Aldosterone promotes atrial fibrillation. Eur. Heart J. 2012, 33, 2098–2108. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.C.; Waters, D.D.; et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Zannad, F.; McMurray, J.J.; Krum, H.; van Veldhuisen, D.J.; Swedberg, K.; Shi, H.; Vincent, J.; Pocock, S.J.; Pitt, B.; Group, E.-H.S. Eplerenone in patients with systolic heart failure and mild symptoms. N. Engl. J. Med. 2011, 364, 11–21. [Google Scholar] [CrossRef] [PubMed]
- MacFadyen, R.J.; Barr, C.S.; Struthers, A.D. Aldosterone blockade reduces vascular collagen turnover, improves heart rate variability and reduces early morning rise in heart rate in heart failure patients. Cardiovasc. Res. 1997, 35, 30–34. [Google Scholar] [CrossRef] [Green Version]
- Gerling, I.C.; Sun, Y.; Ahokas, R.A.; Wodi, L.A.; Bhattacharya, S.K.; Warrington, K.J.; Postlethwaite, A.E.; Weber, K.T. Aldosteronism: An immunostimulatory state precedes proinflammatory/fibrogenic cardiac phenotype. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H813–H821. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, J.; Lu, L.; Chen, S.S.; Quinn, M.T.; Weber, K.T. Aldosterone-induced inflammation in the rat heart: Role of oxidative stress. Am. J. Pathol. 2002, 161, 1773–1781. [Google Scholar] [CrossRef]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marvar, P.J.; Thabet, S.R.; Guzik, T.J.; Lob, H.E.; McCann, L.A.; Weyand, C.; Gordon, F.J.; Harrison, D.G. Central and peripheral mechanisms of t-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ. Res. 2010, 107, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Usher, M.G.; Duan, S.Z.; Ivaschenko, C.Y.; Frieler, R.A.; Berger, S.; Schutz, G.; Lumeng, C.N.; Mortensen, R.M. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J. Clin. Investig. 2010, 120, 3350–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selye, H.; Stone, H.; Timiras, P.S.; Schaffenburg, C. Influence of sodium chloride upon the actions of desoxycorticosteron acetate. Am. Heart J. 1949, 37, 1009–1016. [Google Scholar] [CrossRef]
- Funder, J.W. Mineralocorticoid receptor activation and oxidative stress. Hypertension 2007, 50, 840–841. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.J. Aldosterone and vascular inflammation. Hypertension 2008, 51, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Moraes, L.A.; Paul-Clark, M.J.; Rickman, A.; Flower, R.J.; Goulding, N.J.; Perretti, M. Ligand-specific glucocorticoid receptor activation in human platelets. Blood 2005, 106, 4167–4175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lother, A.; Furst, D.; Bergemann, S.; Gilsbach, R.; Grahammer, F.; Huber, T.B.; Hilgendorf, I.; Bode, C.; Moser, M.; Hein, L. Deoxycorticosterone acetate/salt-induced cardiac but not renal injury is mediated by endothelial mineralocorticoid receptors independently from blood pressure. Hypertension 2016, 67, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Marzolla, V.; Armani, A.; Mammi, C.; Moss, M.E.; Pagliarini, V.; Pontecorvo, L.; Antelmi, A.; Fabbri, A.; Rosano, G.; Jaffe, I.Z.; et al. Essential role of ICAM-1 in aldosterone-induced atherosclerosis. Int. J. Cardiol. 2017, 232, 233–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caprio, M.; Mammi, C.; Jaffe, I.Z.; Zennaro, M.C.; Aversa, A.; Mendelsohn, M.E.; Fabbri, A.; Rosano, G.M. The mineralocorticoid receptor in endothelial physiology and disease: Novel concepts in the understanding of erectile dysfunction. Curr. Pharm. Des. 2008, 14, 3749–3757. [Google Scholar] [CrossRef] [PubMed]
- Young, M.J.; Rickard, A.J. Mechanisms of mineralocorticoid salt-induced hypertension and cardiac fibrosis. Mol. Cell. Endocrinol. 2012, 350, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Rickard, A.J.; Morgan, J.; Chrissobolis, S.; Miller, A.A.; Sobey, C.G.; Young, M.J. Endothelial cell mineralocorticoid receptors regulate deoxycorticosterone/salt-mediated cardiac remodeling and vascular reactivity but not blood pressure. Hypertension 2014, 63, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Tesch, G.H.; Young, M.J. Mineralocorticoid receptor signaling as a therapeutic target for renal and cardiac fibrosis. Front. Pharmacol. 2017, 8, 313. [Google Scholar] [CrossRef] [PubMed]
- Kadoya, H.; Satoh, M.; Sasaki, T.; Taniguchi, S.; Takahashi, M.; Kashihara, N. Excess aldosterone is a critical danger signal for inflammasome activation in the development of renal fibrosis in mice. FASEB J. 2015, 29, 3899–3910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, M.; Chen, Y.; Zhao, M.; Zhang, Y.; He, J.C.; Huang, S.; Jia, Z.; Zhang, A. NLRP3 inflammasome activation contributes to aldosterone-induced podocyte injury. Am. J. Physiol. Renal. Physiol. 2017, 312, F556–F564. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Ishikawa, A.; Watanabe, E.; Nakamura, Y.; Aruga, Y.; Hasegawa, H.; Onogi, Y.; Honda, H.; Nagai, Y.; Takatsu, K.; et al. Eplerenone prevented obesity-induced inflammasome activation and glucose intolerance. J. Endocrinol. 2017, 235, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, U.G. Evidence for an initial, thymus independent and a chronic, thymus dependent phase of doca and salt hypertension in mice. Acta Pathol. Microbiol. Scand. A 1976, 84, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Besedovsky, L.; Linz, B.; Born, J.; Lange, T. Mineralocorticoid receptor signaling reduces numbers of circulating human naïve T cells and increases their CD62L, CCR7, and CXCR4 expression. Eur. J. Immunol. 2014, 44, 1759–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhur, M.S.; Lob, H.E.; McCann, L.A.; Iwakura, Y.; Blinder, Y.; Guzik, T.J.; Harrison, D.G. Interleukin 17 Promotes Angiotensin II-Induced Hypertension and Vascular Dysfunction. Hypertension 2010, 55, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.C.; Yu, H.T.; Lim, B.J.; Koh, M.J.; Lee, J.; Chang, D.Y.; Choi, Y.S.; Lee, S.H.; Kang, S.M.; Jang, Y.; et al. Immunosenescent CD8+ T Cells and C-X-C Chemokine Receptor Type 3 Chemokines are Increased in Human Hypertension. Hypertension 2013, 62, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Amador, C.A.; Barrientos, V.; Pena, J.; Herrada, A.A.; Gonzalez, M.; Valdes, S.; Carrasco, L.; Alzamora, R.; Figueroa, F.; Kalergis, A.M.; et al. Spironolactone decreases doca-salt-induced organ damage by blocking the activation of T helper 17 and the downregulation of regulatory T lymphocytes. Hypertension 2014, 63, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Sun, X.N.; Zeng, M.R.; Zheng, X.J.; Zhang, Y.Y.; Wan, Q.; Zhang, W.C.; Shi, C.; Du, L.J.; Ai, T.J.; et al. Mineralocorticoid receptor deficiency in T cells attenuates pressure overload-induced cardiac hypertrophy and dysfunction through modulating T-cell activation. Hypertension 2017, 70, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Herrada, A.A.; Contreras, F.J.; Marini, N.P.; Amador, C.A.; Gonzalez, P.A.; Cortes, C.M.; Riedel, C.A.; Carvajal, C.A.; Figueroa, F.; Michea, L.F.; et al. Aldosterone promotes autoimmune damage by enhancing Th17-mediated immunity. J. Immunol. 2010, 184, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Steinman, R.M. Dendritic cells: Specialized and regulated antigen processing machines. Cell 2001, 106, 255–258. [Google Scholar] [CrossRef]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory t cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chung, Y.; Bishop, C.; Daugherty, B.; Chute, H.; Holst, P.; Kurahara, C.; Lott, F.; Sun, N.; Welcher, A.A.; et al. Regulation of T cell activation and tolerance by PDL2. Proc. Natl. Acad. Sci. USA 2006, 103, 11695–11700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elrefaei, M.; Baker, C.A.; Jones, N.G.; Bangsberg, D.R.; Cao, H. Presence of suppressor HIV-specific CD8+ T cells is associated with increased PD-1 expression on effector CD8+ T cells. J. Immunol. 2008, 180, 7757–7763. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Gavrilova, O.; Pack, S.; Jou, W.; Mullen, S.; Sumner, A.E.; Cushman, S.W.; Periwal, V. Hypertrophy and/or hyperplasia: Dynamics of adipose tissue growth. PLoS Comput. Biol. 2009, 5, e1000324. [Google Scholar] [CrossRef] [PubMed]
- Sartipy, P.; Loskutoff, D.J. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 7265–7270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruun, J.M.; Pedersen, S.B.; Richelsen, B. Regulation of interleukin 8 production and gene expression in human adipose tissue in vitro. J. Clin. Endocrinol. Metab. 2001, 86, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Obin, M.S. Obesity and the role of adipose tissue in inflammation and metabolism. Am. J. Clin. Nutr. 2006, 83, 461S–465S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Gao, Z.; Ye, J. Regulation of 11β-HSD1 expression during adipose tissue expansion by hypoxia through different activities of NF-κB and HIF-1α. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1035–E1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppmann, J.; Perwitz, N.; Meier, B.; Fasshauer, M.; Hadaschik, D.; Lehnert, H.; Klein, J. The balance between gluco- and mineralo-corticoid action critically determines inflammatory adipocyte responses. J. Endocrinol. 2010, 204, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Hirata, A.; Maeda, N.; Hiuge, A.; Hibuse, T.; Fujita, K.; Okada, T.; Kihara, S.; Funahashi, T.; Shimomura, I. Blockade of mineralocorticoid receptor reverses adipocyte dysfunction and insulin resistance in obese mice. Cardiovasc. Res. 2009, 84, 164–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labuzek, K.; Liber, S.; Buldak, L.; Machnik, G.; Liber, J.; Okopien, B. Eplerenone promotes alternative activation in human monocyte-derived macrophages. Pharmacol. Rep. 2013, 65, 226–234. [Google Scholar] [CrossRef]
- Armani, A.; Cinti, F.; Marzolla, V.; Morgan, J.; Cranston, G.A.; Antelmi, A.; Carpinelli, G.; Canese, R.; Pagotto, U.; Quarta, C.; et al. Mineralocorticoid receptor antagonism induces browning of white adipose tissue through impairment of autophagy and prevents adipocyte dysfunction in high-fat-diet-fed mice. FASEB J. 2014, 28, 3745–3757. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Ricchiuti, V.; Lian, B.Q.; Yao, T.M.; Coutinho, P.; Romero, J.R.; Li, J.; Williams, G.H.; Adler, G.K. Mineralocorticoid Receptor Blockade Reverses Obesity-Related Changes in Expression of Adiponectin, Peroxisome Proliferator-Activated Receptor-gamma, and Proinflammatory Adipokines. Circulation 2008, 117, 2253–2261. [Google Scholar] [CrossRef] [PubMed]
- Caprio, M.; Feve, B.; Claes, A.; Viengchareun, S.; Lombes, M.; Zennaro, M.C. Pivotal role of the mineralocorticoid receptor in corticosteroid-induced adipogenesis. FASEB J. 2007, 21, 2185–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, E.D.; Hsu, C.H.; Wang, X.; Sakai, S.; Freeman, M.W.; Gonzalez, F.J.; Spiegelman, B.M. C/EBPalpha induces adipogenesis through PPARgamma: A unified pathway. Genes Dev. 2002, 16, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Caprio, M.; Antelmi, A.; Chetrite, G.; Muscat, A.; Mammi, C.; Marzolla, V.; Fabbri, A.; Zennaro, M.C.; Feve, B. Antiadipogenic effects of the mineralocorticoid receptor antagonist drospirenone: Potential implications for the treatment of metabolic syndrome. Endocrinology 2011, 152, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Urbanet, R.; Pilon, C.; Calcagno, A.; Peschechera, A.; Hubert, E.L.; Giacchetti, G.; Gomez-Sanchez, C.; Mulatero, P.; Toffanin, M.; Sonino, N.; et al. Analysis of insulin sensitivity in adipose tissue of patients with primary aldosteronism. J. Clin. Endocrinol. Metab. 2010, 95, 4037–4042. [Google Scholar] [CrossRef] [PubMed]
- Armani, A.; Marzolla, V.; Rosano, G.; Caprio, M. Mineralocorticoid vs glucocorticoid receptors: Solo players or team mates in the control of adipogenesis? Int. J. Obes. (Lond.) 2014, 38, 1580–1581. [Google Scholar] [CrossRef] [PubMed]
- Urbanet, R.; Nguyen Dinh Cat, A.; Feraco, A.; Venteclef, N.; El Mogrhabi, S.; Sierra-Ramos, C.; Alvarez de la Rosa, D.; Adler, G.K.; Quilliot, D.; Rossignol, P.; et al. Adipocyte mineralocorticoid receptor activation leads to metabolic syndrome and induction of prostaglandin D2 synthase. Hypertension 2015, 66, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Lavie, C.J.; Milani, R.V.; Ventura, H.O. Obesity and cardiovascular disease: Risk factor, paradox, and impact of weight loss. J. Am. Coll. Cardiol. 2009, 53, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Bochud, M.; Nussberger, J.; Bovet, P.; Maillard, M.R.; Elston, R.C.; Paccaud, F.; Shamlaye, C.; Burnier, M. Plasma aldosterone is independently associated with the metabolic syndrome. Hypertension 2006, 48, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Adler, G.K. Role of mineralocorticoid receptor in insulin resistance. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, A.; Garg, R.; Adler, G.K. Mineralocorticoid receptor antagonists and the metabolic syndrome. Curr. Hypertens. Rep. 2010, 12, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Min, S.H.; Kim, S.H.; Jeong, I.K.; Cho, H.C.; Jeong, J.O.; Lee, J.H.; Kang, H.J.; Kim, H.S.; Park, K.S.; Lim, S. Independent association of serum aldosterone level with metabolic syndrome and insulin resistance in korean adults. Korean Circ. J. 2018, 48, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Bjorntorp, P.; Rosmond, R. Obesity and cortisol. Nutrition 2000, 16, 924–936. [Google Scholar] [CrossRef]
- Kawarazaki, W.; Fujita, T. The role of aldosterone in obesity-related hypertension. Am. J. Hypertens. 2016, 29, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, D.A.; Sharma, K. The role of aldosteronism in causing obesity-related cardiovascular risk. Cardiol. Clin. 2010, 28, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Infante, M.; Armani, A.; Mammi, C.; Fabbri, A.; Caprio, M. Impact of adrenal steroids on regulation of adipose tissue. Compr. Physiol. 2017, 7, 1425–1447. [Google Scholar] [PubMed]
- Ehrhart-Bornstein, M.; Lamounier-Zepter, V.; Schraven, A.; Langenbach, J.; Willenberg, H.S.; Barthel, A.; Hauner, H.; McCann, S.M.; Scherbaum, W.A.; Bornstein, S.R. Human adipocytes secrete mineralocorticoid-releasing factors. Proc. Natl. Acad. Sci. USA 2003, 100, 14211–14216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzolla, V.; Armani, A.; Zennaro, M.C.; Cinti, F.; Mammi, C.; Fabbri, A.; Rosano, G.M.; Caprio, M. The role of the mineralocorticoid receptor in adipocyte biology and fat metabolism. Mol. Cell. Endocrinol. 2012, 350, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Huby, A.C.; Antonova, G.; Groenendyk, J.; Gomez-Sanchez, C.E.; Bollag, W.B.; Filosa, J.A.; Belin de Chantemele, E.J. Adipocyte-derived hormone leptin is a direct regulator of aldosterone secretion, which promotes endothelial dysfunction and cardiac fibrosis. Circulation 2015, 132, 2134–2145. [Google Scholar] [CrossRef] [PubMed]
- Diez, J. Arterial hypertension in patients with heart failure. Heart Fail. Clin. 2014, 10, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, D.; van Almen, G.C.; Van Aelst, L.N.; Van Cleemput, J.; Droogne, W.; Jin, Y.; Van de Werf, F.; Carmeliet, P.; Vanhaecke, J.; Papageorgiou, A.P.; et al. Matricellular proteins and matrix metalloproteinases mark the inflammatory and fibrotic response in human cardiac allograft rejection. Eur. Heart J. 2013, 34, 1930–1941. [Google Scholar] [CrossRef] [PubMed]
- Zia, A.A.; Kamalov, G.; Newman, K.P.; McGee, J.E.; Bhattacharya, S.K.; Ahokas, R.A.; Sun, Y.; Gerling, I.C.; Weber, K.T. From aldosteronism to oxidative stress: The role of excessive intracellular calcium accumulation. Hypertens. Res. 2010, 33, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Tsybouleva, N.; Zhang, L.; Chen, S.; Patel, R.; Lutucuta, S.; Nemoto, S.; DeFreitas, G.; Entman, M.; Carabello, B.A.; Roberts, R.; et al. Aldosterone, through novel signaling proteins, is a fundamental molecular bridge between the genetic defect and the cardiac phenotype of hypertrophic cardiomyopathy. Circulation 2004, 109, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Sowers, J.; Tuck, M.; Asp, N.D.; Sollars, E. Plasma aldosterone and corticosterone responses to adrenocorticotropin, angiotensin, potassium, and stress in spontaneously hypertensive rats. Endocrinology 1981, 108, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Iacobone, M.; Citton, M.; Viel, G.; Rossi, G.P.; Nitti, D. Approach to the surgical management of primary aldosteronism. Gland Surg. 2015, 4, 69–81. [Google Scholar] [PubMed]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized aldactone evaluation study investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M.; et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Latouche, C.; El Moghrabi, S.; Messaoudi, S.; Nguyen Dinh Cat, A.; Hernandez-Diaz, I.; Alvarez de la Rosa, D.; Perret, C.; Lopez Andres, N.; Rossignol, P.; Zannad, F.; et al. Neutrophil gelatinase-associated lipocalin is a novel mineralocorticoid target in the cardiovascular system. Hypertension 2012, 59, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Ott, K.M.; Mori, K.; Li, J.Y.; Kalandadze, A.; Cohen, D.J.; Devarajan, P.; Barasch, J. Dual action of neutrophil gelatinase-associated lipocalin. J. Am. Soc. Nephrol. 2007, 18, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Yang, H.; Chen, H.; Zhang, M.; Ma, Q. High expression of neutrophil gelatinase-associated lipocalin (NGAL) in the kidney proximal tubules of diabetic rats. Adv. Med. Sci. 2015, 60, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Hamzic, N.; Blomqvist, A.; Nilsberth, C. Immune-induced expression of lipocalin-2 in brain endothelial cells: Relationship with interleukin-6, cyclooxygenase-2 and the febrile response. J. Neuroendocrinol. 2013, 25, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Eilenberg, W.; Stojkovic, S.; Piechota-Polanczyk, A.; Kaun, C.; Rauscher, S.; Groger, M.; Klinger, M.; Wojta, J.; Neumayer, C.; Huk, I.; et al. Neutrophil gelatinase-associated lipocalin (NGAL) is associated with symptomatic carotid atherosclerosis and drives pro-inflammatory state in vitro. Eur. J. Vasc. Endovasc. Surg. 2016, 51, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, L.; Johnsen, A.H.; Sengelov, H.; Borregaard, N. Isolation and primary structure of ngal, a novel protein associated with human neutrophil gelatinase. J. Biol. Chem. 1993, 268, 10425–10432. [Google Scholar] [PubMed]
- Flo, T.H.; Smith, K.D.; Sato, S.; Rodriguez, D.J.; Holmes, M.A.; Strong, R.K.; Akira, S.; Aderem, A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 2004, 432, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Floderer, M.; Prchal-Murphy, M.; Vizzardelli, C. Dendritic cell-secreted lipocalin2 induces CD8+ T-cell apoptosis, contributes to t-cell priming and leads to a TH1 phenotype. PLoS ONE 2014, 9, e101881. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, P. Neutrophil gelatinase-associated lipocalin: A promising biomarker for human acute kidney injury. Biomark. Med. 2010, 4, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, V.M.; Damman, K.; Voors, A.A.; van der Wal, M.H.; Jaarsma, T.; van Veldhuisen, D.J.; Hillege, H.L. Prognostic value of plasma neutrophil gelatinase-associated lipocalin for mortality in patients with heart failure. Circ. Heart Fail. 2014, 7, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Li, H.; Fang, Q.; Jiang, S.; Zhang, L.; Zhang, J.; Hou, X.; Lu, J.; Bao, Y.; Xu, A.; et al. Elevated circulating lipocalin-2 levels independently predict incident cardiovascular events in men in a population-based cohort. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2457–2464. [Google Scholar] [CrossRef] [PubMed]
- Tarjus, A.; Martinez-Martinez, E.; Amador, C.; Latouche, C.; El Moghrabi, S.; Berger, T.; Mak, T.W.; Fay, R.; Farman, N.; Rossignol, P.; et al. Neutrophil gelatinase-associated lipocalin, a novel mineralocorticoid biotarget, mediates vascular profibrotic effects of mineralocorticoids. Hypertension 2015, 66, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martinez, E.; Buonafine, M.; Boukhalfa, I.; Ibarrola, J.; Fernandez-Celis, A.; Kolkhof, P.; Rossignol, P.; Girerd, N.; Mulder, P.; Lopez-Andres, N.; et al. Aldosterone target NGAL (neutrophil gelatinase-associated lipocalin) is involved in cardiac remodeling after myocardial infarction through NFκβ pathway. Hypertension 2017, 70, 1148–1156. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Li, Y.; Liu, M.; Li, Y.; Cong, B. Renal neutrophil gelatinase associated lipocalin expression in lipopolysaccharide-induced acute kidney injury in the rat. BMC Nephrol. 2012, 13, 25. [Google Scholar] [CrossRef] [PubMed]
- Shashidharamurthy, R.; Machiah, D.; Aitken, J.D.; Putty, K.; Srinivasan, G.; Chassaing, B.; Parkos, C.A.; Selvaraj, P.; Vijay-Kumar, M. Differential role of lipocalin 2 during immune complex-mediated acute and chronic inflammation in mice. Arthritis Rheum. 2013, 65, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Buonafine, M.; Martinez-Martinez, E.; Amador, C.; Gravez, B.; Ibarrola, J.; Fernandez-Celis, A.; El Moghrabi, S.; Rossignol, P.; Lopez-Andres, N.; Jaisser, F. Neutrophil gelatinase-associated lipocalin from immune cells is mandatory for aldosterone-induced cardiac remodeling and inflammation. J. Mol. Cell. Cardiol. 2018, 115, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Azibani, F.; Benard, L.; Schlossarek, S.; Merval, R.; Tournoux, F.; Fazal, L.; Polidano, E.; Launay, J.M.; Carrier, L.; Chatziantoniou, C.; et al. Aldosterone inhibits antifibrotic factors in mouse hypertensive heart. Hypertension 2012, 59, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martinez, E.; Jurado-Lopez, R.; Valero-Munoz, M.; Bartolome, M.V.; Ballesteros, S.; Luaces, M.; Briones, A.M.; Lopez-Andres, N.; Miana, M.; Cachofeiro, V. Leptin induces cardiac fibrosis through galectin-3, mtor and oxidative stress: Potential role in obesity. J. Hypertens. 2014, 32, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, V.L.; Hulsmans, S.; Griffioen, A.W. The galectin profile of the endothelium: Altered expression and localization in activated and tumor endothelial cells. Am. J. Pathol. 2008, 172, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Papaspyridonos, M.; McNeill, E.; de Bono, J.P.; Smith, A.; Burnand, K.G.; Channon, K.M.; Greaves, D.R. Galectin-3 is an amplifier of inflammation in atherosclerotic plaque progression through macrophage activation and monocyte chemoattraction. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Calvier, L.; Miana, M.; Reboul, P.; Cachofeiro, V.; Martinez-Martinez, E.; de Boer, R.A.; Poirier, F.; Lacolley, P.; Zannad, F.; Rossignol, P.; et al. Galectin-3 mediates aldosterone-induced vascular fibrosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Calvier, L.; Martinez-Martinez, E.; Miana, M.; Cachofeiro, V.; Rousseau, E.; Sadaba, J.R.; Zannad, F.; Rossignol, P.; Lopez-Andres, N. The impact of galectin-3 inhibition on aldosterone-induced cardiac and renal injuries. JACC Heart Fail. 2015, 3, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Glinsky, V.V.; Raz, A. Modified citrus pectin anti-metastatic properties: One bullet, multiple targets. Carbohydr. Res. 2009, 344, 1788–1791. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martinez, E.; Calvier, L.; Fernandez-Celis, A.; Rousseau, E.; Jurado-Lopez, R.; Rossoni, L.V.; Jaisser, F.; Zannad, F.; Rossignol, P.; Cachofeiro, V.; et al. Galectin-3 blockade inhibits cardiac inflammation and fibrosis in experimental hyperaldosteronism and hypertension. Hypertension 2015, 66, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Andres, N.; Rossignol, P.; Iraqi, W.; Fay, R.; Nuee, J.; Ghio, S.; Cleland, J.G.; Zannad, F.; Lacolley, P. Association of galectin-3 and fibrosis markers with long-term cardiovascular outcomes in patients with heart failure, left ventricular dysfunction, and dyssynchrony: Insights from the CARE-HF (cardiac resynchronization in heart failure) trial. Eur. J. Heart Fail. 2012, 14, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Maiolino, G.; Rossitto, G.; Pedon, L.; Cesari, M.; Frigo, A.C.; Azzolini, M.; Plebani, M.; Rossi, G.P. Galectin-3 predicts long-term cardiovascular death in high-risk patients with coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martinez, E.; Lopez-Andres, N.; Jurado-Lopez, R.; Rousseau, E.; Bartolome, M.V.; Fernandez-Celis, A.; Rossignol, P.; Islas, F.; Antequera, A.; Prieto, S.; et al. Galectin-3 participates in cardiovascular remodeling associated with obesity. Hypertension 2015, 66, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Hirata, A.; Maeda, N.; Nakatsuji, H.; Hiuge-Shimizu, A.; Okada, T.; Funahashi, T.; Shimomura, I. Contribution of glucocorticoid-mineralocorticoid receptor pathway on the obesity-related adipocyte dysfunction. Biochem. Biophys. Res. Commun. 2012, 419, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Kenmochi, H.; Miyashita, Y.; Sasaki, M.; Ojima, M.; Sasahara, M.; Koya, D.; Tsuneki, H.; Sasaoka, T. Spironolactone improves glucose and lipid metabolism by ameliorating hepatic steatosis and inflammation and suppressing enhanced gluconeogenesis induced by high-fat and high-fructose diet. Endocrinology 2010, 151, 2040–2049. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martinez, E.; Calvier, L.; Rossignol, P.; Rousseau, E.; Fernandez-Celis, A.; Jurado-Lopez, R.; Laville, M.; Cachofeiro, V.; Lopez-Andres, N. Galectin-3 inhibition prevents adipose tissue remodelling in obesity. Int. J. Obes. (Lond.) 2016, 40, 1034–1038. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; Malavazos, A.E.; Corsi, M.M. Epicardial fat: From the biomolecular aspects to the clinical practice. Int. J. Biochem. Cell Biol. 2011, 43, 1651–1654. [Google Scholar] [CrossRef] [PubMed]
- Rahmouni, K. Leptin-induced sympathetic nerve activation: Signaling mechanisms and cardiovascular consequences in obesity. Curr. Hypertens. Rev. 2010, 6, 104–209. [Google Scholar] [CrossRef] [PubMed]
- Rahmouni, K.; Morgan, D.A.; Morgan, G.M.; Mark, A.L.; Haynes, W.G. Role of selective leptin resistance in diet-induced obesity hypertension. Diabetes 2005, 54, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Schram, K.; Sweeney, G. Implications of myocardial matrix remodeling by adipokines in obesity-related heart failure. Trends Cardiovasc. Med. 2008, 18, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, J.; Hyun, J.W.; Park, J.W.; Joo, H.G.; Shin, T. Expression and immunohistochemical localization of galectin-3 in various mouse tissues. Cell Biol. Int. 2007, 31, 655–662. [Google Scholar] [CrossRef] [PubMed]
- De Boer, R.A.; van Veldhuisen, D.J.; Gansevoort, R.T.; Muller Kobold, A.C.; van Gilst, W.H.; Hillege, H.L.; Bakker, S.J.; van der Harst, P. The fibrosis marker galectin-3 and outcome in the general population. J. Intern. Med. 2012, 272, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Weigert, J.; Neumeier, M.; Wanninger, J.; Bauer, S.; Farkas, S.; Scherer, M.N.; Schnitzbauer, A.; Schaffler, A.; Aslanidis, C.; Scholmerich, J.; et al. Serum galectin-3 is elevated in obesity and negatively correlates with glycosylated hemoglobin in type 2 diabetes. J. Clin. Endocrinol. Metab. 2010, 95, 1404–1411. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Chou, C.H.; Wu, X.M.; Chang, Y.Y.; Hung, C.S.; Chen, Y.H.; Tzeng, Y.L.; Wu, V.C.; Ho, Y.L.; Hsieh, F.J.; et al. Aldosterone induced galectin-3 secretion in vitro and in vivo: From cells to humans. PLoS ONE 2014, 9, e95254. [Google Scholar] [CrossRef] [PubMed]
- Jowsey, I.R.; Murdock, P.R.; Moore, G.B.; Murphy, G.J.; Smith, S.A.; Hayes, J.D. Prostaglandin D2 synthase enzymes and PPARγ are co-expressed in mouse 3T3-L1 adipocytes and human tissues. Prostaglandins Other Lipid Mediat. 2003, 70, 267–284. [Google Scholar] [CrossRef]
- Ragolia, L.; Palaia, T.; Hall, C.E.; Maesaka, J.K.; Eguchi, N.; Urade, Y. Accelerated Glucose Intolerance, Nephropathy, and Atherosclerosis in Prostaglandin D2 Synthase Knock-out Mice. J. Biol. Chem. 2005, 280, 29946–29955. [Google Scholar] [CrossRef] [PubMed]
- Ragolia, L.; Hall, C.E.; Palaia, T. Lipocalin-type prostaglandin D2 synthase stimulates glucose transport via enhanced GLUT4 translocation. Prostaglandins Other Lipid Mediat. 2008, 87, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.A.; Hossain, M.S.; Rahman, M.S.; Nishimura, K.; Jisaka, M.; Nagaya, T.; Shono, F.; Yokota, K. Sustained expression of lipocalin-type prostaglandin D synthase in the antisense direction positively regulates adipogenesis in cloned cultured preadipocytes. Biochem. Biophys. Res. Commun. 2011, 411, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Virtue, S.; Masoodi, M.; Velagapudi, V.; Tan, C.Y.; Dale, M.; Suorti, T.; Slawik, M.; Blount, M.; Burling, K.; Campbell, M.; et al. Lipocalin prostaglandin D synthase and PPARγ2 coordinate to regulate carbohydrate and lipid metabolism in vivo. PLoS ONE 2012, 7, e39512. [Google Scholar] [CrossRef] [PubMed]
- Emdin, M.; Fatini, C.; Mirizzi, G.; Poletti, R.; Borrelli, C.; Prontera, C.; Latini, R.; Passino, C.; Clerico, A.; Vergaro, G. Biomarkers of activation of renin-angiotensin-aldosterone system in heart failure: How useful, how feasible? Clin. Chim. Acta 2015, 443, 85–93. [Google Scholar] [CrossRef] [PubMed]
- De Buyzere, M.; Gruson, D. Biomarkers in heart failure. Clin. Chim. Acta 2015, 443, 1–2. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorini, S.; Marzolla, V.; Mammi, C.; Armani, A.; Caprio, M. Mineralocorticoid Receptor and Aldosterone-Related Biomarkers of End-Organ Damage in Cardiometabolic Disease. Biomolecules 2018, 8, 96. https://doi.org/10.3390/biom8030096
Gorini S, Marzolla V, Mammi C, Armani A, Caprio M. Mineralocorticoid Receptor and Aldosterone-Related Biomarkers of End-Organ Damage in Cardiometabolic Disease. Biomolecules. 2018; 8(3):96. https://doi.org/10.3390/biom8030096
Chicago/Turabian StyleGorini, Stefania, Vincenzo Marzolla, Caterina Mammi, Andrea Armani, and Massimiliano Caprio. 2018. "Mineralocorticoid Receptor and Aldosterone-Related Biomarkers of End-Organ Damage in Cardiometabolic Disease" Biomolecules 8, no. 3: 96. https://doi.org/10.3390/biom8030096
APA StyleGorini, S., Marzolla, V., Mammi, C., Armani, A., & Caprio, M. (2018). Mineralocorticoid Receptor and Aldosterone-Related Biomarkers of End-Organ Damage in Cardiometabolic Disease. Biomolecules, 8(3), 96. https://doi.org/10.3390/biom8030096